
Irreversible Electroporation and Nivolumab Combined with Intratumoral Administration of a Toll-Like Receptor Ligand, as a Means of In Vivo Vaccination for Metastatic Pancreatic Ductal Adenocarcinoma (PANFIRE-III). A Phase-I Study Protocol

 , , ,
, , ,  , , ,
, , , 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Metastatic Disease
1.2. Immune Escape
1.3. Anenestic Effects
1.4. Pre-Clinical Evidence IRE Induced Immune Modulation
1.5. Clinical Evidence IRE Induced Immune Modulation
1.6. Pro-Oncogenic Effects
1.7. Synergy with Immunotherapy
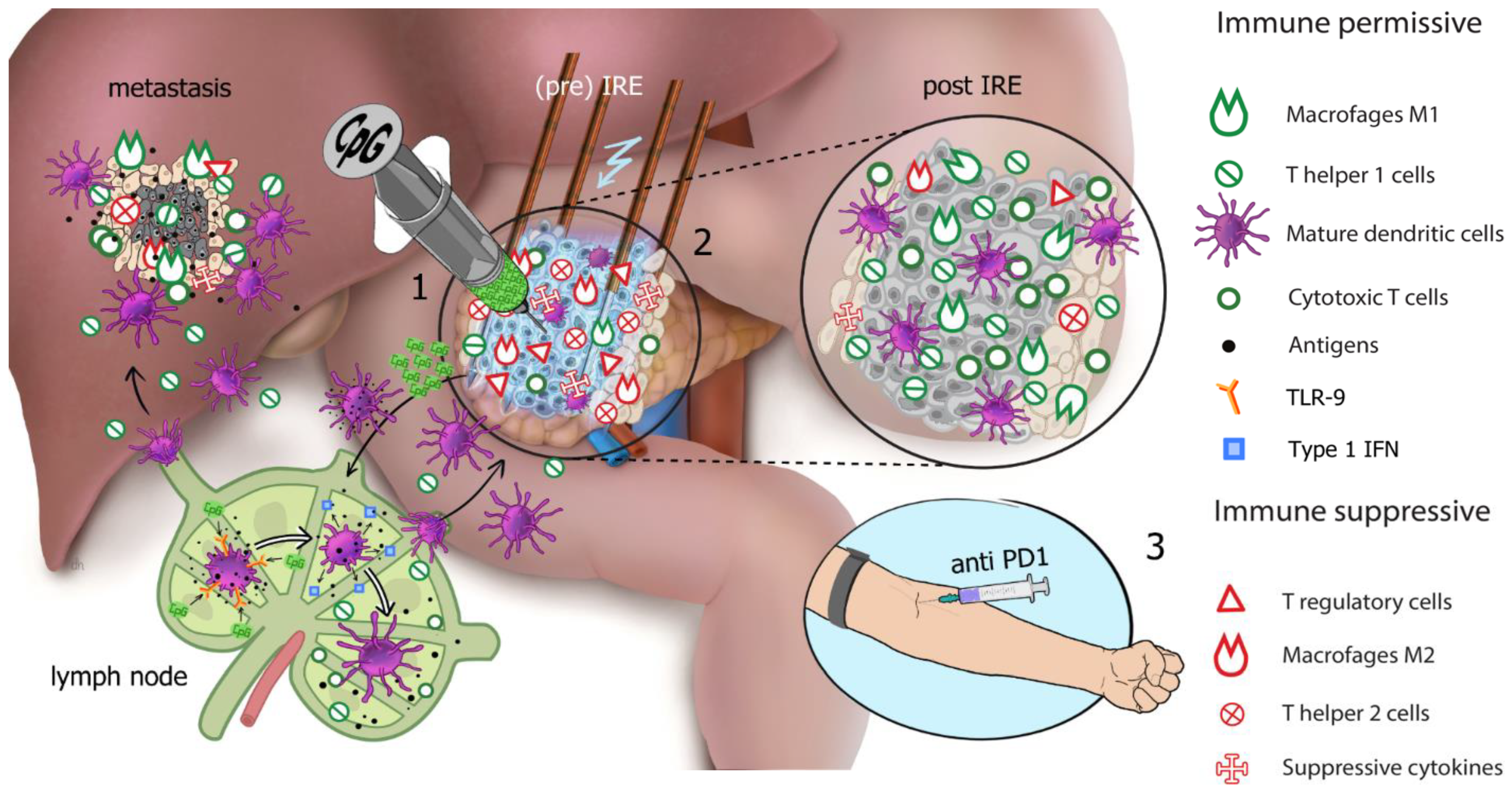
1.8. Hypothesis
2. Materials and Methods
2.1. Objectives
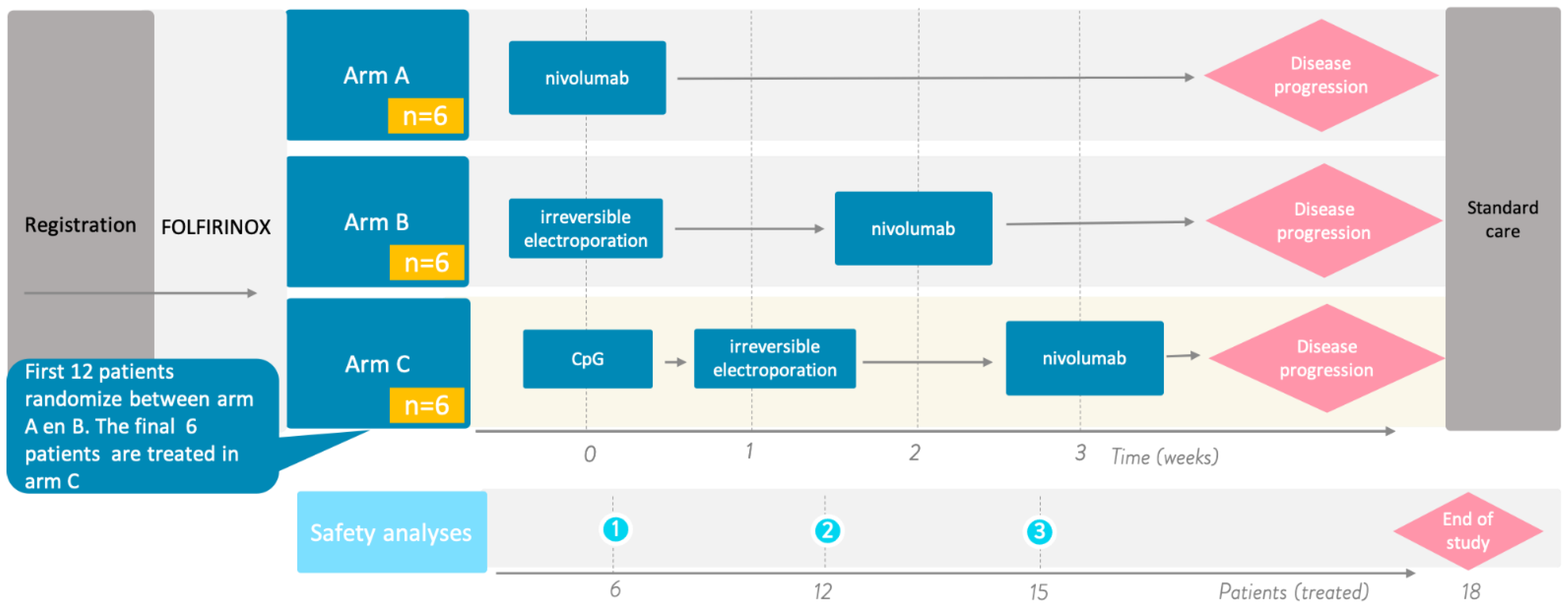
2.2. Design
- Arm A (control arm): intravenous administration of 240 mg nivolumab every 2 weeks for the first 3 doses followed by intravenous administration of 480 mg every 4 weeks until disease progression.
- Arm B: percutaneous CT-guided (partial) IRE of the primary pancreatic tumor. After 2 weeks, this will be followed by the intravenous administration of 240 mg nivolumab every 2 weeks for 2 doses followed by intravenous administration of 480 mg every 4 weeks until disease progression.
- Arm C: single intratumoral (i.t.) injection of 8 mg IMO-2125, which will be followed by percutaneous CT-guided (partial) IRE of the primary pancreatic tumor after one week. A 240 mg dose of nivolumab is administered intravenously every 2 weeks for 2 doses, which will begin two weeks after IRE, followed by the intravenous administration of 480 mg every 4 weeks until disease progression.
