
Chromosomal Translocations in NK-Cell Lymphomas Originate from Inter-Chromosomal Contacts of Active rDNA Clusters Possessing Hot Spots of DSBs

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Determination of rDNA-Mediated Translocation Sites in Paired-End WGS Reads
2.2. Genome-Wide rDNA-Mediated Translocation Mapping
2.3. 4C-rDNA Experiments
2.4. RNA-Seq Analysis
2.5. 4C-rDNA Genome-Wide Mapping
2.6. Genome-Wide Profiles
2.7. Epigenome Statistics
2.8. Statistics
3. Results
3.1. rDNA-Mediated Translocations Are Present in Both T Cells and in NK-Cell Lymphomas
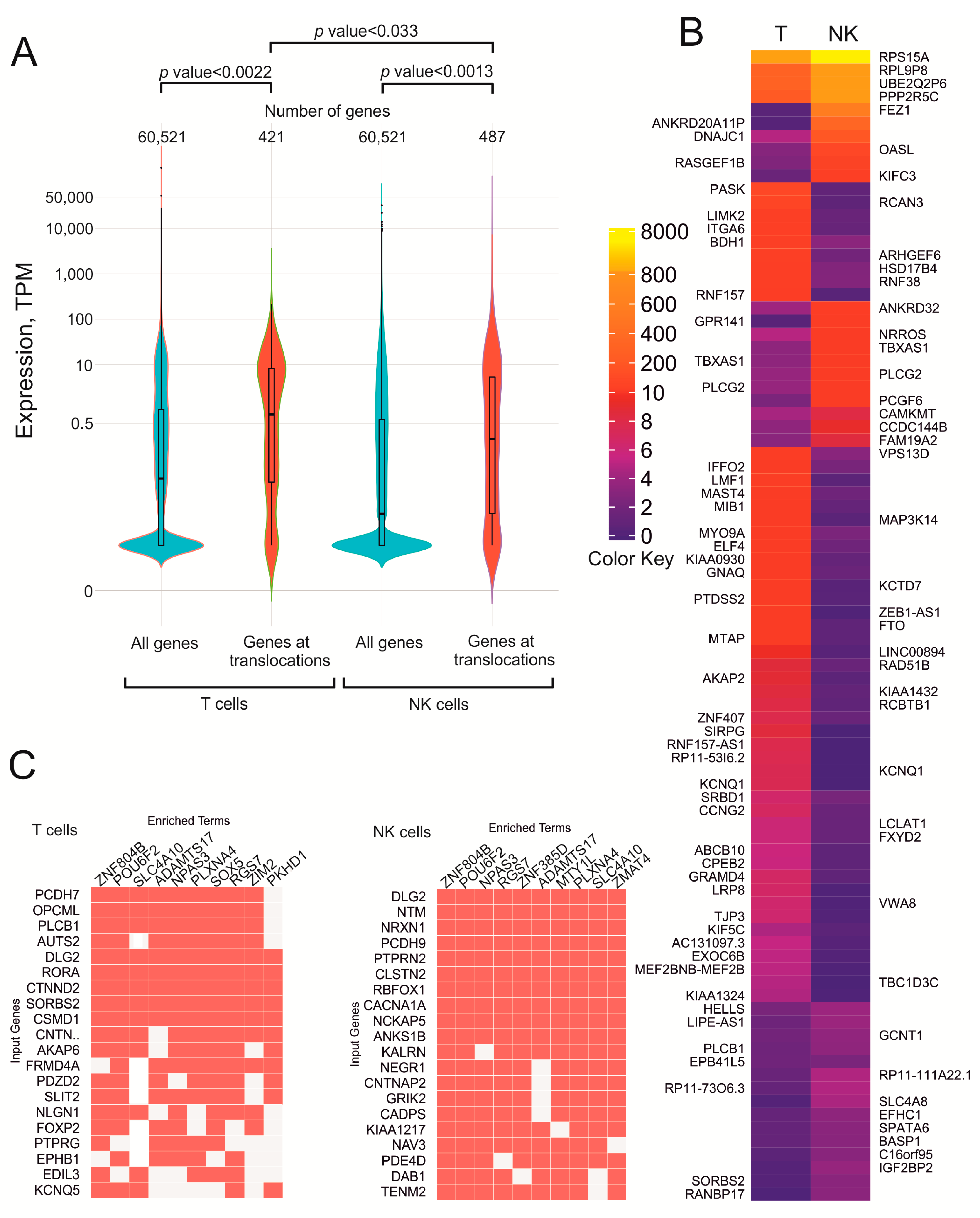
3.2. In T Cells and NK-Cell Lymphomas, Translocations Involve Different Sets of Genes Controlling Development
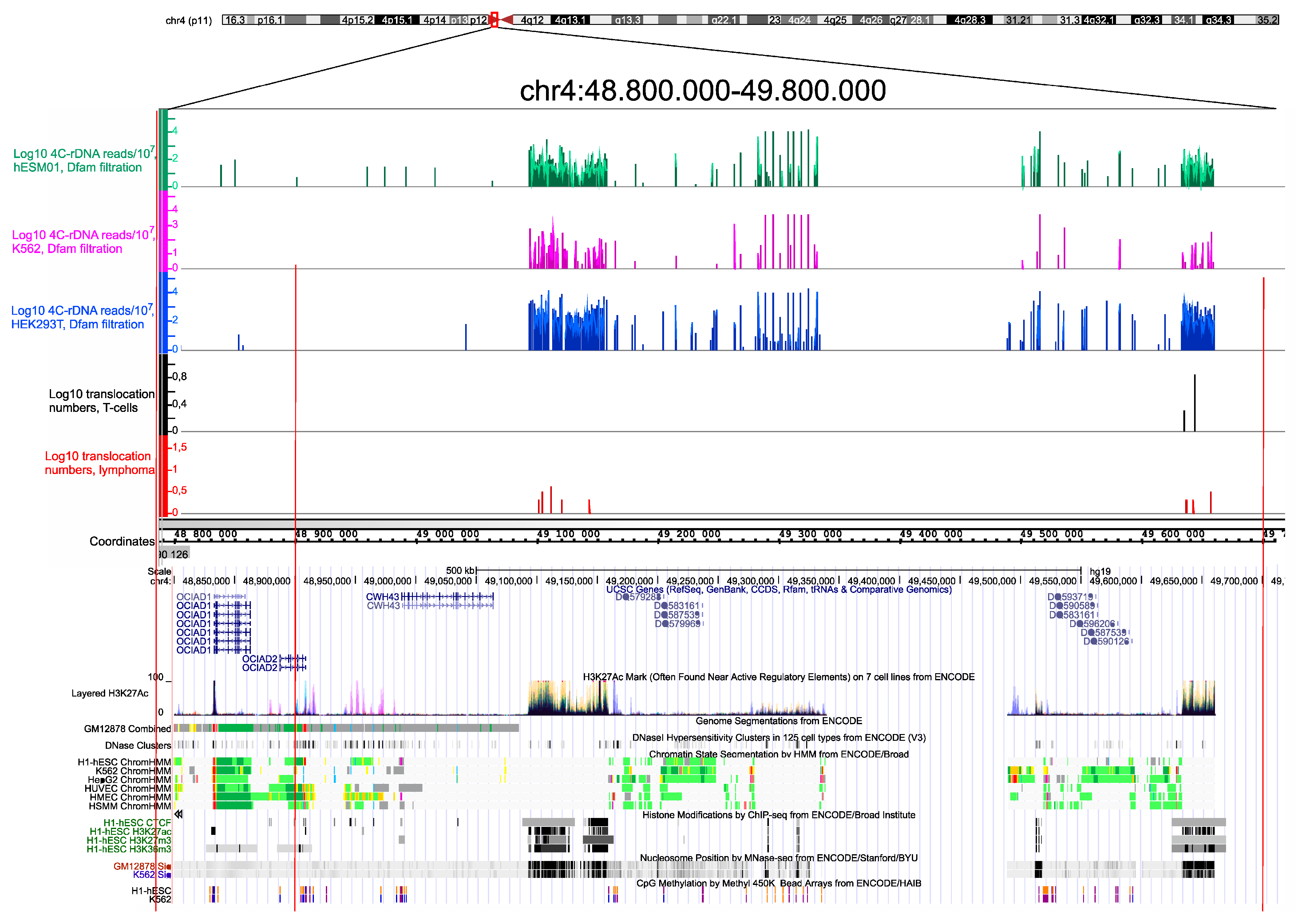
3.3. There Are Hot Spots of rDNA-Mediated Translocations in T Cells and NK-Cell Lymphomas
