
De-Palmitoylation of Tissue Factor Regulates Its Activity, Phosphorylation and Cellular Functions

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Site-Directed Mutagenesis
2.2. Cell Culture, Transfection and Cellular Activation
2.3. Cell Proliferation and Apoptosis Assays
2.4. Determination of Microvesicle Density and TF Antigen
2.5. Cell-Based Factor Xa-Generation Assay
2.6. Factor VIIa Binding Assay
2.7. Immunoprecipitation of TF and Western Blot Analysis of TF Phosphorylation
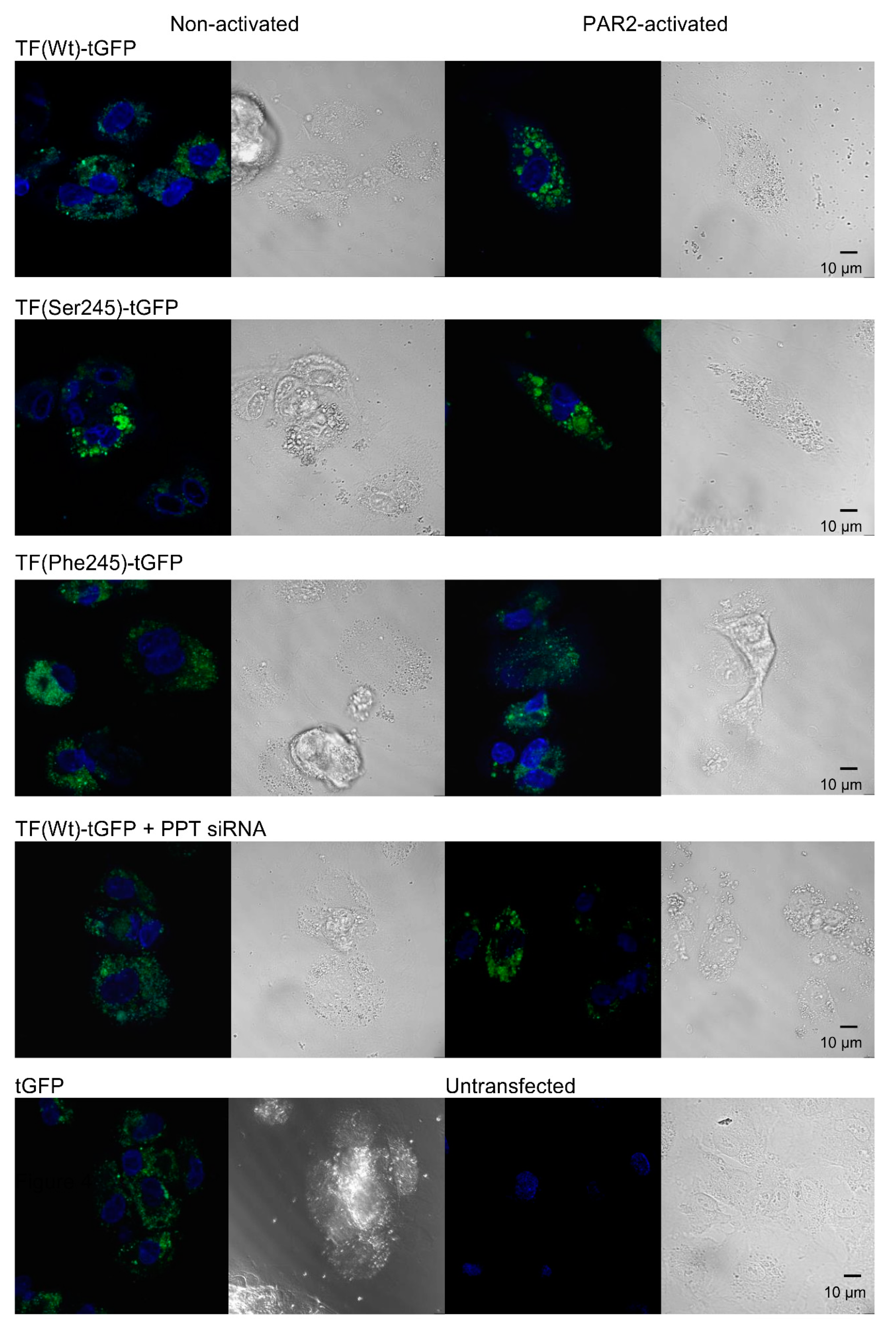
2.8. Confocal Microscopy
2.9. Statistical Analysis
3. Results
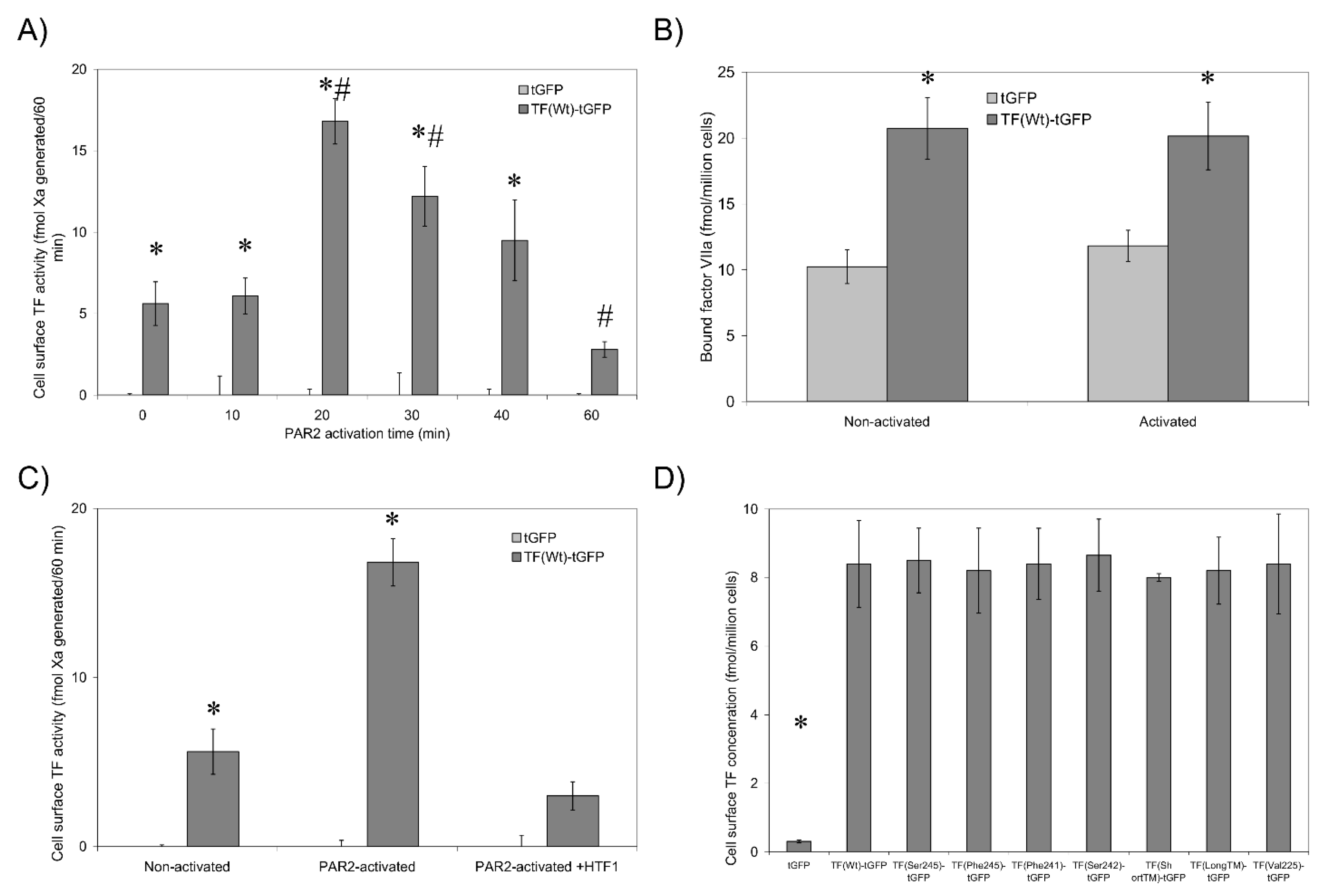
3.1. Validation of the fVIIa-HRP Conjugation and fVIIa Activity
3.2. Non-Active/Encrypted TF Is Capable of Binding fVIIa without Increasing Procoagulant Activity
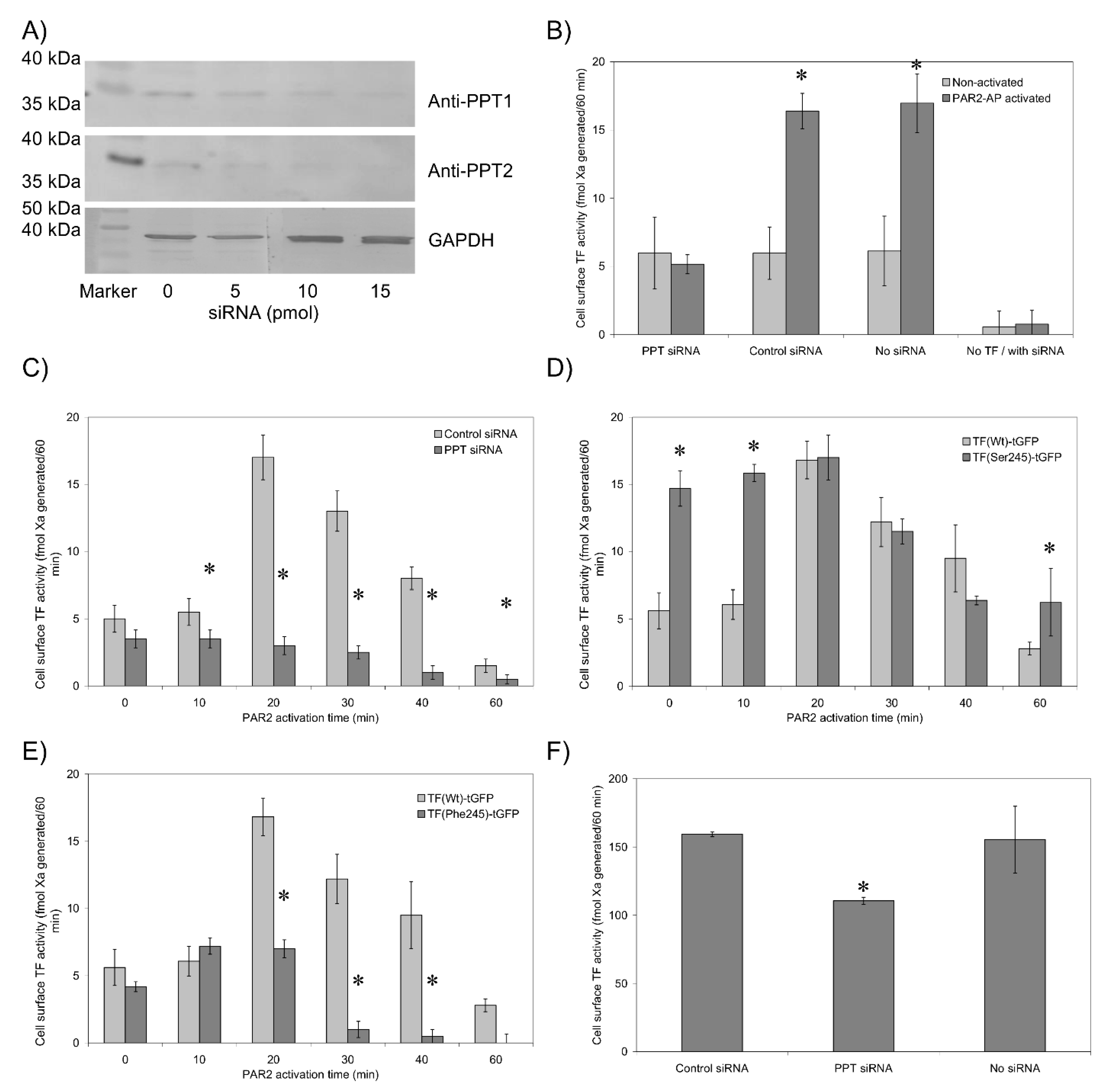
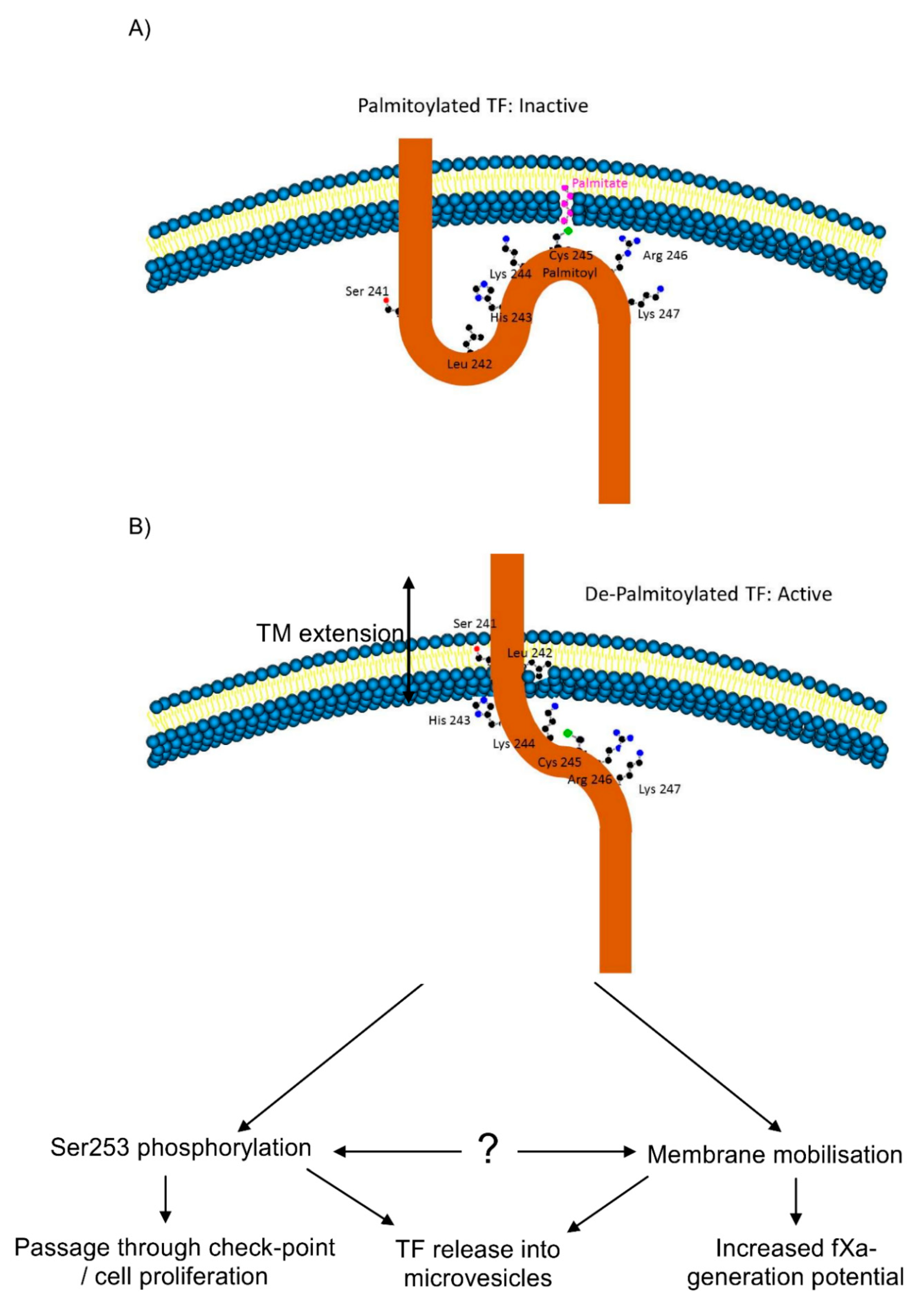
3.3. De-Palmitoylation of Cys245 Is Required for TF-fVIIa Activity but Not Complex Formation
3.4. De-Palmitoylation of Cys245 Promotes the Aggregation of TF on the Cell Membrane
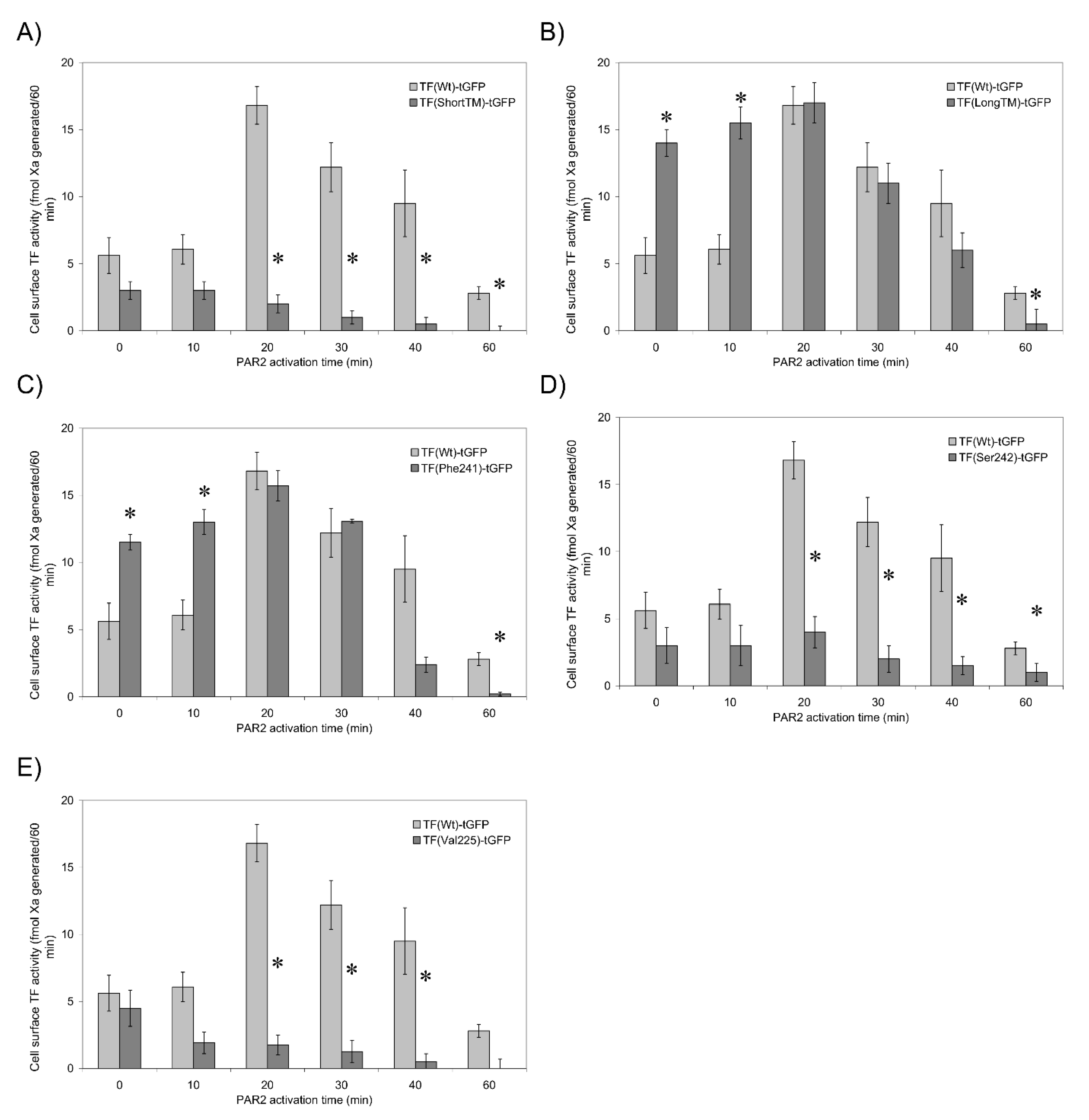
3.5. TF Activity Is Regulated by the Length and Orientation of the Transmembrane Domain
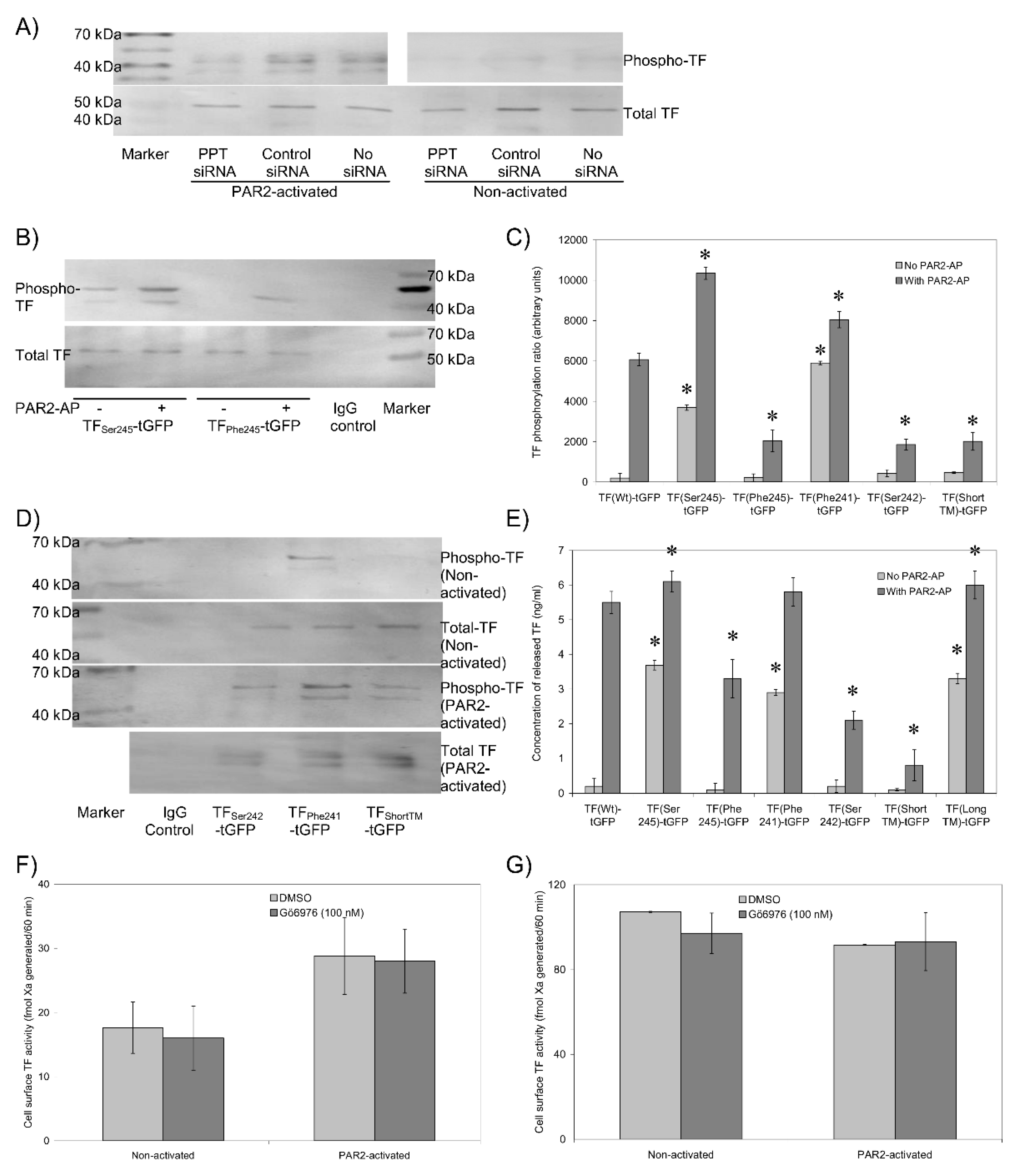
3.6. The Transmembrane Domain Regulates TF Phosphorylation at Ser253 and Incorporation into Microvesicles
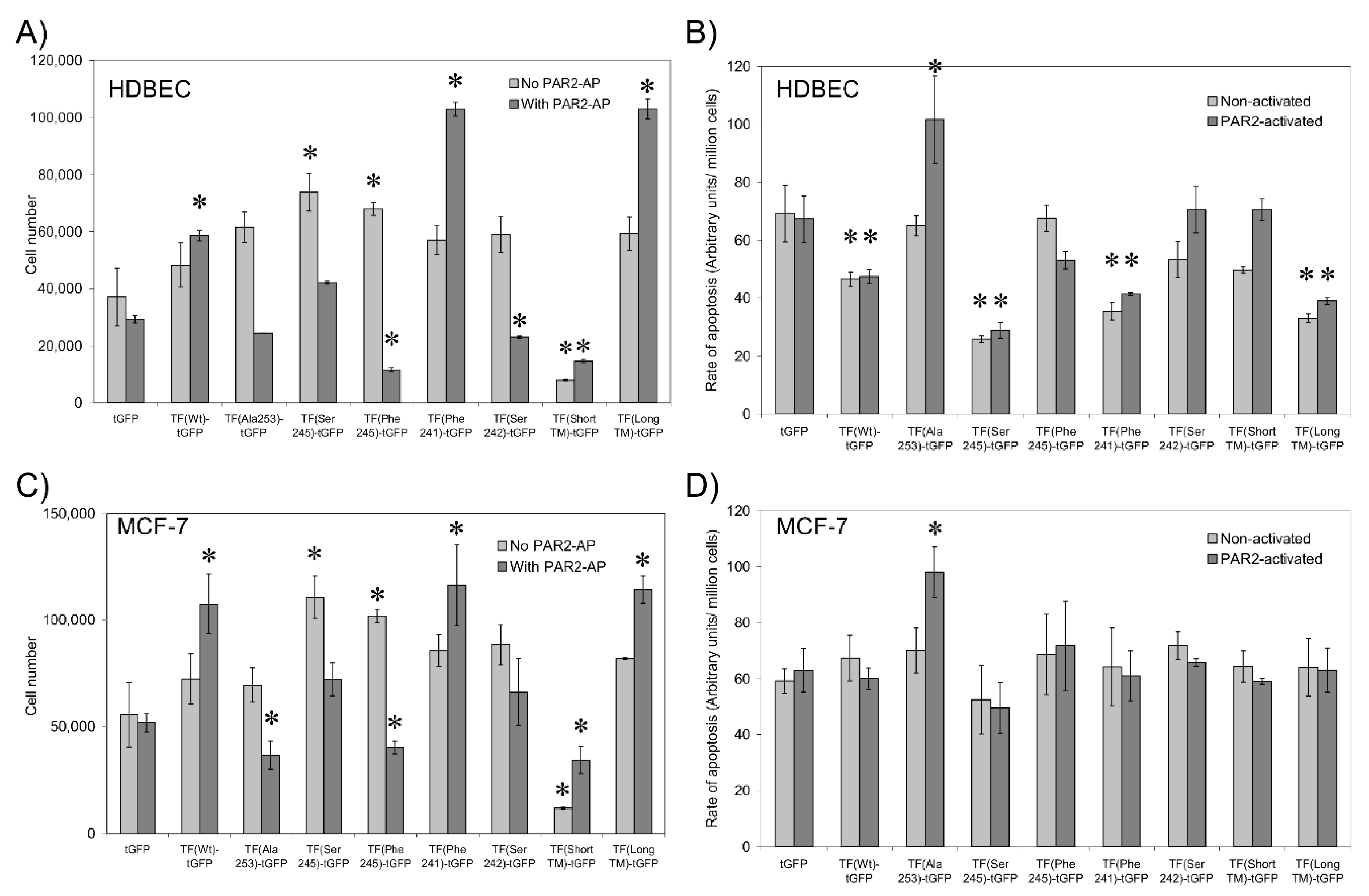
3.7. Sequential De-Palmitoylation and Phosphorylation of TF Can Co-Ordinate Cell Proliferation and Apoptosis Following PAR2 Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Branchford, B.R.; Carpenter, S.L. The Role of Inflammation in Venous Thromboembolism. Front. Pediatr. 2018, 6, 142. [Google Scholar] [CrossRef] [PubMed]
- Kothari, H.; Pendurthi, U.R.; Rao, L.V. Analysis of tissue factor expression in various cell model systems: Cryptic vs. active. J. Thromb. Haemost. 2013, 11, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Bach, R.R. Tissue factor encryption. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Butenas, S.; Amblo-Krudysz, J.; Mann, K.G. Posttranslational modifications of tissue factor. Front. Biosci. 2012, 4, 381–391. [Google Scholar] [CrossRef]