2.3. Eligibility Criteria
2.4. Interventions
2.4.1. Percutaneous CT-Guided IRE
2.4.2. Anti-PD-1 Monoclonal Antibody (mAb) (Nivolumab)
2.4.3. CpG Oligodeoxynucleotide (IMO-2125)
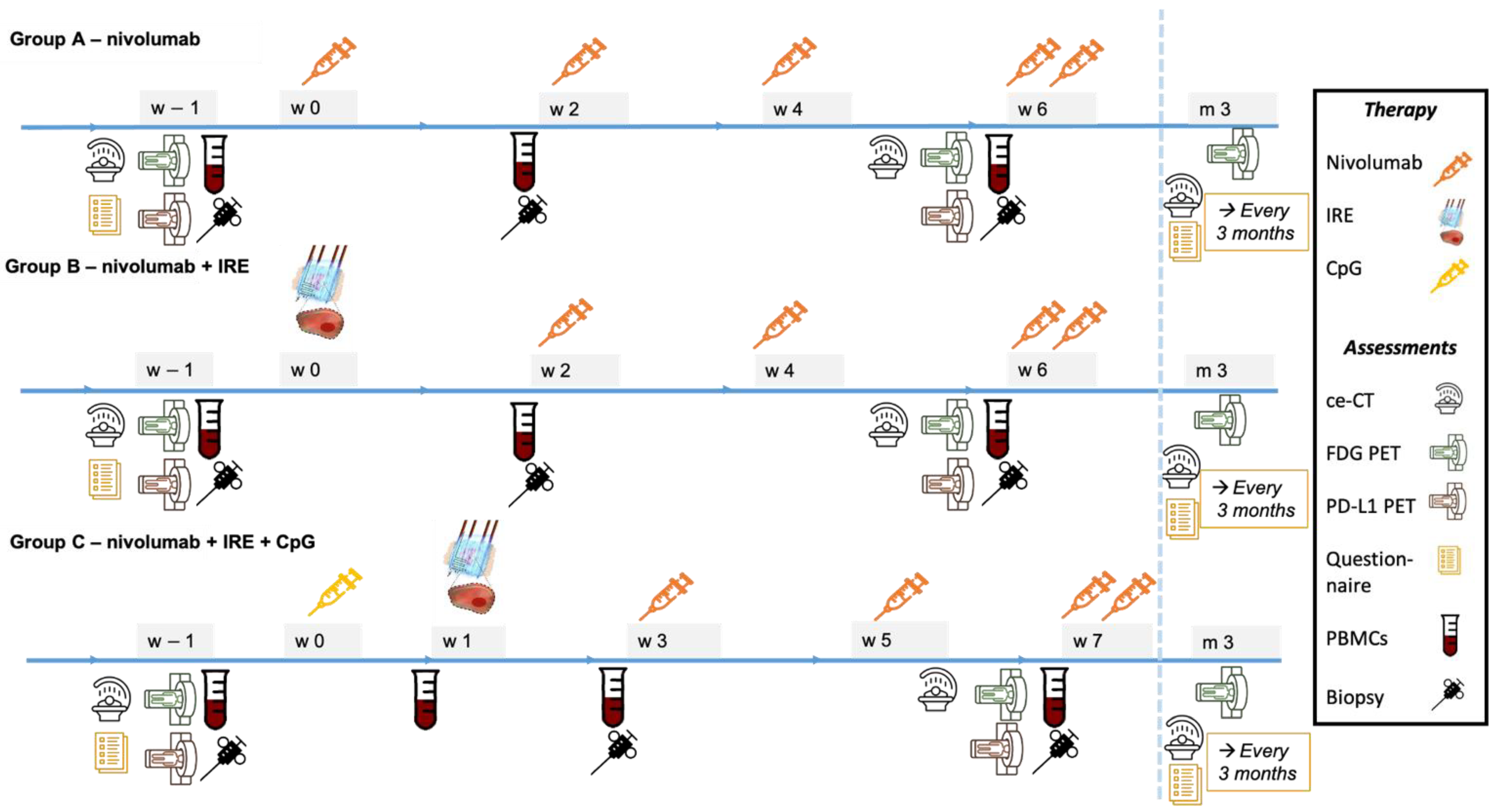
2.5. Outcome Measures
2.6. Data Collection and Analysis
2.6.1. Interim Safety Analysis
2.6.2. Survival
2.6.3. Blood and Tissue
2.6.4. Imaging
2.6.5. Questionnaires
2.7. Follow-Up
2.8. Data Collection and Handling
2.9. Sample Size Calculation and Statistical Considerations
3. Discussion
3.1. Preclinical Evidence of Synergy
3.2. Clinical Evidence of Synergy
3.3. Timing of Study Interventions
3.4. Immune Response
3.5. Imaging
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, S.J.; Walma, M.S.; Vogel, J.A.; van Rijssen, L.B.; Wilmink, J.W.; Mohammad, N.H.; van Santvoort, H.C.; Molenaar, I.Q.; Besselink, M.G. Systematic Review of Resection Rates and Clinical Outcomes After FOLFIRINOX-Based Treatment in Patients with Locally Advanced Pancreatic Cancer. Ann. Surg. Oncol. 2016, 23, 4352–4360. [Google Scholar] [CrossRef] [Green Version]
- Suker, M.; Beumer, B.R.; Sadot, E.; Marthey, L.; Faris, J.E.; Mellon, E.A.; El-Rayes, B.F.; Wang-Gillam, A.; Lacy, J.; Hosein, P.J.; et al. FOLFIRINOX for locally advanced pancreatic cancer: A systematic review and patient-level meta-analysis. Lancet Oncol. 2016, 17, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Ruarus, A.; Vroomen, L.; Puijk, R.; Scheffer, H.; Meijerink, M. Locally Advanced Pancreatic Cancer: A Review of Local Ablative Therapies. Cancers 2018, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Mercadal, B.; Beitel-White, N.; Aycock, K.N.; Castellví, Q.; Davalos, R.V.; Ivorra, A. Dynamics of Cell Death After Conventional IRE and H-FIRE Treatments. Ann. Biomed. Eng. 2020, 48, 1451–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, R.M.; Beitel-White, N.; Davalos, R.V.; Allen, I.C. Starting a Fire Without Flame: The Induction of Cell Death and Inflammation in Electroporation-Based Tumor Ablation Strategies. Front. Oncol. 2020, 10, 1235. [Google Scholar] [CrossRef]
- Vogel, J.A.; Vroomen, L.G.P.H.; Srimathveeravalli, G. The Effect of Irreversible Electroporation on Blood Vessels, Bile Ducts, Urinary Tract, Intestines, and Nerves, in Irreversible Electroporation in Clinical Practice; Meijerink, M.R., Scheffer, H.J., Narayanan, G., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 81–94. [Google Scholar]
- Ruarus, A.H.; Vroomen, L.; Puijk, R.S.; Scheffer, H.J.; Zonderhuis, B.M.; Kazemier, G.; van den Tol, M.P.; Berger, F.H.; Meijerink, M.R. Irreversible Electroporation in Hepatopancreaticobiliary Tumours. Can. Assoc. Radiol. J. 2018, 69, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.C.; Philips, P.; Ellis, S.; Hayes, D.; Bagla, S. Irreversible electroporation of unresectable soft tissue tumors with vascular invasion: Effective palliation. BMC Cancer 2014, 14, 540. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, G.; Belfiore, M.P.; Reginelli, A.; Capasso, R.; Romano, F.; Ianniello, G.P.; Cappabianca, S.; Brunese, L. Concurrent chemotherapy alone versus irreversible electroporation followed by chemotherapy on survival in patients with locally advanced pancreatic cancer. Med. Oncol. 2017, 34, 38. [Google Scholar] [CrossRef] [PubMed]