3.4. Translocation Sites in T Cells and NK-Cell Lymphomas Have Fewer Silenced Genes
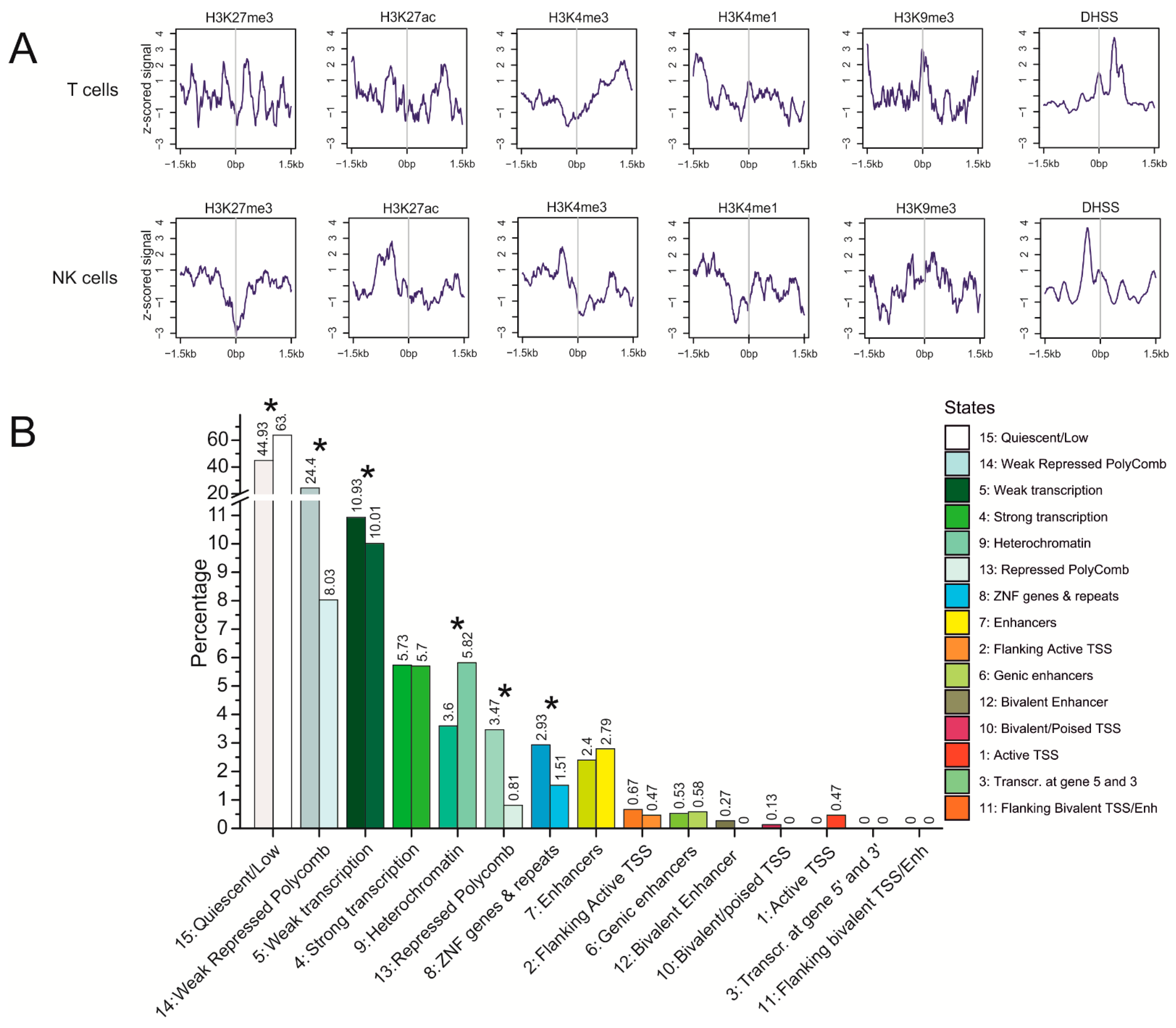
3.5. Epigenetic Features at Translocation Sites in T Cells and NK Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meaburn, K.J.; Misteli, T.; Soutoglou, E. Spatial genome organization in the formation of chromosomal translocations. Semin Cancer Biol. 2007, 17, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roix, J.J.; McQueen, P.G.; Munson, P.J.; Parada, L.A.; Misteli, T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 2003, 34, 287–291. [Google Scholar] [CrossRef]
- Nambiar, M.; Kari, V.; Raghavan, S.C. Chromosomal translocations in cancer. Biochim. Biophys. Acta 2008, 1786, 139–152. [Google Scholar] [CrossRef]