- Egorina, E.M.; Sovershaev, M.A.; Osterud, B. Regulation of tissue factor procoagulant activity by post-translational modifications. Thromb. Res. 2008, 122, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S.; Monroe, D.M.; Roberts, H.R.; Hoffman, M.R. Tissue factor de-encryption: Ionophore treatment induces changes in tissue factor activity by phosphatidylserine-dependent and -independent mechanisms. Blood Coagul. Fibrinolysis 1999, 10, 201–210. [Google Scholar] [CrossRef]
- Chen, V.M.; Hogg, P.J. Encryption and decryption of tissue factor. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 277–284. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S.; Kon, R.H.; Monroe, D.M.; Ezban, M.; Roberts, H.R.; Hoffman, M. Deencryption of cellular tissue factor is independent of its cytoplasmic domain. Biochem. Biophys. Res. Commun. 2000, 272, 332–336. [Google Scholar] [CrossRef]
- Carson, S.D.; Bromberg, M.E. Tissue factor encryption/de-encryption is not altered in the absence of the cytoplasmic domain. Thromb. Haemost. 2000, 84, 657–663. [Google Scholar]
- Dorfleutner, A.; Ruf, W. Regulation of tissue factor cytoplasmic domain phosphorylation by palmitoylation. Blood 2003, 102, 3998–4005. [Google Scholar] [CrossRef] [Green Version]
- Zioncheck, T.F.; Roy, S.; Vehar, G.A. The cytoplasmic domain of tissue factor is phosphorylated by a protein kinase C-dependent mechanism. J. Biol. Chem. 1992, 267, 3561–3564. [Google Scholar] [CrossRef]
- Collier, M.E.W.; Ettelaie, C. Regulation of the incorporation of tissue factor into microparticles by serine phosphorylation of the cytoplasmic domain of tissue factor. J. Biol. Chem. 2011, 286, 11977–11984. [Google Scholar] [CrossRef] [Green Version]
- Collier, M.E.; Maraveyas, A.; Ettelaie, C. Filamin-A is required for the incorporation of tissue factor into cell-derived microvesicles. Thromb. Haemost. 2014, 111, 647–655. [Google Scholar] [PubMed] [Green Version]
- Collier, M.E.W.; Ettelaie, C.; Goult, B.T.; Maraveyas, A.; Goodall, A.H. Investigation of the Filamin A-Dependent Mechanisms of Tissue Factor Incorporation into Microvesicles. Thromb. Haemost. 2017, 117, 2034–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizume, S.; Ito, S.; Yoshioka, Y.; Kanayama, T.; Nakamura, Y.; Yoshihara, M.; Yamada, R.; Ochiya, T.; Ruf, W.; Miyagi, E.; et al. High-level secretion of tissue factor-rich extracellular vesicles from ovarian cancer cells mediated by filamin-A and protease-activated receptors. Thromb. Haemost. 2016, 115, 299–310. [Google Scholar] [PubMed] [Green Version]
