
Protective and Therapeutic Effects of an IL-15:IL-15Rα-Secreting Cell-Based Cancer Vaccine Using a Baculovirus System

Abstract
:Simple Summary
Abstract
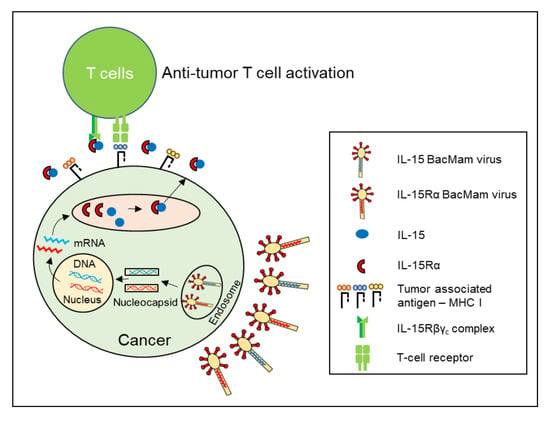
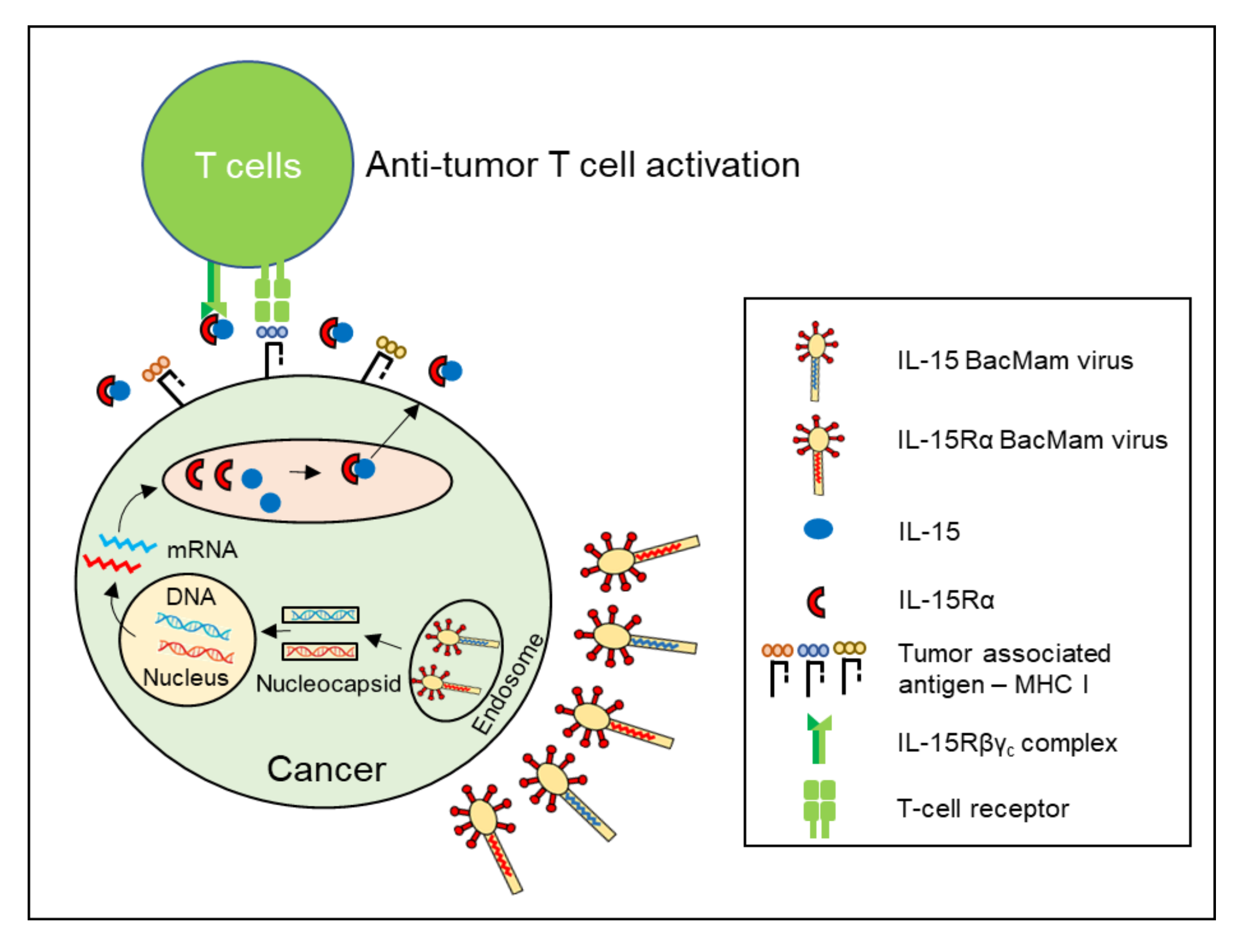{kind=link}