- Fröhling, S.; Döhner, H. Chromosomal abnormalities in cancer. N. Engl. J. Med. 2008, 359, 722–734. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Fedoseeva, D.M.; Sosin, D.V.; Snezhkina, A.V.; Melnikova, N.V.; Kudryavtseva, A.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks and genomic contacts of human rDNA units are involved in epigenetic regulation. J. Mol. Cell. Biol. 2015, 7, 366–382. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Yudkin, D.V.; Gorbacheva, M.A.; Kulemzina, A.I.; Grischenko, I.V.; Fedoseeva, D.M.; Sosin, D.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks in human rDNA units are produced in vivo. Sci. Rep. 2016, 6, 25866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchurikov, N.A.; Fedoseeva, D.M.; Klushevskaya, E.S.; Slovohotov, I.Y.; Chechetkin, V.R.; Kravatsky, Y.V.; Kretova, O.V. rDNA Clusters Make Contact with Genes that Are Involved in Differentiation and Cancer and Change Contacts after Heat Shock Treatment. Cells 2019, 8, 1393. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, I.; Almeida-King, J.; Kumanduri, V.; Senf, A.; Spalding, J.D.; Ur-Rehman, S.; Saunders, G.; Kandasamy, J.; Caccamo, M.; Leinonen, R.; et al. The European Genome-phenome Archive of human data consented for biomedical research. Nat. Genet. 2015, 47, 692–695. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Cherry, J.M. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic. Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Stempor, P.; Ahringer, J. SeqPlots-Interactive software for exploratory data analyses, pattern discovery and visualization in genomics. Wellcome Open Res. 2016, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH Roadmap Epigenomics. Available online: https://egg2.wustl.edu/roadmap/web_portal (accessed on 1 August 2021).