- Rothmeier, A.S.; Marchese, P.; Petrich, B.G.; Furlan-Freguia, C.; Ginsberg, M.H.; Ruggeri, Z.M.; Ruf, W. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. J. Clin. Investig. 2015, 125, 1471–1484. [Google Scholar] [CrossRef] [Green Version]
- Bach, R.R.; Moldow, C.F. Mechanism of tissue factor activation on HL-60 cells. Blood 1997, 89, 3270–3276. [Google Scholar] [CrossRef] [PubMed]
- Bach, R.; Konigsberg, W.H.; Nemerson, Y. Human tissue factor contains thioester-linked palmitate and stearate on the cytoplasmic half-cystine. Biochemistry 1988, 27, 4227–4231. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; López, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Böing, A.N.; Hau, C.M.; Hajji, N.; Ruf, W.; Sturk, A.; Nieuwland, R. Tissue factor coagulant activity is regulated by the plasma membrane microenvironment. Thromb. Hasmost. 2018, 118, 990–1000. [Google Scholar] [CrossRef]
- Mandal, S.K.; Iakhiaev, A.; Pendurthi, U.R.; Rao, L.V. Acute cholesterol depletion impairs functional expression of tissue factor in fibroblasts: Modulation of tissue factor activity by membrane cholesterol. Blood 2005, 105, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, D.J.; Page, K.L.; Tetzloff, T.A. Lipid rafts are necessary for tonic inhibition of cellular tissue factor procoagulant activity. Blood 2004, 103, 3038–3044. [Google Scholar] [CrossRef]
- Ettelaie, C.; Collier, M.E.; Featherby, S.; Greenman, J.; Maraveyas, A. Peptidyl-prolyl isomerase 1 (Pin1) preserves the phosphorylation state of tissue factor and prolongs its release within microvesicles. Biochim. Biophys. Acta 2017, 1865, 12–24. [Google Scholar] [CrossRef]
- Ettelaie, C.; Collier, M.E.; Featherby, S.; Greenman, J.; Maraveyas, A. Oligoubiquitination of tissue factor on Lys255 promotes Ser253-dephosphorylation, terminates TF release. Biochim. Biophys. Acta 2016, 1863, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Ettelaie, C.; Collier, M.E.; Mei, M.P.; Xiao, Y.P.; Maraveyas, A. Enhanced binding of tissue factor-microparticles to collagen-IV and fibronectin leads to increased tissue factor activity in vitro. Thromb. Haemost. 2013, 109, 61–71. [Google Scholar] [PubMed]