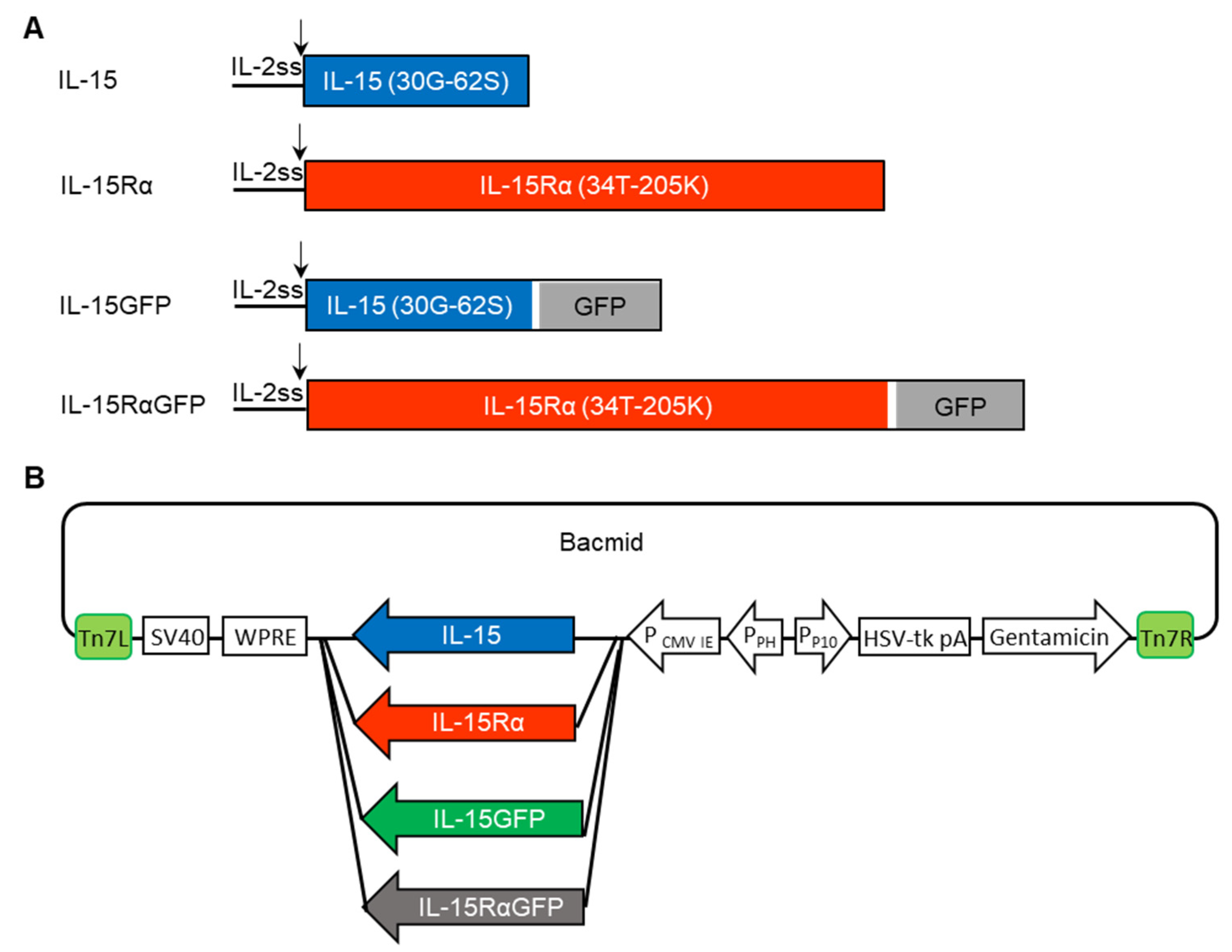{kind=link}
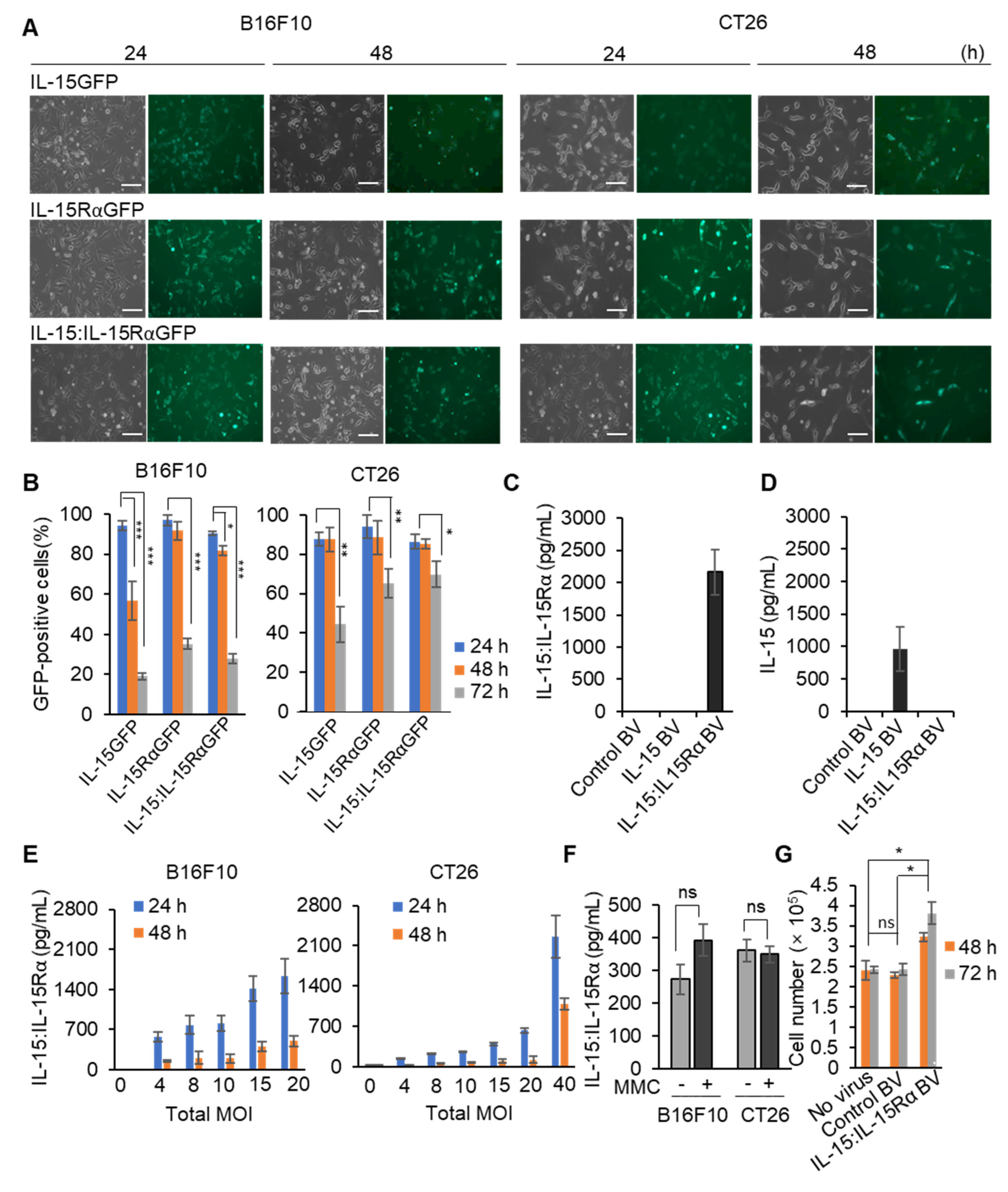{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Tumor Cell Lines
2.2. Antibodies and Reagents
2.3. Generation of OVA-Expressing Cancer Cell Lines
