1. Introduction
Cancer is a major public health problem worldwide and therefore, continuous scientific efforts in this field are clearly needed [
1]. Therefore, in recent decades, thanks mainly to increasing knowledge on cancer molecular biology and growing interest in applying bio- and nano-technology techniques against this pathology, cancer therapeutic strategies have notably changed [
2,
3]. However, despite these advancements, the overall cancer death rate still remains high as the critical issue of targeting cancer cells with high selectivity has not been reached for most cancers [
4]. For this reason, novel and challenging therapeutic approaches must be explored to improve selectivity for cancer cells over normal cells to avoid systemic toxicity and to overcome drug resistance [
5].
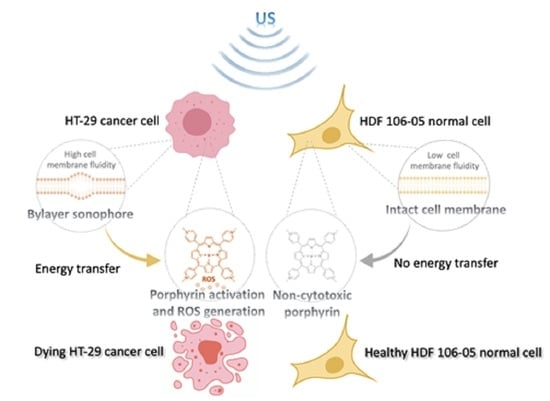
In order to investigate new therapeutic anticancer approaches able to improve cancer selectivity, we have investigated sonodynamic therapy (SDT), a therapeutic strategy derived from photodynamic therapy (PDT), based on the synergistic effect triggered by combining a suitable chemical compound (sonosensitizer) and low-intensity ultrasound (US), which is used to kill cancer cells and microbial cells [
6,
7]. The anticancer effectiveness of SDT has been demonstrated in in vitro and in vivo animal models [
8,
9,
10,
11]. Despite the promising results of this approach, however, only a few clinical reports have described the use of SDT [
6]. In our opinion, for SDT to be quickly turned into the clinical setting, a deeper understanding of its mechanism of action and its potentiality is needed. Therefore, the aim of our work was to shed new light on the SDT mechanism of action and to understand if selectivity towards cancer could be one of the main features of this approach. To this end, in our previous investigations, different outcomes of cancer cells and normal cells to SDT were reported, supporting the finding that cancer cells and normal cells behave differently in response to SDT [
12]. Indeed, it has been suggested that cell responsiveness to US is different between cancer cells and normal cells as the resistance of these cell types to physical stress is not the same [
13]. Moreover, recently, Geltmeier and colleagues reported that a tension/compression force of US can provoke damage preferentially on cancer cells, compared to normal cells [
14].
To achieve our goals, SDT experiments were carried out in cancer and normal cell lines, namely the human colon cancer HT-29 cell line and the dermal fibroblast HDF 106-05 cell line, in co-culture to provide a more representative in vivo-like tissue model [
15] and to avoid any misinterpretation of the results when the same experiments are performed separately.
Since the generation of reactive oxygen species (ROS) is considered a pivotal step in SDT anticancer activity, we performed our STD experiments with a palladium (II) porphyrin complex (Pd-P) as a sonosensitizer because, under US exposure, it resulted in being the most efficient sonosensitizer, compared to other metal–porphyrin complexes such as iron (III) and zinc (II) porphyrin complexes, in generating singlet oxygen (
1O
2) and hydroxyl radicals (
•OH), and in US-mediated cancer cell killing [
12]. Therefore, the selective cytotoxicity of SDT towards cancer HT-29 cells co-cultured with normal HDF 106-05 cells was evaluated in terms of cell death and ROS generation. In addition, we tried to elucidate the SDT mechanism of action by investigating the possible process responsible for selective responsiveness of cancer cells to SDT treatment. Firstly, we hypothesized that the intracellular activation of the sonosensitizer was probably due to a US-mediated energy transfer in the form of intramembrane cavitation across the cytoplasmic membrane according to the bilayer sonophore (BLS) theory, suggesting that the cell type-dependent responsiveness to SDT could rely on specific plasma membrane features. Secondly, we hypothesized that mitochondria were involved as the main intracellular target of SDT in the cell type-dependent SDT response [
16,
17]. For this reason, the plasma membrane diversity between cancer cells and normal cells was investigated in terms of SDT impact on plasma membrane fluidity, while the mitochondrial membrane potential was studied to confirm the pivotal role of these intracellular organelles in the efficacy of SDT.