- Bonnekoh, B.; Wevers, A.; Jugert, F.; Merk, H.; Mahrle, F. Colorimetric growth assay for epidermal cell cultures by their crystal violet binding capacity. Arch. Dermatol. Res. 1989, 281, 487–490. [Google Scholar] [CrossRef] [PubMed]
- ElKeeb, A.M.; Collier, M.E.; Maraveyas, A.; Ettelaie, C. Accumulation of tissue factor in endothelial cells induces cell apoptosis, mediated through p38 and p53 activation. Thromb. Haemost. 2015, 114, 364–378. [Google Scholar]
- Madkhali, Y.; Featherby, S.; Collier, M.E.W.; Maraveyas, A.; Greenman, J.; Ettelaie, C. The ratio of factor VIIa: Tissue factor content within microvesicles determines the differential influence on endothelial cells. TH Open 2019, 3, 2019e132. [Google Scholar]
- Ettelaie, C.; Collier, M.E.; Maraveyas, A.; Ettelaie, R. Characterization of physical properties of tissue factor-containing microvesicles and a comparison of ultracentrifuge-based recovery procedures. J. Extracell. Vesicles 2014, 3, 23592. [Google Scholar] [CrossRef]
- Collier, M.E.; Mei, P.M.; Xiao, Y.P.; Maraveyas, A.; Ettelaie, C. The uptake of tumour cell-derived microparticles by microvascular endothelial cells results in the recycling of tissue factor to the cell surface with enhanced activity, in vitro. Thromb. Haemost. 2013, 110, 966–976. [Google Scholar] [PubMed] [Green Version]
- Lin, D.T.; Conibear, E. Enzymatic protein depalmitoylation by acyl protein thioesterases. Biochem. Soc. Trans. 2015, 43, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Blaskovic, S.; Blanc, M.; van der Goot, F.G. What does S-palmitoylation do to membrane proteins? FEBS J. 2013, 280, 2766–2774. [Google Scholar] [CrossRef] [Green Version]
- Abrami, L.; Kunz, B.; Iacovache, I.; van der Goot, F.G. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 5384–5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, M.; Nagaraj, R. Interaction of peptides corresponding to fatty acylation sites in proteins with model membranes. J. Biol. Chem. 1995, 270, 16749–16755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorent, J.H.; Diaz-Rohrer, B.; Lin, X.; Spring, K.; Gorfe, A.A.; Levental, K.R.; Levental, I. Structural determinants and functional consequences of protein affinity for membrane rafts. Nat. Commun. 2018, 8, 1219. [Google Scholar] [CrossRef]