- Coelen, R.J.S.; Vogel, J.A.; Vroomen, L.; Roos, E.; Busch, O.R.C.; van Delden, O.M.; Delft, F.V.; Heger, M.; van Hooft, J.E.; Kazemier, G.; et al. Ablation with irreversible electroporation in patients with advanced perihilar cholangiocarcinoma (ALPACA): A multicentre phase I/II feasibility study protocol. BMJ Open 2017, 7, e015810. [Google Scholar] [CrossRef] [PubMed]
- Leen, E.; Picard, J.; Stebbing, J.; Abel, M.; Dhillon, T.; Wasan, H. Percutaneous irreversible electroporation with systemic treatment for locally advanced pancreatic adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Paiella, S.; Butturini, G.; Frigerio, I.; Salvia, R.; Armatura, G.; Bacchion, M.; Fontana, M.; D’Onofrio, M.; Martone, E.; Bassi, C. Safety and feasibility of Irreversible Electroporation (IRE) in patients with locally advanced pancreatic cancer: Results of a prospective study. Dig. Surg. 2015, 32, 90–97. [Google Scholar] [CrossRef]
- Martin, R.C., 2nd; Kwon, D.; Chalikonda, S.; Sellers, M.; Kotz, E.; Scoggins, C.; McMasters, K.M.; Watkins, K. Treatment of 200 locally advanced (stage III) pancreatic adenocarcinoma patients with irreversible electroporation: Safety and efficacy. Ann. Surg. 2015, 262, 486–494. [Google Scholar] [CrossRef]
- Narayanan, G.; Bhatia, S.; Echenique, A.; Suthar, R.; Barbery, K.; Yrizarry, J. Vessel patency post irreversible electroporation. Cardiovasc. Intervent. Radiol. 2014, 37, 1523–1529. [Google Scholar] [CrossRef]
- Mansson, C.; Bergenfeldt, M.; Brahmstaedt, R.; Karlson, B.M.; Nygren, P.; Nilsson, A. Safety and preliminary efficacy of ultrasound-guided percutaneous irreversible electroporation for treatment of localized pancreatic cancer. Anticancer Res. 2014, 34, 289–293. [Google Scholar]
- Kluger, M.D.; Epelboym, I.; Schrope, B.A.; Mahendraraj, K.; Hecht, E.M.; Susman, J.; Weintraub, J.L.; Chabot, J.A. Single-Institution Experience with Irreversible Electroporation for T4 Pancreatic Cancer: First 50 Patients. Ann. Surg. Oncol. 2016, 23, 1736–1743. [Google Scholar] [CrossRef]
- Scheffer, H.J.; Vroomen, L.G.; de Jong, M.C.; Melenhorst, M.C.; Zonderhuis, B.M.; Daams, F.; Vogel, J.A.; Besselink, M.G.; van Kuijk, C.; Witvliet, J.; et al. Ablation of Locally Advanced Pancreatic Cancer with Percutaneous Irreversible Electroporation: Results of the Phase I/II PANFIRE Study. Radiology 2017, 282, 585–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Veldhuisen, E.; Vroomen, L.G.; Ruarus, A.H.; Derksen, T.C.; Busch, O.R.; de Jong, M.C.; Kazemier, G.; Puijk, R.S.; Sorgedrager, N.S.; Vogel, J.A.; et al. Value of CT-Guided Percutaneous Irreversible Electroporation Added to FOLFIRINOX Chemotherapy in Locally Advanced Pancreatic Cancer: A Post Hoc Comparison. J. Vasc. Interv. Radiol. 2020, 31, 1600–1608. [Google Scholar] [CrossRef]
- He, C.; Wang, J.; Zhang, Y.; Lin, X.; Li, S. Irreversible electroporation after induction chemotherapy versus chemotherapy alone for patients with locally advanced pancreatic cancer: A propensity score matching analysis. Pancreatology 2020, 20, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nederlandse Kankerregistratie (NKR), IKNL. Available online: iknl.nl/nkr-cijfers (accessed on 28 March 2019).