2. Materials and Methods
2.1. Preparation of Porphyrin-Metal Complex Solution
5,10,15,20-tetrakis(
N-methylpyridinium-4-yl) porphyrinato palladium (II) tetrachloride (Pd-P) was synthesized according to the method described by Giuntini et al. [
12]. In order to obtain a 2 mM solution, 9.2 mg of Pd-P powder (MW 925.05 g/mol) was reconstituted in 5 mL cell medium without serum that was then filtered with sterile syringe filter membrane 0.2 μm (Enrico Bruno S.r.l., Torino, Italy) and stored in 1 mL aliquots at −20 °C away from the light.
2.2. Cell Culture
The human Caucasian colon adenocarcinoma cell line, HT-29 (Interlab Cell Line Collection, Genova, Italy), was cultured as a monolayer in RPMI-1640 growth medium (Sigma-Aldrich, Milano, Italy). The normal adult human primary dermal fibroblast cell line, HDF 106-05 (ECACC, Salisbury, UK), was cultured as a monolayer in DMEM growth medium (Sigma-Aldrich). Both cell culture media were supplemented with 10% decomplemented fetal bovine serum (FBS) (Lonza, Verviers, Belgium), 2 mM-glutamine, 100 UI/mL penicillin and 100 μg/mL streptomycin antibiotic (Sigma-Aldrich). Both cell lines were maintained in a dark incubator (Thermo Fisher Scientific, Waltham, MA, USA) in a humidified atmosphere containing 5% CO2 at 37 °C.
HT-29 and HDF 106-05 Cell Co-Culture
For the co-culture sonodynamic and photodynamic experiments, equal amounts of HT-29 and HDF 106-05 cells were used (1:1), with a final concentration of 5 × 105 cells. In particular, HT-29 and HDF 106-05 cells were maintained in co-culture with an equal mix of the respective cell culture medium.
2.3. Intracellular Porphyrin Fluorescence Quantification
According to our previous work [
12], the non-cytotoxic Pd-P concentration required to perform sonodynamic and photodynamic treatment on HT-29 and HDF 106-05 cells was 500 µM for P-Pd. In order to ascertain the correct time of sonosensitizer incubation before performing sonodynamic and photodynamic treatment, semiquantitative intracellular uptake of Pd-P in HT-29 and HDF 106-05 cells was investigated using the fluorescence plate reader EnSight (PerkinElmer, Waltham, MA, USA). Briefly, 5 × 10
3 HT-29 and HDF 106-05 cells were cultured in total black 96-well plates and, 24 h after seeding, cells were incubated for 1, 6, 12 and 24 h with Pd-P at 500 μM, respectively. Prior to analysis, culture media was removed, cells were washed with PBS, and nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI) 2 μL/mL for 10 min, then fixed with paraformaldehyde (PAF) 4% solution (Sigma-Aldrich) for 10 min at room temperature. Each well was then supplemented with 200 µL PBS for plate reading according to the plate normalization procedure for top reading setting.
