
PKM2 Regulates HSP90-Mediated Stability of the IGF-1R Precursor Protein and Promotes Cancer Cell Survival during Hypoxia

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Cells
2.2. cDNA and siRNA Transfection
2.3. RT-PCR and Real-Time PCR
2.4. Immunoprecipitation
2.5. Cell Viability
2.6. FACS Analysis
2.7. Immunofluorescence
2.8. Tissue Microarray and Immunohistochemistry
2.9. Statistical Analysis
3. Results
3.1. Expression of PKM2 Is Associated with Cell Survival during Hypoxia
3.2. PKM2 Depletion Attenuates the IGF-1R-AKT Signaling Pathway by Suppressing IGF-1R Expression
3.3. PKM2 Physically Binds to and Promotes the Stability of IGF-1R Protein
3.4. PKM2 Maintains IGF-1R Protein Stability by Mediating the Binding of IGF-1R to HSP90
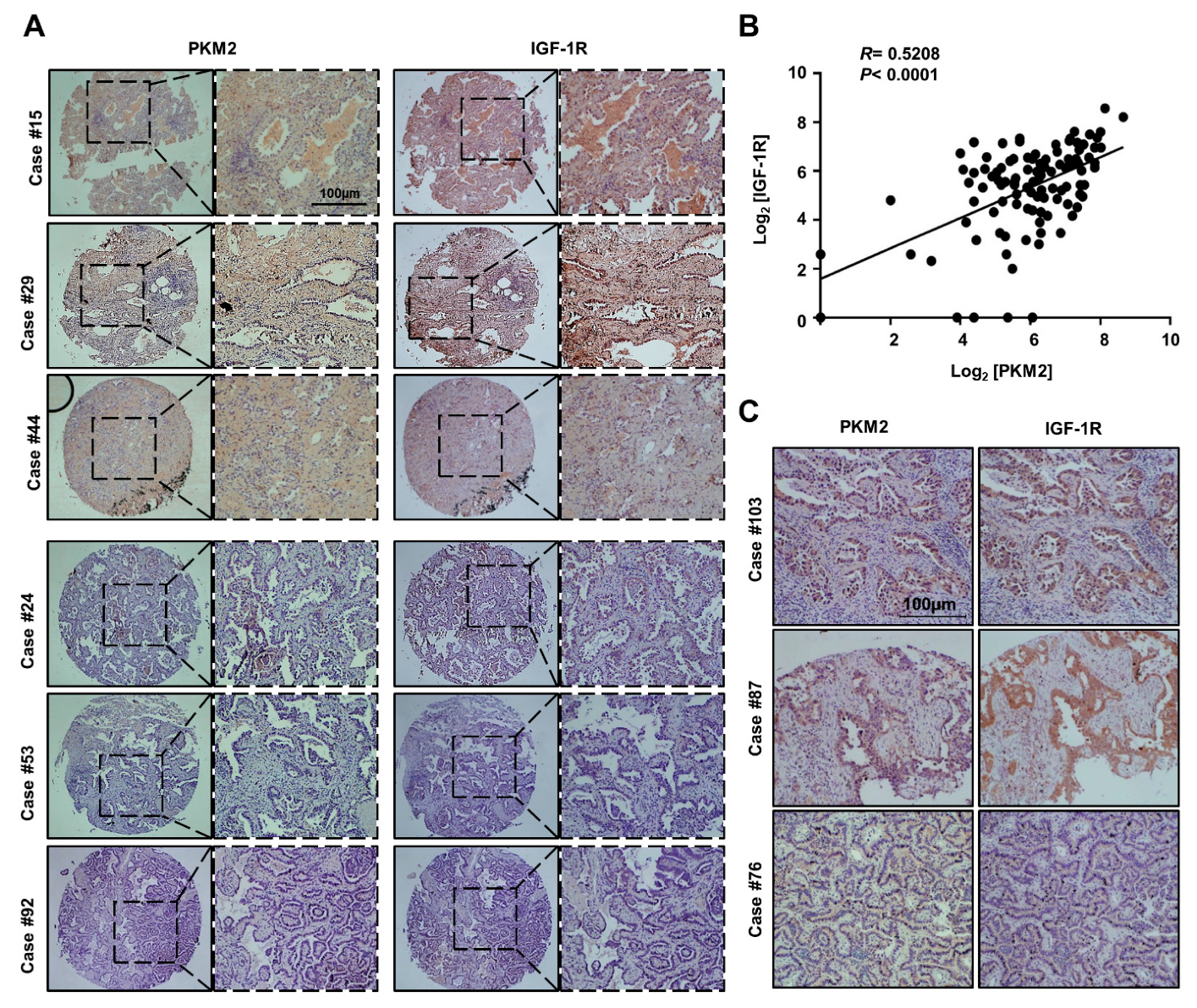
3.5. PKM2 Expression Positively Correlated with IGF-1R Expression in Human Lung Adenocarcinoma Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- Iams, W.T.; Lovly, C.M. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef] [Green Version]
- Kooijman, R. Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev. 2006, 17, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Wu, Z.F.; Cao, Q.H.; Chen, C.; Chen, Z.W.; Xu, Z.; Li, W.S.; Liu, F.K.; Yao, X.Q.; Li, G. Insulin-like growth factor receptor-1 overexpression is associated with poor response of rectal cancers to radiotherapy. World J. Gastroenterol. 2014, 20, 16268–16274. [Google Scholar] [CrossRef] [PubMed]
- All-Ericsson, C.; Girnita, L.; Seregard, S.; Bartolazzi, A.; Jager, M.J.; Larsson, O. Insulin-like growth factor-1 receptor in uveal melanoma: A predictor for metastatic disease and a potential therapeutic target. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1–8. [Google Scholar]
- DiGiovanni, J.; Bol, D.K.; Wilker, E.; Beltran, L.; Carbajal, S.; Moats, S.; Ramirez, A.; Jorcano, J.; Kiguchi, K. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000, 60, 1561–1570. [Google Scholar] [PubMed]
- Eliasz, S.; Liang, S.; Chen, Y.; De Marco, M.A.; Machek, O.; Skucha, S.; Miele, L.; Bocchetta, M. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene 2010, 29, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahra, K.; Dey, T.; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Luo, H.; Zhu, X.; Hu, X.; Zheng, L.; Zhu, X. Pyruvate kinase M2 (PKM2) expression correlates with prognosis in solid cancers: A meta-analysis. Oncotarget 2017, 8, 1628–1640. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, H.; Mei, Y.; Ding, X.; Yang, X.; Li, C.; Deng, M.; Gong, J. Clinicopathological and prognostic significance of PKM2 protein expression in cirrhotic hepatocellular carcinoma and non-cirrhotic hepatocellular carcinoma. Sci. Rep. 2017, 7, 15294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Pan, X.; Zhu, D.; Deng, Z.; Jiang, R.; Wang, X. Circular RNA MAT2B Promotes Glycolysis and Malignancy of Hepatocellular Carcinoma Through the miR-338-3p/PKM2 Axis Under Hypoxic Stress. Hepatology 2019, 70, 1298–1316. [Google Scholar] [CrossRef]