- Tchurikov, N.A.; Klushevskaya, E.S.; Kravatsky, Y.V.; Kravatskaya, G.I.; Fedoseeva, D.M. Interchromosomal Contacts of rDNA Clusters in Three Human Cell Lines Are Associated with Silencing of Genes Controlling Morphogenesis. Dokl. Biochem. Biophys. 2021, 496, 22–26. [Google Scholar] [CrossRef]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression.Nat Rev Genet. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Lagarkova, M.A.; Shutova, M.V.; Bogomazova, A.N.; Vassina, E.M.; Glazov, E.A.; Zhang, P.; Rizvanov, A.A.; Chestkov, I.V.; Kiselev, S.L. Induction of pluripotency in human endothelial cells resets epigenetic profile on genome scale. Cell Cycle. 2010, 9, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.H.; Li, Y.W.; LI, P. Regulatory Effects of Linc00486 on Biological Characteristics of Breast Cancer Cells. Rev. Argent. De Clínica Psicológica 2020, 29, 356–363. [Google Scholar] [CrossRef]
- Wang, C.; Yin, R.; Dai, J.; Gu, Y.; Cui, S.; Ma, H.; Zhang, Z.; Huang, J.; Qin, N.; Jiang, T.; et al. Whole-genome sequencing reveals genomic signatures associated with the inflammatory microenvironments in Chinese NSCLC patients. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Belaghzal, H.; Tyler Borrman, T.; Stephens, A.D.; Lafontaine, D.L.; Venev, S.; Zhiping Weng, Z.; Marko, J.F.; Dekkerl, J. Liquid chromatin Hi-C characterizes compartment-dependent chromatin interaction dynamics. Nat. Genet. 2021, 53, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Kretova, O.V.; Fedoseeva, D.M.; Kravatsky, Y.V.; Alembekov, I.R.; Slovohotov, I.Y.; Tchurikov, N.A. Homeotic DUX4 Genes that Control Human Embryonic Development at the Two-Cell Stage Are Surrounded by Regions Contacting with rDNA Gene Clusters. Mol. Biol. 2019, 53, 268–273. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Klushevskaya, E.S.; Kravatsky, Y.V.; Kravatskaya, G.I.; Fedoseeva, D.M. Kretova. Interchromosomal Contacts of rDNA Clusters with DUX Genes in Human Chromosome 4 Are Very Sensitive to Heat Shock Treatment. Dokl. Biochem. Biophys. 2020, 490, 50–53. [Google Scholar] [CrossRef]
- Smith, S.L.; Kennedy, P.R.; Stacey, K.B.; Worboys, J.D.; Yarwood, A.; Seo, S.; Solloa, E.H.; Mistretta, B.; Chatterjee, S.S.; Gunaratne, P.; et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 2020, 4, 1388–1406. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Kretova, O.V.; Fedoseeva, D.M.; Sosin, D.V.; Grachev, S.A.; Serebraykova, M.V.; Romanenko, S.A.; Vorobieva, N.V.; Kravatsky, Y.V. DNA double-strand breaks coupled with PARP1 and HNRNPA2B1 binding sites flank coordinately expressed domains in human chromosomes. PLoS Genet. 2013, 9, e1003429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shalaby, N.A.; Buszczak, M. Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 2014, 343, 298–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchurikov, N.A.; Klushevskaya, E.S.; Fedoseeva, D.M.; Alembekov, I.R.; Kravatskaya, G.I.; Chechetkin, V.R.; Kravatsky, Y.V.; Kretova, O.V. Dynamics of Whole-Genome Contacts of Nucleoli in Drosophila Cells Suggests a Role for rDNA Genes in Global Epigenetic Regulation. Cells 2020, 9, 2587. [Google Scholar] [CrossRef]