DAPI is a blue, fluorescent, nucleic acid stain that preferentially stains double-stranded DNA. The detected fluorescence is directly proportional to the amount of DNA present in the cell, and as DAPI is rapidly taken up into cellular DNA, it is easy to discriminate fluorescent nuclei with no detectable cytoplasmic fluorescence. Fluorescence was read according to the parameters derived from relative spectra: λex 358 nm–λem 461 nm for DAPI and λex 416 nm–λem 683 nm for P-Pd. DAPI fluorescence from nuclei staining was used to normalize the fluorescence signal from the plate reader according to the cell number in each well. Porphyrin uptake was expressed as Intensity of Fluorescence DAPI Related Index (IFDR).
2.4. Confocal Microscopy
Qualitative uptake of Pd-P in HT-29 and HDF 106-05 cells was performed by confocal microscopy to investigate its intracellular localization. Specifically, confocal porphyrins uptake was performed at 24 h after the incubation. A total of 1 × 105 HT-29 and HDF 106-05 cells were seeded in 24-well plate with glass coverslips on the bottom, and 24 h after the seeding, cells were then incubated with Pd-P (500 μM) for 24 h. Cells on slides were fixed with 4% PAF (Sigma-Aldrich) for 15 min at room temperature. Cells were washed, and glass coverslips were placed on slides with Fluoroshield (Sigma-Aldrich) mounting medium for preserving and preventing rapid photobleaching of the fluorescent probe.
Confocal images were acquired using a laser scanning (λ
ex 405 nm and λ
em 633 nm) confocal microscope (Zeiss LSM5 Pascal, Oberkochen, Germany) with a 40× oil immersion objective using the multi-track mode. Images were analyzed with ImageJ software (FiJi, Bristol, UK, version 2.0). In order to quantify the porphyrin fluorescence in HT-29 and HDF 106-05 cells obtained by confocal acquisitions, five cells per each sample in three independent regions of the slide were considered, and the following formula for the corrected total cell fluorescence (CTCF), calculated with Image J, was then used [
18]: CTCF = integrated density − (area of selected cell × mean fluorescence of background readings).
Data are expressed as CTCF mean of three independent regions.
2.5. GSH Evaluation
The total glutathione level, glutathione disulfide (GSSG) and reduced glutathione (GSH) of HT-29 and HDF 106-05 as single cell lines was determined using the Glutathione Assay Kit (Sigma-Aldrich), according to the manufacturer’s instructions. The GSH content (nmol) was normalized to protein content in each sample by quantifying cell protein concentration (μg/mL) using the Quant-iT Protein Assay Kit by using the fluorimeter Qubit (Invitrogen-Life Technologies, Milano, Italy). Calibration was performed by applying a two-point standard curve, according to the manufacturer’s instructions. Briefly, GSH reacts with 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB) in a recycling assay and produces GSSG and the 1,3,5-trinitrobenzene (TNB) anion, which can be detected by absorbance. In turn, the enzyme glutathione reductase then reduces GSSG, which releases GSH that can react with another DTNB molecule. Therefore, the rate of TNB production is measured rather than a single determination of how much DTNB reacts with GSH, as it is proportional to the initial amount of GSH [
8]. The plate was read at 412 nm on a microplate reader Asys UV 340 (Biochrom, Cambridge, UK), and the amount of GSH was expressed in nmol/µg protein.
2.6. Sonodynamic Treatment
HT-29 and HDF 106-05 cells in the exponential growth phase were preincubated in the dark for 24 h with the sonosensitizer, Pd-P (500 μM), in 6-well plates. For the co-culture experiments, cells were then washed with PBS, trypsinized with 0.05% trypsin—0.02% EDTA solution and normalized to 5.0 × 10
5 cells consisting of an equal amount of HT-29 cells and HDF 106-05 cells (1:1) in 2.5 mL PBS in polystyrene tubes (TPP, Trasadingen, Switzerland) away from the light. The polystyrene tube (10 mm diameter) containing the cell suspension was connected to the US transducer by means of a specific mechanical adaptor, with a chamber filled with temperature controlled ultrapure water, in order to fix the distance from the transducer at 17 mm, to control the temperature variance and to have reproducible measurement conditions [
19].