- Awasthi, V.; Mandal, S.K.; Papanna, V.; Rao, L.V.; Pendurthi, U.R. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1447–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.T.; Conibear, E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 2015, 4, e11306. [Google Scholar] [CrossRef]
- Yeh, D.; Duncan, J.; Yamashita, S.; Michel, T. Depalmitoylation of Endothelial Nitric-oxide Synthase by Acyl-protein Thioesterase 1 Is Potentiated by Ca2+-Calmodulin. J. Biol. Chem. 1999, 274, 33148–33154. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.A.; Gilman, A.G. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS). J. Biol. Chem. 1998, 273, 15830–15837. [Google Scholar] [CrossRef] [Green Version]
- Tomatis, V.M.; Trenchi, A.; Gomez, G.A.; Daniotti, J.L. Acyl-protein thioesterase 2 catalyzes the deacylation of peripheral membrane-associated GAP-43. PLoS ONE 2010, 5, e15045. [Google Scholar] [CrossRef] [Green Version]
- Segal-Salto, M.; Sapir, T.; Reiner, O. Reversible Cysteine Acylation Regulates the Activity of Human Palmitoyl-Protein Thioesterase 1 (PPT1). PLoS ONE 2016, 11, e0146466. [Google Scholar] [CrossRef] [Green Version]
- Hellsten, E.; Vesa, J.; Olkkonen, V.M.; Jalanko, A.; Peltonen, L. Human palmitoyl protein thioesterase: Evidence for lysosomal targeting of the enzyme and disturbed cellular routing in infantile neuronal ceroid lipofuscinosis. EMBO J. 1996, 15, 5240–5245. [Google Scholar] [CrossRef]
- Sevinsky, J.R.; Rao, L.V.; Ruf, W. Ligand-induced protease receptor translocation into caveolae: A mechanism for regulating cell surface proteolysis of the tissue factor-dependent coagulation pathway. J. Cell Biol. 1996, 133, 293–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, W.L.; Lee, M.J.; Cummins, T.D.; Schultz, D.J.; Powell, D.W. Proteomic and functional characterisation of platelet microparticle size classes. Thromb. Haemost. 2009, 102, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Davizon, P.; Munday, A.D.; López, J.A. Tissue factor, lipid rafts, and microparticles. Semin. Thromb. Hemost. 2010, 36, 857–864. [Google Scholar] [CrossRef]