- Tse, E.; Kwong, Y.L. How I treat NK/T-cell lymphomas. Blood 2013, 121, 4997–5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Cui, B.W.; Wang, N.; Dai, Y.T.; Zhang, H.; Wang, C.F.; Zhong, H.J.; Cheng, S.; Ou-Yang, B.S.; Hu, Y.; et al. Genomic and Transcriptomic Characterization of Natural Killer T Cell Lymphoma. Cancer Cell 2020, 37, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, 1502–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.M.; Martinez-Fundichely, A.; Diaz, B.J.; Aronson, B.; Cuykendall, T.; MacKay, M.; Dhingra, P.; Wong, E.W.P.; Chi, P.; Apostolou, E.; et al. Identification of Cancer Drivers at CTCF Insulators in 1,962 Whole Genomes. Cell Syst. 2019, 8, 446–455. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anchors | Sequences, 5′–3′ | Numbering in rDNA Unit (Accession # U13369.1) | Average Translocation Number in T Cells (102 Samples) | Average Translocation Number in NK Cells Lymphomas (102 Samples) |
|---|---|---|---|---|
| R1 | GCAGTGCGGTGGCGCGATCTTGGCTCACCGCAACCTCTGCCTCCCGGTTTCAAGCGATTCTCCTGCATCG | 19,641–19,710 | 575.4 | 430.2 |
| R2 | CCTATTTTCAGTAGAGACGGGGTTTCTCCACGTTGGCCACGCTGGTCTCGAACTCCTGACCTCAAATGAT | 21,261–21,330 | 829.6 | 881.9 |
| R3 | CTACTCGGGAGGCTGGGGTGGAAGAATTGCTTGAACCTGGCAGGCGGAGGCTGCAGTGAC | 21,801–21,860 | 2015.5 | 2384.0 |
| R4 | TTTCTGAGATGGAGTCTTGCTCTGTCCCCCAGGCTGGAGTGCAGTGGCGT | 31,701–31,750 | 1452.8 | 1723.9 |
| R5 | TGTCGCCCAGGCTGGAGTGCGATGGTGTGATCTCGGCTCACTGCAACCGCCACCTCCCTG | 32,721–32,780 | 1733.0 | 1980.4 |
| R6 | CACCGCAACCTCCACCTCCCGCGTTCAAGCGATTCTCCTGCCTCAGCCTCCTGAGTAGCT | 36,601–36,660 | 1154.2 | 1308.8 |
| R7 | CACTGCAACGTCCGCCTCCCGGGTTCACGCCATTCTCCTGCCTCAGCCTCCCAAGTAGCT | 37,001–37,060 | 1.1 | 1.0 |
| R8 | AATCGCTTGAACCTGGGAAGCGGAGGTTGCAGTGAGCCGAGATTGCGCCATCGCACTCCA | 38,551–38,610 | 856.7 | 885.1 |
| R9 | AGAGGGCAATGGCGCGATCTCGGCTCACCGCACCCTCCGCCTCCCAGGTTCAAGCGATTC | 39,601–39,660 | 651.5 | 674.6 |
| Total translocations in 102 samples | 9269.8 | 10,269.9 | ||
| Number of translocations per genome | 90.9 | 100.68 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchurikov, N.A.; Uroshlev, L.A.; Klushevskaya, E.S.; Alembekov, I.R.; Lagarkova, M.A.; Kravatskaya, G.I.; Makeev, V.Y.; Kravatsky, Y.V. Chromosomal Translocations in NK-Cell Lymphomas Originate from Inter-Chromosomal Contacts of Active rDNA Clusters Possessing Hot Spots of DSBs. Cancers 2021, 13, 3889. https://doi.org/10.3390/cancers13153889
Tchurikov NA, Uroshlev LA, Klushevskaya ES, Alembekov IR, Lagarkova MA, Kravatskaya GI, Makeev VY, Kravatsky YV. Chromosomal Translocations in NK-Cell Lymphomas Originate from Inter-Chromosomal Contacts of Active rDNA Clusters Possessing Hot Spots of DSBs. Cancers. 2021; 13(15):3889. https://doi.org/10.3390/cancers13153889
Chicago/Turabian StyleTchurikov, Nickolai A., Leonid A. Uroshlev, Elena S. Klushevskaya, Ildar R. Alembekov, Maria A. Lagarkova, Galina I. Kravatskaya, Vsevolod Y. Makeev, and Yuri V. Kravatsky. 2021. "Chromosomal Translocations in NK-Cell Lymphomas Originate from Inter-Chromosomal Contacts of Active rDNA Clusters Possessing Hot Spots of DSBs" Cancers 13, no. 15: 3889. https://doi.org/10.3390/cancers13153889
APA StyleTchurikov, N. A., Uroshlev, L. A., Klushevskaya, E. S., Alembekov, I. R., Lagarkova, M. A., Kravatskaya, G. I., Makeev, V. Y., & Kravatsky, Y. V. (2021). Chromosomal Translocations in NK-Cell Lymphomas Originate from Inter-Chromosomal Contacts of Active rDNA Clusters Possessing Hot Spots of DSBs. Cancers, 13(15), 3889. https://doi.org/10.3390/cancers13153889