US field was generated using a piezoelectric plane wave transducer (25.4 mm diameter), which operates in continuous wave mode at a frequency of 1.866 MHz, connected to a function generator (Type 33250; Agilent, Santa Clara, CA, USA) and a power amplifier (Type AR 100A250A; Amplifier Research, Souderton, PA, USA). All the conditions were then exposed to US generated at a 1.5 W/cm2 intensity for 5 min. An important aspect to be taken into consideration during US exposure is temperature control to avoid the hyperthermic effect. Therefore, the temperature of the cell suspension exposed to US was monitored by a thermocouple sensor (RS Components, Milano, Italy), and the maximum temperature recorded during the experiments in the US exposed samples was below 29 °C.
2.7. Ultrasound Field Characterization
US intensity (I) was measured as I = P/A where P is the US beam power and A is the cross-sectional area of US beam (A). The standardized method used to measure P is based on the radiation force balance principle, which is defined as the time averaged force exerted by an acoustic field on a target that intercepts the US beam. The measuring system was developed by the National Institute of Metrological Research (INRIM, Torino, Italy) and is based on a commercial balance (Mettler Toledo, model SAG-285, Columbus, OH, USA). Moreover, the spatial distribution of the US pressure has been evaluated by a scanning tank system (Onda Corp., Sunnyvale, CA, USA) with needle hydrophone (HNA-0400, Onda Corp.) and a preamplifier (AH-2020, Onda Corp.). During the 5 min insonation process at our experimental parameters, into the polystyrene tube, a value of maximum root-mean-square (rms) acoustic pressure of 300 kPa was measured.
2.8. Cytotoxicity Evaluation
At the end of the sonodynamic treatment, 8000 cells were plated in 24-well culture plates and then manually counted at 24, 48 and 72 h after the treatment, considering five different regions per well. Cytotoxicity was expressed as percentage (%) according to the following equation: % cytotoxicity = 100 × (untreated cell number − treated cell number).
2.9. Photodynamic Treatment
HT-29 and HDF 106-05 cells in the exponential growth phase were preincubated in the dark for 24 h with the sonosensitizer, Pd-P (500 μM), in 6-well plates. For the co-culture experiments, cells were then washed with PBS, trypsinized with 0.05% trypsin—0.02% EDTA solution and normalized to 5.0 × 105 cells consisting of an equal amount of colon cancer cells and fibroblast cells (1:1) in 2.5 mL PBS in polystyrene tubes (TPP) protected from the light. The light-emitting source of the system is based on InGaN light-emitting diodes (Cree Inc, Durham, NC, USA) with 20 mW maximum radiant power (emitted flux), and a central wavelength of 405 nm. The system allows continuous radiant flux regulation from 0 to 20 mW, with a programmable non-switching diode current source and energy fluency rates were adjusted to 15 mW/cm2. In particular, cell suspensions were illuminated for 5 min in a dark box. Following the photodynamic treatment, 8000 cells were plated in 24-well culture plates, and then manually counted at 24, 48 and 72 h after the treatment, considering five different regions per well. Cytotoxicity was expressed as a percentage according to the following equation: % cytotoxicity = 100 × (untreated cell number − treated cell number).
2.10. Flow Cytometry Assay
Cytofluorimetric assays were performed using a C6 flow cytometer (Accuri Cytometers, Annarbor, MI, USA) in order to investigate intracellular ROS production and mitochondrial membrane potential.
2.10.1. Intracellular ROS Production
Intracellular ROS generation after sonodynamic and photodynamic treatment in HT-29 or HDF 106-05 cells was investigated by using the 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich) probe. Briefly, cells in the exponential growth phase were incubated with Pd-P 500 μM for 24 h, and 10 µM DCFH-DA for the last 30 min of incubation at 37 °C protected from light. Cells were then washed with PBS, trypsinized and exposed to sonodynamic and photodynamic treatment as previously described. ROS production was assessed at 1, 5, 15, 30 and 60 min after treatments, and 10,000 events were considered for analysis by the FL1 channel (λ
ex 532 nm). ROS production was expressed as the integrated mean fluorescence intensity (iMFI), which was calculated as the product of the mean fluorescence intensity of the cells and the frequency of ROS-producing cells. The iMFI ratio was then calculated in order to yield the ratiometric increase in fluorescence per time point related to the iMFI of untreated cells, i.e., control cells [
20].