- Das, M.; Zhou, X.; Liu, Y.; Das, A.; Vincent, B.G.; Li, J.; Liu, R.; Huang, L. Tumor neoantigen heterogeneity impacts bystander immune inhibition of pancreatic cancer growth. Transl. Oncol. 2020, 13, 100856. [Google Scholar] [CrossRef] [PubMed]
- Sideras, K.; Braat, H.; Kwekkeboom, J.; van Eijck, C.H.; Peppelenbosch, M.P.; Sleijfer, S.; Bruno, M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat. Rev. 2014, 40, 513–522. [Google Scholar] [CrossRef]
- Bowers, J.; Bailey, S.; Rubinstein, M.; Paulos, C.; Camp, E.R. Genomics meets immunity in pancreatic cancer: Current research and future directions for pancreatic adenocarcinoma immunotherapy. Oncol. Rev. 2019, 13, 102–113. [Google Scholar] [CrossRef]
- Li, M.; Bharadwaj, U.; Zhang, R.; Zhang, S.; Mu, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol. Cancer Ther. 2008, 7, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Erinjeri, J.P.; Fine, G.C.; Adema, G.J.; Ahmed, M.; Chapiro, J.; den Brok, M.; Duran, R.; Hunt, S.J.; Johnson, D.T.; Ricke, J.; et al. Immunotherapy and the Interventional Oncologist: Challenges and Opportunities-A Society of Interventional Oncology White Paper. Radiology 2019, 292, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Mole, R.H. Whole Body Irradiation—Radiobiology or Medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef]
- Marabelle, A.; Andtbacka, R.; Harrington, K.; Melero, I.; Leidner, R.; de Baere, T.; Robert, C.; Ascierto, P.A.; Baurain, J.F.; Imperiale, M.; et al. Starting the fight in the tumor: Expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann. Oncol. 2018, 29, 2163–2174. [Google Scholar] [CrossRef]
- Van den Bijgaart, R.J.E.; Schuurmans, F.; Fütterer, J.J.; Verheij, M.; Cornelissen, L.A.M.; Adema, G.J. Immune Modulation Plus Tumor Ablation: Adjuvants and Antibodies to Prime and Boost Anti-Tumor Immunity In Situ. Front. Immunol. 2021, 12, 617365. [Google Scholar] [CrossRef]
- Shao, Q.; O’Flanagan, S.; Lam, T.; Roy, P.; Pelaez, F.; Burbach, B.J.; Azarin, S.M.; Shimizu, Y.; Bischof, J.C. Engineering T cell response to cancer antigens by choice of focal therapeutic conditions. Int. J. Hyperth. 2019, 36, 130–138. [Google Scholar] [CrossRef] [Green Version]
- White, S.B.; Zhang, Z.; Chen, J.; Gogineni, V.R.; Larson, A.C. Early Immunologic Response of Irreversible Electroporation versus Cryoablation in a Rodent Model of Pancreatic Cancer. J. Vasc. Interv. Radiol. 2018, 29, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Rubinsky, B.; Onik, G.; Mikus, P. Irreversible electroporation: A new ablation modality—clinical implications. Technol. Cancer Res. Treat. 2007, 6, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Jose, A.; Sobrevals, L.; Ivorra, A.; Fillat, C. Irreversible electroporation shows efficacy against pancreatic carcinoma without systemic toxicity in mouse models. Cancer Lett. 2012, 317, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, H.; Hong, Y.K.; Li, Y.; Rostas, J.; Pulliam, Z.; Li, S.P.; Martin, R.C.G. Evaluating the Regulatory Immunomodulation Effect of Irreversible Electroporation (IRE) in Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2019, 26, 800–806. [Google Scholar] [CrossRef]
- Scheffer, H.J.; Stam, A.G.M.; Geboers, B.; Vroomen, L.; Ruarus, A.; de Bruijn, B.; van den Tol, M.P.; Kazemier, G.; Meijerink, M.R.; de Gruijl, T.D. Irreversible electroporation of locally advanced pancreatic cancer transiently alleviates immune suppression and creates a window for antitumor T cell activation. Oncoimmunology 2019, 8, 1652532. [Google Scholar] [CrossRef] [Green Version]
- Ruarus, A.H.; Vroomen, L.; Geboers, B.; van Veldhuisen, E.; Puijk, R.S.; Nieuwenhuizen, S.; Besselink, M.G.; Zonderhuis, B.M.; Kazemier, G.; de Gruijl, T.D.; et al. Percutaneous Irreversible Electroporation in Locally Advanced and Recurrent Pancreatic Cancer (PANFIRE-2): A Multicenter, Prospective, Single-Arm, Phase II Study. Radiology 2020, 294, 212–220. [Google Scholar] [CrossRef]
- Geboers, B.; Scheffer, H.J.; Graybill, P.M.; Ruarus, A.H.; Nieuwenhuizen, S.; Puijk, R.S.; van den Tol, P.M.; Davalos, R.V.; Rubinsky, B.; de Gruijl, T.D.; et al. High-Voltage Electrical Pulses in Oncology: Irreversible Electroporation, Electrochemotherapy, Gene Electrotransfer, Electrofusion, and Electroimmunotherapy. Radiology 2020, 295, 254–272. [Google Scholar] [CrossRef]