- Lu, J.Y.; Verkruyse, L.A.; Hofmann, S.L. Lipid thioesters derived from acylated proteins accumulate in infantile neuronal ceroid lipofuscinosis: Correction of the defect in lymphoblasts by recombinant palmitoyl-protein thioesterase. Proc. Natl. Acad. Sci. USA 1996, 93, 10046–10050. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Soyombo, A.A.; Atashband, A.; Wisniewski, K.E.; Shelton, J.M.; Richardson, J.A.; Hammer, R.E.; Hofmann, S.L. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 13566–13571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.K.; Becerra, C.H.; Yi, W.; Lu, J.Y.; Siakotos, A.N.; Wisniewski, K.E.; Hofmann, S.L. Molecular genetics of palmitoyl-protein thioesterase deficiency in the U.S. J. Clin. Investig. 1998, 102, 361–370. [Google Scholar] [CrossRef]
- Ladygina, N.; Martin, B.R.; Altman, A. Dynamic palmitoylation and the role of DHHC proteins in T cell activation and anergy. Adv. Immunol. 2011, 109, 1–44. [Google Scholar]
- Wang, R.; Borazjani, A.; Matthews, A.T.; Mangum, L.C.; Edelmann, M.J.; Ross, M.K. Identification of palmitoyl protein thioesterase 1 in human THP1 monocytes and macrophages and characterization of unique biochemical activities for this enzyme. Biochemistry 2013, 52, 7559–7574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardy, C.; Sabourdy, F.; Garcia, V.; Jalanko, A.; Therville, N.; Levade, T.; Andrieu-Abadie, N. Palmitoyl protein thioesterase 1 modulates tumor necrosis factor alpha-induced apoptosis. Biochim. Biophys. Acta 2009, 1793, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Ojha, R.; Noguera-Ortega, E.; Rebecca, V.W.; Attanasio, J.; Liu, S.; Piao, S.; Lee, J.J.; Nicastri, M.C.; Harper, S.L.; et al. PPT1 inhibition enhances the antitumor activity of anti-PD-1 antibody in melanoma. JCI Insight 2020, 5, e133225. [Google Scholar] [CrossRef]
- Blom, T.; Schmiedt, M.L.; Wong, A.M.; Kyttälä, A.; Soronen, J.; Jauhiainen, M.; Tyynelä, J.; Cooper, J.D.; Jalanko, A. Exacerbated neuronal ceroid lipofuscinosis phenotype in Cln1/5 double-knockout mice. Dis. Model. Mech. 2013, 6, 342–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, W. Roles of factor Xa beyond coagulation. J. Thromb. Thrombolysis 2021. [Google Scholar] [CrossRef]
- Butenas, S. Tissue factor structure and function. Scientifica 2012, 2012, 964862. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, M.A.; Greenman, J.; Maraveyas, A.; Ettelaie, C. Activation of PAR2 by tissue factor induces the release of the PTEN from MAGI proteins and regulates PTEN and Akt activities. Sci. Rep. 2020, 10, 20908. [Google Scholar] [CrossRef] [PubMed]