2.10.2. Mitochondrial Membrane Potential
The effects that sonodynamic treatment had on HT-29 or HDF 106-05 cell mitochondrial membrane potential was investigated using the Membrane Potential Detection Kit (BD Bioscience, San Jose, CA, USA) by flow cytometry. Mitochondrial functionality is related to optimal vital cellular status. Changes in mitochondrial membrane potential, particularly depolarization, were reported to occur during many cellular processes, such as production of ROS, activation of apoptosis and necrosis. 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) is a membrane-permeable lipophilic cationic fluorochrome that penetrates cells, and its fluorescence is a reflection of mitochondrial membrane potential (Δψ). The fluorescence emission spectrum of JC-1 is dependent on the status of Δψ. JC-1 excites at λex 488–490 nm and can exist in two different states, aggregates (λem 590 nm) or monomers (λem 527 nm), each with a different emission spectrum; both states exhibit fluorescence by FL-1 channel (λem 532 nm, green) but aggregates are also detectable by the FL-2 channel (λem 585 nm, red). When live cells are incubated with JC-1, the JC-1 penetrates the plasma membrane and mitochondria membrane. In polarized membranes of functional mitochondria conditions JC-1, accumulates in aggregate form, conversely, in depolarized membranes of less functional mitochondria JC-1 leaks out of the mitochondria and accumulates into the cytoplasm in monomeric form.
Briefly, HT-29 or HDF 106-05 cells were incubated for 24 h with P-Pd 500 μM at 37 °C and then exposed to US, as previously described. Moreover, a positive control was performed by exposing cells to 500 μM H2O2 for 3 h. JC-1 aggregates and monomers were investigated for each condition, and 10,000 events were recorded by the FL1 channel (λem 532 nm) and the FL2 channel (λem 585 nm); compensation was required due to fluorophore monomers and aggregates with partially overlapping emission spectra. Regions were placed around HT-29 or HDF 106-05 cell populations with high JC-1 aggregates and high monomer concentration (functional mitochondria); other regions were placed around HT-29 or HDF 106-05 cell populations with low JC-1 aggregates and high JC-1 monomer concentration (less functional mitochondria). For quantitative analysis, Δψ was expressed as the ratio between JC-1 aggregate and monomer mean fluorescence emission.
2.11. Membrane Fluidity/Phospholipid Polarization
The influence that US exposure had on HT-29 or HDF 106-05 cell membrane fluidity/phospholipid polarization was investigated by 1,6-diphenyl-1,3,5-hexatriene (DPH) fluorescence anisotropy using a polarized fluorescence spectrometer (LS 55 PerkinElmer, USA). DPH powder (MW = 232.32 g/mol) was dissolved to a final concentration of 2 mM in tetrahydrofuran (Sigma-Aldrich). Then, the pH of DPH final working solution (final concentration of 2 μM DPH in PBS) used for cells was measured and a value of pH 7.00 was obtained. HT-29 or HDF 106-05 cells were unexposed or exposed to US, as previously described. Positive controls for the reduction in anisotropy (increase in membrane fluidity and polarization) were obtained by exposing cells at 45 °C for 30 min [
21,
22]. Immediately after treatments, cells were incubated with working solution (2 μM DPH) at room temperature and protected by light. DPH anisotropy was evaluated at 30 and 90 min after being treated five times. Data are expressed as mean values ± standard deviation. The excitation wavelength and the detection wavelength were set at λ
ex 365 nm and λ
em 420 nm, respectively, and the excitation and emission split widths were set at 5 nm and 16 nm, respectively. Anisotropy (r) was calculated using the following equation:
where
I is the fluorescence emission intensity and subscript
V and
H represent the vertical and horizontal excitation and emission polarized light orientations.