2.4. Recombinant Baculovirus Generation
2.5. In Vitro Viral Delivery
2.6. In Vivo Immunization
2.7. ELISA
2.8. Splenocyte Proliferation Assay
2.9. Flow Cytometric Analysis
2.10. In Vitro Stimulation of Splenocytes and the Cytotoxicity Assay
2.11. In Vitro Cell Proliferation Assay
2.12. Viral Stability Study
2.13. Statistical Analysis
3. Results
3.1. BacMam Viruses Successfully Mediated Multiple Gene Delivery to Murine Cancer Cell Lines
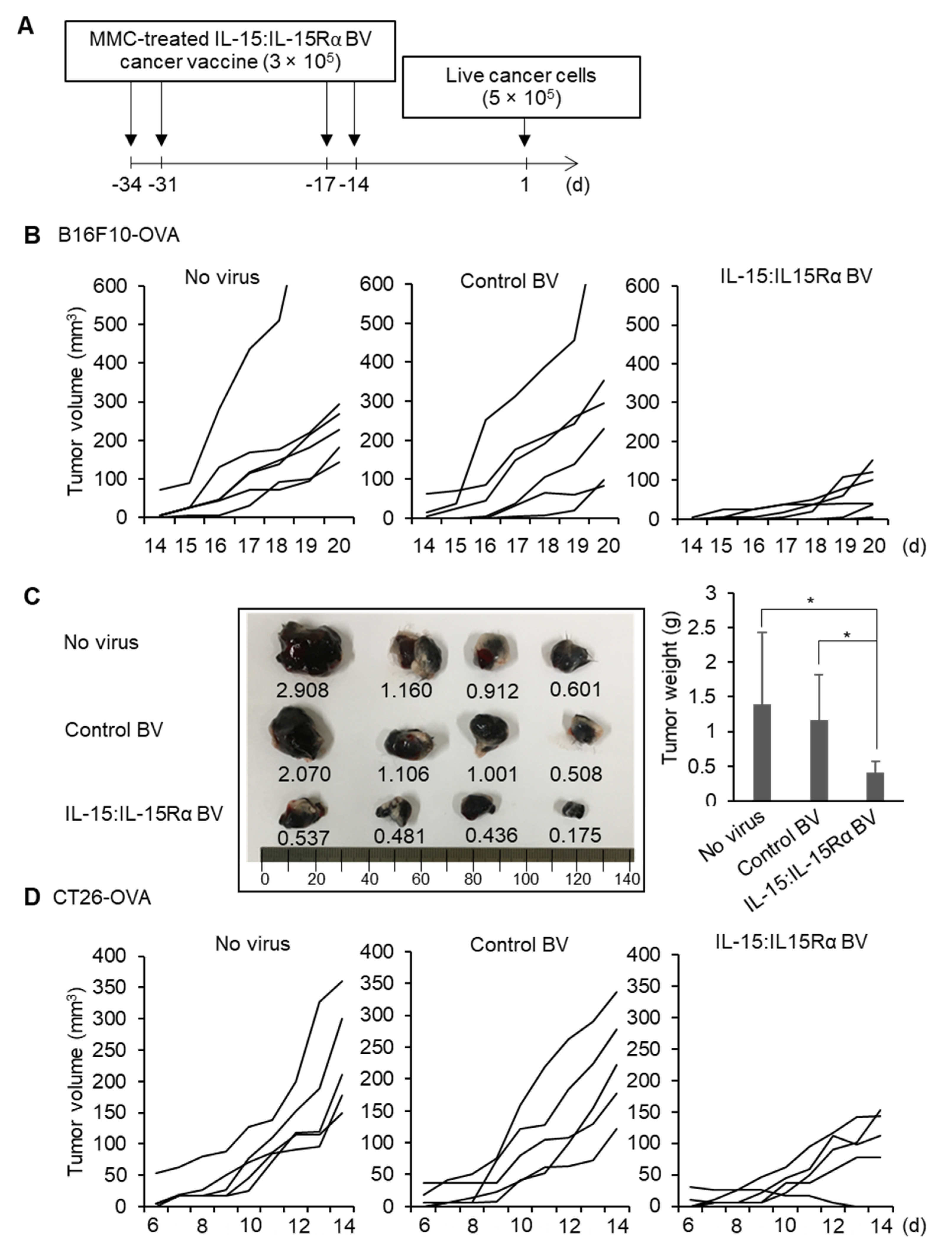
3.2. BacMam-Based IL-15:IL-15Rα-Secreting Autologous Cancer Cells Triggered Antitumor Protection in Mice
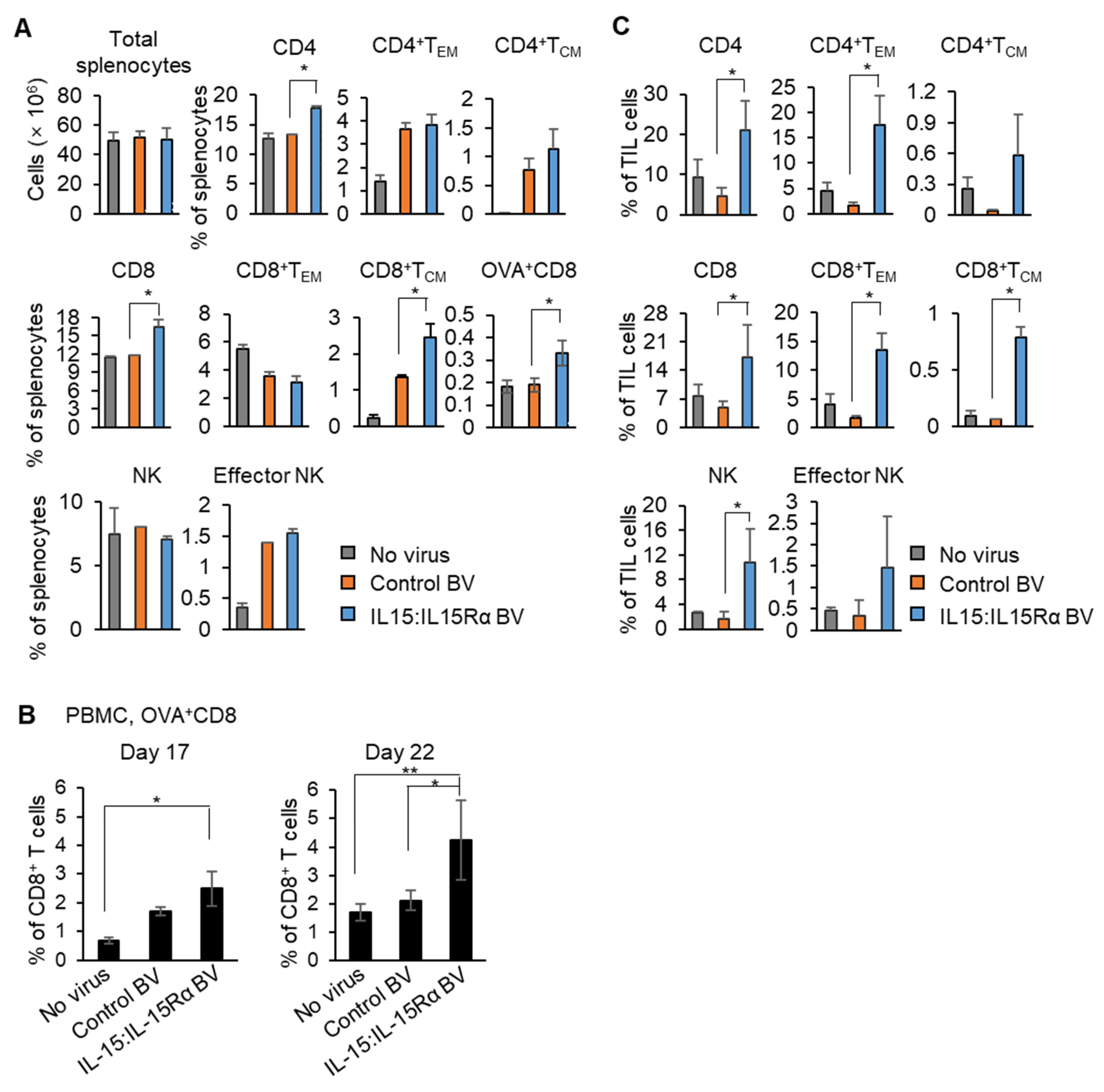
3.3. Immunization with IL-15:IL-15Rα-B16F10-OVA Induced a Robust