- Zhang, L.F.; Lou, J.T.; Lu, M.H.; Gao, C.; Zhao, S.; Li, B.; Liang, S.; Li, Y.; Li, D.; Liu, M.F. Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J. 2015, 34, 2671–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, function and expression and regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O'Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.S.; Kim, D.J.; Koo, H.; Jang, S.H.; You, Y.M.; Cho, J.H.; Yang, S.J.; Yu, E.S.; Jung, Y.; Lee, D.C.; et al. AKT-induced PKM2 phosphorylation signals for IGF-1-stimulated cancer cell growth. Oncotarget 2016, 7, 48155–48167. [Google Scholar] [CrossRef]
- Puckett, D.L.; Alquraishi, M.; Chowanadisai, W.; Bettaieb, A. The Role of PKM2 in Metabolic Reprogramming: Insights into the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 1171. [Google Scholar] [CrossRef]
- Yang, Y.C.; Cheng, T.Y.; Huang, S.M.; Su, C.Y.; Yang, P.W.; Lee, J.M.; Chen, C.K.; Hsiao, M.; Hua, K.T.; Kuo, M.L. Cytosolic PKM2 stabilizes mutant EGFR protein expression through regulating HSP90-EGFR association. Oncogene 2016, 35, 3387–3398. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Sendoel, A.; Hengartner, M.O. Apoptotic cell death under hypoxia. Physiology 2014, 29, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Guan, J.Z.; Sun, Y.; Le, Z.; Zhang, P.; Yu, D.; Liu, Y. Insulin-like growth factor 1 receptor-mediated cell survival in hypoxia depends on the promotion of autophagy via suppression of the PI3K/Akt/mTOR signaling pathway. Mol. Med. Rep. 2017, 15, 2136–2142. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Henson, E.S.; Xiao, W.; Huang, D.; McMillan-Ward, E.M.; Israels, S.J.; Gibson, S.B. Tyrosine kinase receptor EGFR regulates the switch in cancer cells between cell survival and cell death induced by autophagy in hypoxia. Autophagy 2016, 12, 1029–1046. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwisscha van Scheltinga, A.G.; Berghuis, P.; Nienhuis, H.H.; Timmer-Bosscha, H.; Pot, L.; Gaykema, S.B.; Lub-de Hooge, M.N.; Kosterink, J.G.; de Vries, E.G.; Schroder, C.P. Visualising dual downregulation of insulin-like growth factor receptor-1 and vascular endothelial growth factor-A by heat shock protein 90 inhibition effect in triple negative breast cancer. Eur. J. Cancer 2014, 50, 2508–2516. [Google Scholar] [CrossRef]
- Grimberg, A. Mechanisms by which IGF-I may promote cancer. Cancer Biol. Ther. 2003, 2, 630–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Yin, Z.; Tao, K.; Wang, G.; Gao, J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy. Oncol. Lett. 2018, 15, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Peretz, S.; Kim, C.; Rockwell, S.; Baserga, R.; Glazer, P.M. IGF1 receptor expression protects against microenvironmental stress found in the solid tumor. Radiat. Res. 2002, 158, 174–180. [Google Scholar] [CrossRef]
- Ter Braak, B.; Siezen, C.L.; Lee, J.S.; Rao, P.; Voorhoeve, C.; Ruppin, E.; van der Laan, J.W.; van de Water, B. Insulin-like growth factor 1 receptor activation promotes mammary gland tumor development by increasing glycolysis and promoting biomass production. Breast Cancer Res. 2017, 19, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Tejado, M.; Naranjo-Suarez, S.; Jimenez, C.; Carrera, A.C.; Landazuri, M.O.; del Peso, L. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: Protective role in apoptosis. J. Biol. Chem. 2001, 276, 22368–22374. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF1 signaling pathway in hypoxiaischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuzzahab, M.J.; Schneider, A.; Goddard, A.; Grigorescu, F.; Lautier, C.; Keller, E.; Kiess, W.; Klammt, J.; Kratzsch, J.; Osgood, D.; et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N. Engl. J. Med. 2003, 349, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Ouban, A.; Muraca, P.; Yeatman, T.; Coppola, D. Expression and distribution of insulin-like growth factor-1 receptor in human carcinomas. Hum. Pathol. 2003, 34, 803–808. [Google Scholar] [CrossRef]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Cui, Z.Z.; Zhu, L.; Fu, S.P.; Rossi, M.; Cui, Y.H.; Zhu, B.M. Central IGF1 improves glucose tolerance and insulin sensitivity in mice. Nutr. Diabetes 2017, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Hamilton, A.; Geng, S.; Hong, T.; Kalkum, M.; Momand, J.; Kane, S.E.; Huss, J.M. t-Darpp Activates IGF-1R Signaling to Regulate Glucose Metabolism in Trastuzumab-Resistant Breast Cancer Cells. Clin. Cancer Res. 2018, 24, 1216–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pore, N.; Jiang, Z.; Shu, H.K.; Bernhard, E.; Kao, G.D.; Maity, A. Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol. Cancer Res. 2006, 4, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Mitsiades, C.S.; Laubach, J.P.; Lonial, S.; Chanan-Khan, A.A.; Anderson, K.C. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br. J. Haematol. 2011, 152, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.; Sharp, S.; Workman, P. HSP90 inhibition: Two-pronged exploitation of cancer dependencies. Drug. Discov. Today 2012, 17, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Blagg, B.; Haystead, T.; Trepel, J.B.; Whitesell, L.; Picard, D. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress. Chaperones 2018, 23, 467–482. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koo, H.; Byun, S.; Seo, J.; Jung, Y.; Lee, D.C.; Cho, J.H.; Park, Y.S.; Yeom, Y.I.; Park, K.C. PKM2 Regulates HSP90-Mediated Stability of the IGF-1R Precursor Protein and Promotes Cancer Cell Survival during Hypoxia. Cancers 2021, 13, 3850. https://doi.org/10.3390/cancers13153850
Koo H, Byun S, Seo J, Jung Y, Lee DC, Cho JH, Park YS, Yeom YI, Park KC. PKM2 Regulates HSP90-Mediated Stability of the IGF-1R Precursor Protein and Promotes Cancer Cell Survival during Hypoxia. Cancers. 2021; 13(15):3850. https://doi.org/10.3390/cancers13153850
Chicago/Turabian StyleKoo, Han, Sangwon Byun, Jieun Seo, Yuri Jung, Dong Chul Lee, Jung Hee Cho, Young Soo Park, Young Il Yeom, and Kyung Chan Park. 2021. "PKM2 Regulates HSP90-Mediated Stability of the IGF-1R Precursor Protein and Promotes Cancer Cell Survival during Hypoxia" Cancers 13, no. 15: 3850. https://doi.org/10.3390/cancers13153850
APA StyleKoo, H., Byun, S., Seo, J., Jung, Y., Lee, D. C., Cho, J. H., Park, Y. S., Yeom, Y. I., & Park, K. C. (2021). PKM2 Regulates HSP90-Mediated Stability of the IGF-1R Precursor Protein and Promotes Cancer Cell Survival during Hypoxia. Cancers, 13(15), 3850. https://doi.org/10.3390/cancers13153850