G is a grating factor that accounts for differences in sensitivity to horizontal and vertical polarized light and is calculated as
G =
IHV/
IHH [
23]. In particular, membrane fluidity/phospholipid polarization was expressed as DPH fluorescence anisotropy.
2.12. Statistical Analysis
Data are shown as mean values ± standard deviation of three independent experiments. Statistical analyses were performed using Prism 7.0 software (GraphPad, La Jolla, CA, USA). According to the design of the experiment under analysis, multiple t-tests, two-way ANOVA, one-way ANOVA and Bonferroni’s test were used to calculate the threshold of significance. The statistical significance threshold was set at p < 0.05.
4. Discussion
One of the main goals of anticancer drug therapy is to determine the selective destruction of cancer cells while leaving normal cells unaffected. Indeed, the poor drug selectivity towards cancer cells is one of the main causes of severe side effects associated with cytotoxic chemotherapeutic drugs [
30]. Developing targeted therapies such as small molecule inhibitors and antibody targeted therapies has significantly improved the specificity of cancer treatment [
31], but improvement can also be achieved by studying US-based treatment that takes advantages of structural differences between cancer cells and normal cells [
14]. To this end, the use of US to activate intracellular sonosensitizer, in the so-called sonodynamic treatment, could be very promising. Sonodynamic treatment works with low intensity US, which avoids temperature increases in the target site, to trigger the cytotoxic nature of sensitizers such as porphyrins [
32]. Porphyrins are well-known photosensitizers as their tetrapyrrole ring structure can be activated by light to generate ROS, causing damage to cell structures and cell death [
33]. These features also make porphyrins also suitable sonosensitizers, i.e., compounds responsive to US, due to the hypothesis of the sonoluminescence occurrence under appropriate US exposure [
34].
In order to investigate if the sonodynamic treatment could be a selective cancer treatment, a co-culture of human colon cancer HT-29 and fibroblast HDF 106-05 cells was developed. The palladium (II) porphyrin complex (Pd-P) was chosen as the sonosensitizer as it is well-accepted that the anticancer efficacy of sonodynamic treatment relies on the induction of a strong ROS generation due to the US-mediated activation of the sonosensitizer. Indeed, we previously showed that the insertion of Pd (II) in the porphyrin rings results in highly efficient ROS production under US exposure [
12].
Before investigating the possible differences in response to the SDT by the two cell lines considered in co-culture, i.e., HT-29 and HDF 106-05, Pd-P uptake by the two cell lines was investigated in order to establish the optimal incubation time to perform SDT. Therefore, HT-29 and HDF 106-05 cells were incubated with the same noncytotoxic Pd-P concentration at different time points, and intracellular fluorescence quantification was performed, showing similar Pd-P uptake after 24 h of incubation in both cell lines (
Figure 1). Interestingly, focusing on confocal images (
Figure 2), it was possible to note a preferential Pd-P localization in the perinuclear regions of HT-29 cell cytoplasm, while Pd-P was localized all over the cellular body in the HDF 106-05 cell cytoplasm. Since it has been reported that in cancer cells, mitochondria are mostly localized at a perinuclear level, whereas in normal cells, they are spread all over the cellular body [
35,
36], and it has been considered that porphyrins accumulate preferentially in mitochondria [
37], we suggested that the Pd-P complex preferentially accumulated in the mitochondria in both cell lines.