- Ethaeb, A.M.; Mohammad, M.A.; Madkhali, Y.; Featherby, S.; Maraveyas, A.; Greenman, J.; Ettelaie, C. Accumulation of tissue factor in endothelial cells promotes cellular apoptosis through over-activation of Src1 and involves β1-integrin signalling. Apoptosis 2020, 25, 29–41. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Mutation | Primer Set |
|---|---|---|
| pCMV-Ac-TFSer245-tGFP | Cys245→Ser | 5′-TCTACACAAGAGTAGAAAGGCAG |
| 5′-GATATAGCCAGGATGATG | ||
| pCMV-Ac-TFPhe245-tGFP | Cys245→Phe | 5′-CTACACAAGTTTAGAAAGGCAG |
| 5′-AGATATAGCCAGGATGATG | ||
| pCMV-Ac-TFPhe241-tGFP | Ser241→Phe | 5′-GTGGCTATATTTCTACACAAGTG |
| 5′GATGATGACAAGGATGATG | ||
| pCMV-Ac-TFSer242-tGFP | Leu242→Ser | 5′-GGCTATATCTTCACACAAGTGTAG |
| 5′-AGGATGATGACAAGGATG | ||
| pCMV-Ac-TFShortTM-tGFP | Ser241-Leu242-del | 5′-CACAAGTGTAGAAAGGCAG |
| 5′-TATAGCCAGGATGATGAC | ||
| pCMV-Ac-TFLongTM-tGFP | Ser243-Leu244-add | 5′-TCCCTGCACAAGTGTAGAAAGGCAG |
| 5′-TAGAGATATAGCCAGGATG | ||
| pCMV-Ac-TFVal225-tGFP | Gly225→Val | 5′-TACATCATTGTAGCTGTGGTATTTG |
| 5′-GAATATTTCTCTGAATTCCC |
| TF Variant | Non-Activated (Fmol/Million Cells) | PAR2-Activated (Fmol/Million Cells) |
|---|---|---|
| tGFP | 11.81 ± 1.19 * | 10.2 ± 1.28 * |
| TFWt-tGFP | 20.15 ± 2.56 | 20.7 ± 2.33 |
| TFSer245-tGFP + PPT siRNA | 19.33 ± 1.03 | 19.51 ± 0.92 |
| TFSer245-tGFP + control siRNA | 19.70 ± 0.64 | 20.01 ± 0.38 |
| TFSer245-tGFP | 18.29 ± 2.12 | 19.6 ± 0.99 |
| TFPhe245-tGFP | 17.04 ± 1.59 | 14.4 ± 1.01 * |
| TFShortTM-tGFP | 18.13 ± 4.04 | 8.8 ± 2.87 * |
| TFLongTM-tGFP | 20.80 ± 1.50 | 21.0 ± 1.51 |
| TFPhe241-tGFP | 20.00 ± 1.00 | 20.0 ± 0.82 |
| TFSer242-tGFP | 20.00 ± 2.00 | 19.0 ± 2.50 |
| TFVal225-tGFP | 17.00 ± 2.05 | 12.0 ± 3.10 * |
| Sample | Non-Activated (µm) | PAR2-Activated (µm) |
|---|---|---|
| tGFP | 0.09 ± 0.03 | 0.09 ± 0.03 |
| TFWt-tGFP | 0.15 ± 0.06 | 0.43 ± 0.11 |
| TFSer245-tGFP | 0.41 ± 0.10 | 0.53 ± 0.12 |
| TFPhe245-tGFP | 0.09 ± 0.03 | 0.12 ± 0.05 |
| TFWt-tGFP +PPT siRNA | 0.12 ± 0.04 | 0.12 ± 0.04 |
| Plasmid | Purpose | Observation | Possible Explanation |
|---|---|---|---|