- Zhao, J.; Wen, X.; Tian, L.; Li, T.; Xu, C.; Wen, X.; Melancon, M.P.; Gupta, S.; Shen, B.; Peng, W.; et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat. Commun. 2019, 10, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oji, Y.; Nakamori, S.; Fujikawa, M.; Nakatsuka, S.; Yokota, A.; Tatsumi, N.; Abeno, S.; Ikeba, A.; Takashima, S.; Tsujie, M.; et al. Overexpression of the Wilms’ tumor gene WT1 in pancreatic ductal adenocarcinoma. Cancer Sci. 2004, 95, 583–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulloa-Montoya, F.; Louahed, J.; Dizier, B.; Gruselle, O.; Spiessens, B.; Lehmann, F.F.; Suciu, S.; Kruit, W.H.; Eggermont, A.M.; Vansteenkiste, J.; et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J. Clin. Oncol. 2013, 31, 2388–2395. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Krieg, A.M. CpG still rocks! Update on an accidental drug. Nucleic Acid. Ther. 2012, 22, 77–89. [Google Scholar] [CrossRef]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015, 36, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Geboers, B.; Ruarus, A.H.; Nieuwenhuizen, S.; Puijk, R.S.; Scheffer, H.J.; de Gruijl, T.D.; Meijerink, M.R. Needle-guided ablation of locally advanced pancreatic cancer: Cytoreduction or immunomodulation by in vivo vaccination? Chin. Clin. Oncol. 2019, 8, 61. [Google Scholar] [CrossRef]
- Kakar, S.; Pawlik, T.M.; Allen, P.J. Exocrine Pancreas. Pancreatic adenocarcinoma. In AJCC Cancer Staging Manual, 8th ed.; Amin, M.B., Ed.; Springer: New York, NY, USA, 2016. [Google Scholar]
- Timmer, F.E.F.; Geboers, B.; Ruarus, A.H.; Schouten, E.A.C.; Nieuwenhuizen, S.; Puijk, R.S.; de Vries, J.J.J.; Meijerink, M.R.; Scheffer, H.J. Irreversible Electroporation for Locally Advanced Pancreatic Cancer. Tech. Vasc. Interv. Radiol. 2020, 23, 100675. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services National Institutes of Health; NCI. Common Terminology Criteria for Adverse Events Version 5.0. Available online: https://evs.nci.nih.gov/ftp1/CTCAE/About.html (accessed on 6 December 2019).
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef]
- Ivy, S.P.; Siu, L.L.; Garrett-Mayer, E.; Rubinstein, L. Approaches to Phase 1 Clinical Trial Design Focused on Safety, Efficiency, and Selected Patient Populations: A Report from the Clinical Trial Design Task Force of the National Cancer Institute Investigational Drug Steering Committee. Clin. Cancer Res. 2010, 16, 1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef] [Green Version]
- Wahl, R.L.; Jacene, H.; Kasamon, Y.; Lodge, M.A. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. J. Nucl. Med. 2009, 50, 122s–150s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boellaard, R.; O’Doherty, M.J.; Weber, W.A.; Mottaghy, F.M.; Lonsdale, M.N.; Stroobants, S.G.; Oyen, W.J.; Kotzerke, J.; Hoekstra, O.S.; Pruim, J.; et al. FDG PET and PET/CT: EANM procedure guidelines for tumour PET imaging: Version 1.0. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 181–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Okada, M.; Kaji, Y.; Satouchi, M.; Sato, Y.; Yamabe, Y.; Onaya, H.; Endo, M.; Sone, M.; Arai, Y. New response evaluation criteria in solid tumours-revised RECIST guideline (version 1.1). Gan To Kagaku Ryoho 2009, 36, 2495–2501. [Google Scholar]
- Mackay, T.M.; Smits, F.J.; Latenstein, A.E.J.; Bogte, A.; Bonsing, B.A.; Bos, H.; Bosscha, K.; Brosens, L.A.A.; Hol, L.; Busch, O.R.C.; et al. Impact of nationwide enhanced implementation of best practices in pancreatic cancer care (PACAP-1): A multicenter stepped-wedge cluster randomized controlled trial. Trials 2020, 21, 334. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Romano, E.; Rufo, N.; Agostinis, P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: Mechanisms and clinical translation. Cell. Death Differ. 2016, 23, 938–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Brok, M.H.; Sutmuller, R.P.; Nierkens, S.; Bennink, E.J.; Frielink, C.; Toonen, L.W.; Boerman, O.C.; Figdor, C.G.; Ruers, T.J.; Adema, G.J. Efficient loading of dendritic cells following cryo and radiofrequency ablation in combination with immune modulation induces anti-tumour immunity. Br. J. Cancer 2006, 95, 896–905. [Google Scholar] [CrossRef] [Green Version]