Following the results of the Pd-P uptake experiments in our cell lines, SDT was carried out in a co-culture of HT-29 and HDF 106-05 cells (1:1), showing a very significant cytotoxicity over time on HT-29 cells, but not on HDF 106-05 cells (
Figure 3). To avoid any misinterpretations in our cytotoxic results between HT-29 and HDF 106-05 cells in co-culture, due to a possible difference in responses to the oxidative stress trigger by SDT, the amount of intracellular GSH was investigated. Indeed, cells can have a common antioxidant defensive strategy against ROS action, and one of the most important is the GSH redox cycle, able to metabolize hydrogen peroxide and to minimize its participation in reactions leading to hydroxyl radical formation [
38,
39]. In HT-29 and HDF 106-05 cell lines, no statistically significant difference in the intracellular amount of GSH was observed, suggesting that the response of the GSH system to oxidative stress could be similar in both the considered cell lines (
Figure 4) and, most importantly, that our different cytotoxic results between HT-29 and HDF 106-05 cells in our co-culture model did not appear to be affected by the amount of intracellular GSH.
To understand the different cytotoxicity observed between HT-29 and HDF 106-05 cells in co-culture after the sonodynamic treatment, we investigated the cytotoxic effect of a photodynamic treatment with the same sensitizer, Pd-P, on the same HT-29 and HDF 106-05 co-culture. Indeed, SDT was developed as a novel, promising non-invasive anticancer approach derived from PDT and, furthermore, it is well known that the low selectivity of PDT towards malignant cells is mainly due to the direct transmission of light energy to the sensitizer without involving structures or intracellular organelles that could be different among cells, such as the outer cell membrane [
40]. Therefore, after 24 h of incubation of a noncytotoxic concentration of Pd-P in both cell lines, our HT-29 and HDF 106-05 co-culture was subjected to PDT, and a significant cytotoxicity over time in both cancer HT-29 and normal HDF 106-05 cell lines was observed (
Figure 5). To confirm these findings, the ROS production in our co-culture was investigated both after STD and PDT, since oxidative stress is the main mechanism underlying the cytotoxic effect of both treatments. ROS were strongly generated in HT-29 cells, but not in HDF 106-05 cells when our co-culture was subjected to sonodynamic treatment, whereas a strong ROS generation in both cell lines was observed when our co-culture was subjected to PDT (
Figure 6), mirroring the cytotoxicity data. These data are in line with other reports confirming the strong ROS production as the pivotal step in the sonodynamic and photodynamic cytotoxic effect [
8,
41].
Since the major difference between SDT and PDT is the energy source used to activate the sensitizer (US versus light), this discrepancy between the SDT and PDT results suggested that US and light could activate the intracellular sensitizer differently in HT-29 and HDF 106-05 cells, probably due to the diverse nature of the wave, mechanical and electromagnetic, and therefore a different mechanism and amount of energy deposited to the sensitizer. Specifically, our findings suggested that, in SDT, the US-mediated intracellular sensitizer activation could be more influenced by differences in the cell structure of HT-29 and HDF 106-05 cells compared to PDT because, due to the type of energy released from the light, the selectivity of photodynamic action can be better achieved investigating cell organelles-targeted photosensitizers instead of cell structure differences between normal and cancer cells [
42].
In order to understand whether the cell structure plays a role in the selective cytotoxicity towards cancer cells during SDT, plasma membrane fluidity and mitochondrial membrane potential in both cell lines were investigated. Our choice to initially focus our attention on the plasma cell membrane was based on strong evidence that malignant and normal cells differ not only in their metabolism and morphology, but also in the plasma cell membrane architecture. This latter aspect can lead to differences in terms of mechanical properties between cancer cells and normal cells, being crucial for the success of the sonodynamic approach, as the sonosensitizer intracellular activation is mediated by an US-induced energy transfer across the plasma cell membrane [
43,
44,
45]. Indeed, one of the main interesting hypotheses explaining the US-induced energy transfer across the cell membrane to activate the intracellular sonosensitizer is represented by the bilayer sonophore (BLS) theory. According to this theory, Krasovitski et al. suggested that the cytoplasmic membrane, under appropriate conditions, is capable of transforming the US oscillating acoustic pressure wave into an intramembrane cavitation, which could explain all US-induced bioeffects [
16]. In other words, this cyclic expansion and contraction of the BLS could stimulate cycles of stretch and release in the plasma membrane and in the cytoskeleton, becoming a source of intramembrane cavitation. Therefore, we suggest that this kind of cavitation could generate intracellular submicron-sized gas bubbles that, when collapsed, release very high energy and perhaps also sonoluminescence [
34].