| pCMV-Ac-TFSer245-tGFP | Prevent palmitoylation | ↑TF activation | TF can mobilise within the membrane |
| ↑Proliferation | |||
| (non-activated) | |||
| pCMV-Ac-TFPhe245-tGFP | Mimic palmitoylation | ↓TF activation | TF cannot mobilise or be released–Increased membrane thickness hampers fVIIa binding |
| ↓Proliferation | |||
| ↓fVIIa binding | |||
| (activated) | |||
| pCMV-Ac-TFPhe241-tGFP | Increase hydrophobicity of TMD | ↑TF activation | Facilitates mobilisation to thicker membrane regions |
| ↑Proliferation | |||
| (activated) | |||
| pCMV-Ac-TFSer242-tGFP | Decrease hydrophobicity of TMD | ↓TF activation | Hampers mobilisation to thicker membrane regions |
| ↓Proliferation | |||
| (activated) | |||
| pCMV-Ac-TFShortTM-tGFP | Shorten TMD | ↓TF activation | Cannot mobilise to to thicker membrane regions |
| ↓Proliferation | |||
| ↓fVIIa binding | |||
| pCMV-Ac-TFLongTM-tGFP | Lengthen TMD | ↑TF activation | Facilitates mobilisation to thicker membrane regions |
| ↑Proliferation | |||
| (activated) | |||
| pCMV-Ac-TFVal225-tGFP | Reduce TMD flexibility | ↓TF activation | Cannot accommodate correct structure for interaction with fVIIa |
| ↓fVIIa binding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ettelaie, C.; Featherby, S.; Rondon, A.M.R.; Greenman, J.; Versteeg, H.H.; Maraveyas, A. De-Palmitoylation of Tissue Factor Regulates Its Activity, Phosphorylation and Cellular Functions. Cancers 2021, 13, 3837. https://doi.org/10.3390/cancers13153837
Ettelaie C, Featherby S, Rondon AMR, Greenman J, Versteeg HH, Maraveyas A. De-Palmitoylation of Tissue Factor Regulates Its Activity, Phosphorylation and Cellular Functions. Cancers. 2021; 13(15):3837. https://doi.org/10.3390/cancers13153837
Chicago/Turabian StyleEttelaie, Camille, Sophie Featherby, Araci M. R. Rondon, John Greenman, Henri H. Versteeg, and Anthony Maraveyas. 2021. "De-Palmitoylation of Tissue Factor Regulates Its Activity, Phosphorylation and Cellular Functions" Cancers 13, no. 15: 3837. https://doi.org/10.3390/cancers13153837
APA StyleEttelaie, C., Featherby, S., Rondon, A. M. R., Greenman, J., Versteeg, H. H., & Maraveyas, A. (2021). De-Palmitoylation of Tissue Factor Regulates Its Activity, Phosphorylation and Cellular Functions. Cancers, 13(15), 3837. https://doi.org/10.3390/cancers13153837