- Shan, C.C.; Shi, L.R.; Ding, M.Q.; Zhu, Y.B.; Li, X.D.; Xu, B.; Jiang, J.T.; Wu, C.P. Cytokine-induced killer cells co-cultured with dendritic cells loaded with the protein lysate produced by radiofrequency ablation induce a specific antitumor response. Oncol. Lett. 2015, 9, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestrini, M.T.; Ingham, E.S.; Mahakian, L.M.; Kheirolomoom, A.; Liu, Y.; Fite, B.Z.; Tam, S.M.; Tucci, S.T.; Watson, K.D.; Wong, A.W.; et al. Priming is key to effective incorporation of image-guided thermal ablation into immunotherapy protocols. JCI Insight 2017, 2, e90521. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Alnaggar, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. An important discovery on combination of irreversible electroporation and allogeneic natural killer cell immunotherapy for unresectable pancreatic cancer. Oncotarget 2017, 8, 101795–101807. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, C.; Hayat, T.; Hamm, J.; Healey, M.; Zheng, Q.; Li, Y.; Martin, R.C.G., 2nd. A phase 1b trial of concurrent immunotherapy and irreversible electroporation in the treatment of locally advanced pancreatic adenocarcinoma. Surgery 2020, 168, 610–616. [Google Scholar] [CrossRef]
- Molenkamp, B.G.; van Leeuwen, P.A.; Meijer, S.; Sluijter, B.J.; Wijnands, P.G.; Baars, A.; van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Intradermal CpG-B activates both plasmacytoid and myeloid dendritic cells in the sentinel lymph node of melanoma patients. Clin. Cancer Res. 2007, 13, 2961–2969. [Google Scholar] [CrossRef] [Green Version]
- Molenkamp, B.G.; Sluijter, B.J.; van Leeuwen, P.A.; Santegoets, S.J.; Meijer, S.; Wijnands, P.G.; Haanen, J.B.; van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8+ T-cell reactivity in melanoma patients. Clin. Cancer Res. 2008, 14, 4532–4542. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2015, 13, 143. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of Action of Conventional and Targeted Anticancer Therapies: Reinstating Immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Waypa, J.; Blaydorn, L.; Coats, J.; McGahey, K.; Sangal, A.; Niu, J.; Lynch, C.A.; Farley, J.H.; Khemka, V. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br. J. Cancer 2017, 117, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daamen, L.A.; Groot, V.P.; Goense, L.; Wessels, F.J.; Rinkes, I.H.B.; Intven, M.P.W.; van Santvoort, H.C.; Molenaar, I.Q. The diagnostic performance of CT versus FDG PET-CT for the detection of recurrent pancreatic cancer: A systematic review and meta-analysis. Eur. J. Radiol. 2018, 106, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Schwenck, J.; Schörg, B.; Fiz, F.; Sonanini, D.; Forschner, A.; Eigentler, T.; Weide, B.; Martella, M.; Gonzalez-Menendez, I.; Campi, C.; et al. Cancer immunotherapy is accompanied by distinct metabolic patterns in primary and secondary lymphoid organs observed by non-invasive in vivo (18)F-FDG-PET. Theranostics 2020, 10, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Huisman, M.C.; Niemeijer, A.L.N.; Windhorst, A.D.; Schuit, R.C.; Leung, D.; Hayes, W.; Poot, A.; Bahce, I.; Radonic, T.; Oprea-Lager, D.E.; et al. Quantification of PD-L1 expression with [(18)F]BMS-986192 PET/CT in patients with advanced stage non-small-cell lung cancer. J. Nucl. Med. 2020, 61, 1455–1460. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion | Exclusion |
|---|---|
| Radiologically and histopathologically proven stage IV pancreatic cancer (according to the AJCC staging system for pancreatic cancer [52]). | Brain metastases. |
| Max. 5 unequivocal metastases ≥ 1 cm at the time of inclusion (i.e., after FOLFIRINOX). | Active epilepsy (last convulsion < 5 years). |
| Primary tumor is in situ. | History of cardiac disease:
|
| A minimum of 8 cycles of FOLFIRINOX chemotherapy is required before study inclusion, with at least stable disease according to RECIST. | Known hypersensitivity to any oligodeoxynucleotides. |
| Age ≥ 18 years. | Compromised liver function defined as warning signs of portal hypertension, INR > 1,5 without use of anticoagulants, bilirubin > × 1.5 Upper limit of normal range (ULN) ASAT > 3.0 × ULN, ALAT > 3.0 × ULN. |
| World Health Organization (WHO) scale performance status 0–2. | Compromised kidney function defined as eGFR < 30 mL/min (using the Cockcroft Gault formula). |