Therefore, our hypothesis was that the SDT selective cytotoxicity between HT-29 and HDF 106-05 cells could be based on BLS occurrence, thanks to our US set-up and according to the different mechanical properties of the plasma membrane of the two cell lines considered. Therefore, to investigate our hypothesis, we studied an important plasma membrane feature that could affect, according to BLS hypothesis, the US-induced energy transfer across the plasma membrane namely, the cell membrane fluidity. Cell membrane fluidity was analyzed by using DPH, a lipid probe that is able to link phospholipid chains contained in the cytoplasmic membrane bilayer, where an increase in DPH fluorescence anisotropy occurs in the presence of destabilized phospholipid polarization, being related to increased cell membrane fluidity [
22,
46]. Specifically, HDF 106-05 normal cells at basal conditions, i.e., without US exposure, showed a significantly lower membrane fluidity compared to HT-29 cancer cells, with normal cells being stiffer than cancer cells (
Figure 7).
These data are in agreement with other studies that reported cancer cells as being more compliant to physical stimuli such as acoustic waves than normal cells, and this evidence could support the preferential response of cancer cells to sonodynamic treatment [
14,
47]. Indeed, some researchers demonstrated that normal cells and cancer cells display differences in cell–cell interactions, cytoskeleton organization and subcellular structure, determining a specific elasticity and, therefore, distinct mechanical phenotypes [
44,
45,
47]. Indeed, cancer cells, in general, appear softer than their normal counterparts, while conversely, normal cells show greater stiffness and higher resistance to mechanical stress compared to cancer cells [
13,
14,
43]. Therefore, the observed difference in cell membrane fluidity between the two cell lines under investigation could be related to the selective response of cancer cells to the sonodynamic treatment. The difference highlighted between cancer cells and normal cells in their responsiveness to sonodynamic treatment is also supported by Kosheleva’s work, where it was noticed that the synergistic cytotoxic effects of US and nanoparticles were more pronounced in A549 lung cancer cells than in their normal counterparts, BEAS-2B cells [
48]. Moreover, after US exposure, we observed a decrease in membrane fluidity for HT-29 (
Figure 7), suggesting a bilayer cell membrane separation, as described in Di Giacinto’s work [
49]. Therefore, our results could support the hypothesis that SDT cytotoxic selectivity between HT-29 and HDF 106-05 cells may be based on the BLS theory thanks to our US set-up, and according to a different mechanical property of the plasma membrane between the two cell lines.
Another important aspect that can elucidate and highlight the differences between cancer cells and normal cells under SDT is related to the effect of SDT on HT-29 and HDF 106-05 mitochondria, since mitochondria are one of the main ROS mediators, playing a pivotal role in controlling process that can lead to cell death [
50]. Furthermore, mitochondria are an important subcellular target for sensitizers, and are also sensitive to US exposure [
27,
51,
52,
53]. Therefore, mitochondrial membrane potential was investigated, and a significant mitochondrial membrane potential reduction, along with a subsequent mitochondria dysfunction, was only observed when cancer HT-29 cells underwent sonodynamic treatment (
Figure 8). These data are in line with the sonodynamic-induced cytotoxicity in HT-29 cells and the inefficacy of this treatment in inducing cytotoxicity in HDF 106-05 cells. Therefore, the different SDT responses between the two cell lines in our co-culture model could be also ascribed to a different effect on mitochondria, in line with diverse sensitizer activation under US exposure, occurring in cancer cells and not in normal cells, in accord with the BLS theory.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