| Adequate bile drainage in case of biliary obstruction. | Active autoimmune disease requiring disease-modifying therapy at the time of screening, i.e., >10 mg prednisolone per day or equivalent to this regimen. |
| Uncontrolled hypertension. Blood pressure must be ≤160/95 mmHg at the time of screening on a stable antihypertensive regimen. | |
| Uncontrolled infections (>grade 2 NCI-CTC version 3.0) requiring antibiotics. | |
| Immunotherapy prior to the procedure for the treatment of cancer. | |
| Previous surgical therapy for pancreatic cancer. | |
| Second primary malignancy with median 5-year OS < 90%. This excludes adequately treated cancers such as non-melanoma skin cancer, in situ carcinoma of the cervix uteri, superficial bladder cancer, or other malignancies that have been previously treated without signs of recurrence. | |
| Allergy to contrast agent. | |
| Allergy to PET tracers 18F-FDG and 18F-BMS-986192. | |
| Any implanted stimulation device. | |
| Portal vein or VMS stenosis > 70%, or any arterial stenosis (superior mesenteric artery, celiac artery, common hepatic artery) > 70% unless effectively stented. | |
| Any condition that is unstable or that could jeopardize the safety of the subject and their compliance in the study. |
| Lab Test | Prior to First Cycle of Nivolumab | Prior to Consecutive Cycles of Nivolumab |
|---|---|---|
| Full blood: | hemoglobin/leukocytes and differentiation/thrombocytes | hemoglobin/leukocytes and differentiation/thrombocytes |
| Electrolytes: | natrium/potassium/calcium/magnesium/phosphate | natrium/potassium |
| Liver function: | albumin/glucose/lipase/bilirubin/Alkaline phosphatase/γ-glutamine transferase/aspartate-aminotransferase/alanine-aminotransferase/lactate-dehydrogenase | albumin/glucose/lipase/bilirubin/Alkaline phosphatase/γ-glutamine transferase/aspartate-aminotransferase/alanine-aminotransferase/lactate-dehydrogenase |
| Kidney function: | creatinine/urea | creatinine |
| Thyroid function: | thyroid stimulating hormone/thyroxin | thyroid stimulating hormone/thyroxin |
| Acute phase proteins: | c-reactive protein | c-reactive protein |
| Hormones: | cortisol/luteinizing hormone/follicle stimulating hormone/adrenocorticotropic hormone | |
| Tumor markers: | CA19.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geboers, B.; Timmer, F.E.F.; Ruarus, A.H.; Pouw, J.E.E.; Schouten, E.A.C.; Bakker, J.; Puijk, R.S.; Nieuwenhuizen, S.; Dijkstra, M.; van den Tol, M.P.; et al. Irreversible Electroporation and Nivolumab Combined with Intratumoral Administration of a Toll-Like Receptor Ligand, as a Means of In Vivo Vaccination for Metastatic Pancreatic Ductal Adenocarcinoma (PANFIRE-III). A Phase-I Study Protocol. Cancers 2021, 13, 3902. https://doi.org/10.3390/cancers13153902
Geboers B, Timmer FEF, Ruarus AH, Pouw JEE, Schouten EAC, Bakker J, Puijk RS, Nieuwenhuizen S, Dijkstra M, van den Tol MP, et al. Irreversible Electroporation and Nivolumab Combined with Intratumoral Administration of a Toll-Like Receptor Ligand, as a Means of In Vivo Vaccination for Metastatic Pancreatic Ductal Adenocarcinoma (PANFIRE-III). A Phase-I Study Protocol. Cancers. 2021; 13(15):3902. https://doi.org/10.3390/cancers13153902
Chicago/Turabian StyleGeboers, Bart, Florentine E. F. Timmer, Alette H. Ruarus, Johanna E. E. Pouw, Evelien A. C. Schouten, Joyce Bakker, Robbert S. Puijk, Sanne Nieuwenhuizen, Madelon Dijkstra, M. Petrousjka van den Tol, and et al. 2021. "Irreversible Electroporation and Nivolumab Combined with Intratumoral Administration of a Toll-Like Receptor Ligand, as a Means of In Vivo Vaccination for Metastatic Pancreatic Ductal Adenocarcinoma (PANFIRE-III). A Phase-I Study Protocol" Cancers 13, no. 15: 3902. https://doi.org/10.3390/cancers13153902
APA StyleGeboers, B., Timmer, F. E. F., Ruarus, A. H., Pouw, J. E. E., Schouten, E. A. C., Bakker, J., Puijk, R. S., Nieuwenhuizen, S., Dijkstra, M., van den Tol, M. P., de Vries, J. J. J., Oprea-Lager, D. E., Menke-van der Houven van Oordt, C. W., van der Vliet, H. J., Wilmink, J. W., Scheffer, H. J., de Gruijl, T. D., Meijerink, M. R., & on behalf of the Dutch Pancreatic Cancer Group. (2021). Irreversible Electroporation and Nivolumab Combined with Intratumoral Administration of a Toll-Like Receptor Ligand, as a Means of In Vivo Vaccination for Metastatic Pancreatic Ductal Adenocarcinoma (PANFIRE-III). A Phase-I Study Protocol. Cancers, 13(15), 3902. https://doi.org/10.3390/cancers13153902