Antitumor Response Depending on Tumor Antigen-Specific CD8+ T Cells
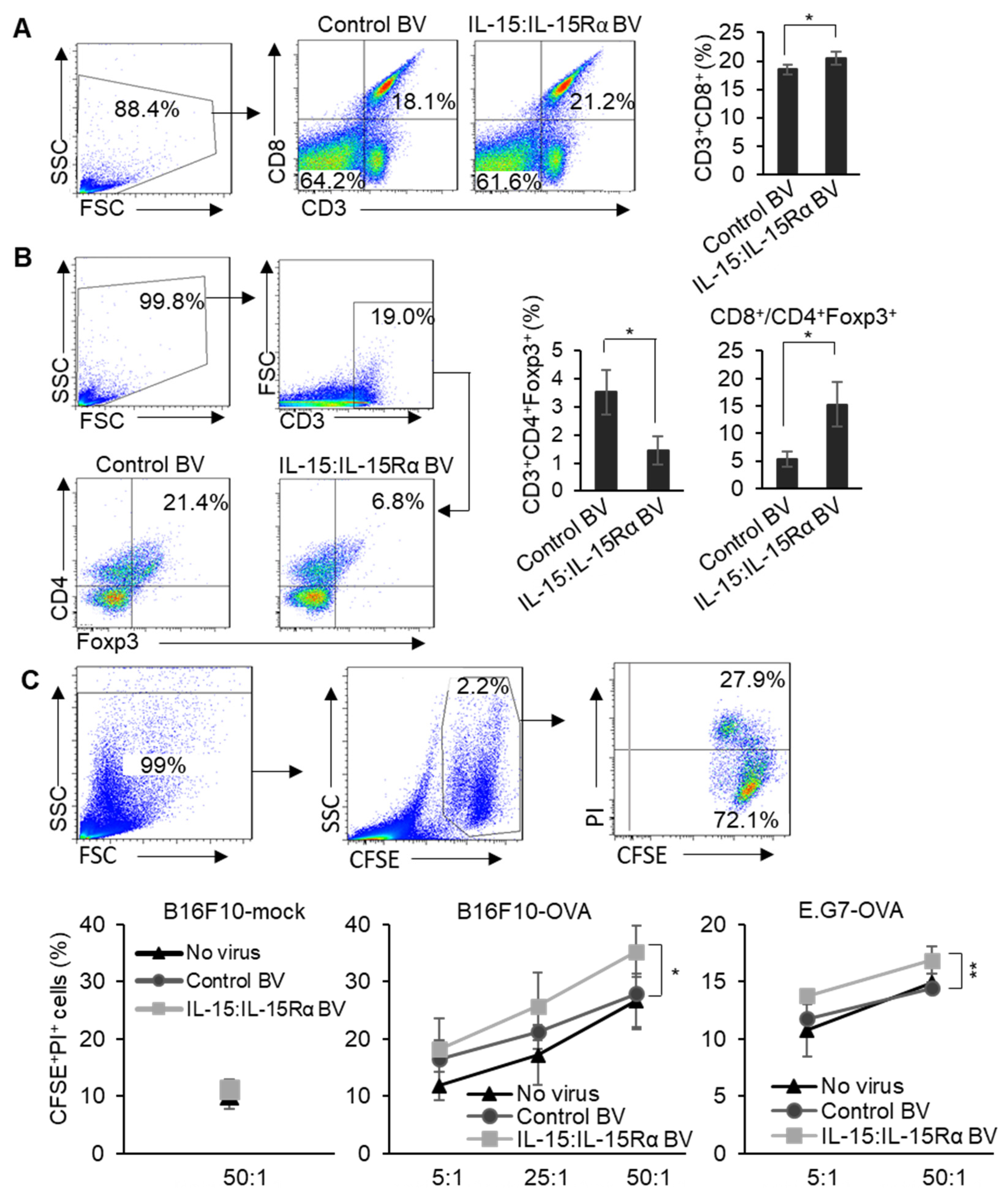
3.4. Immunizing with the IL-15:IL-15Rα-B16F10-OVA Vaccine Specifically Enhanced Cytotoxic T Cell Activity against B16F10-OVA Cells
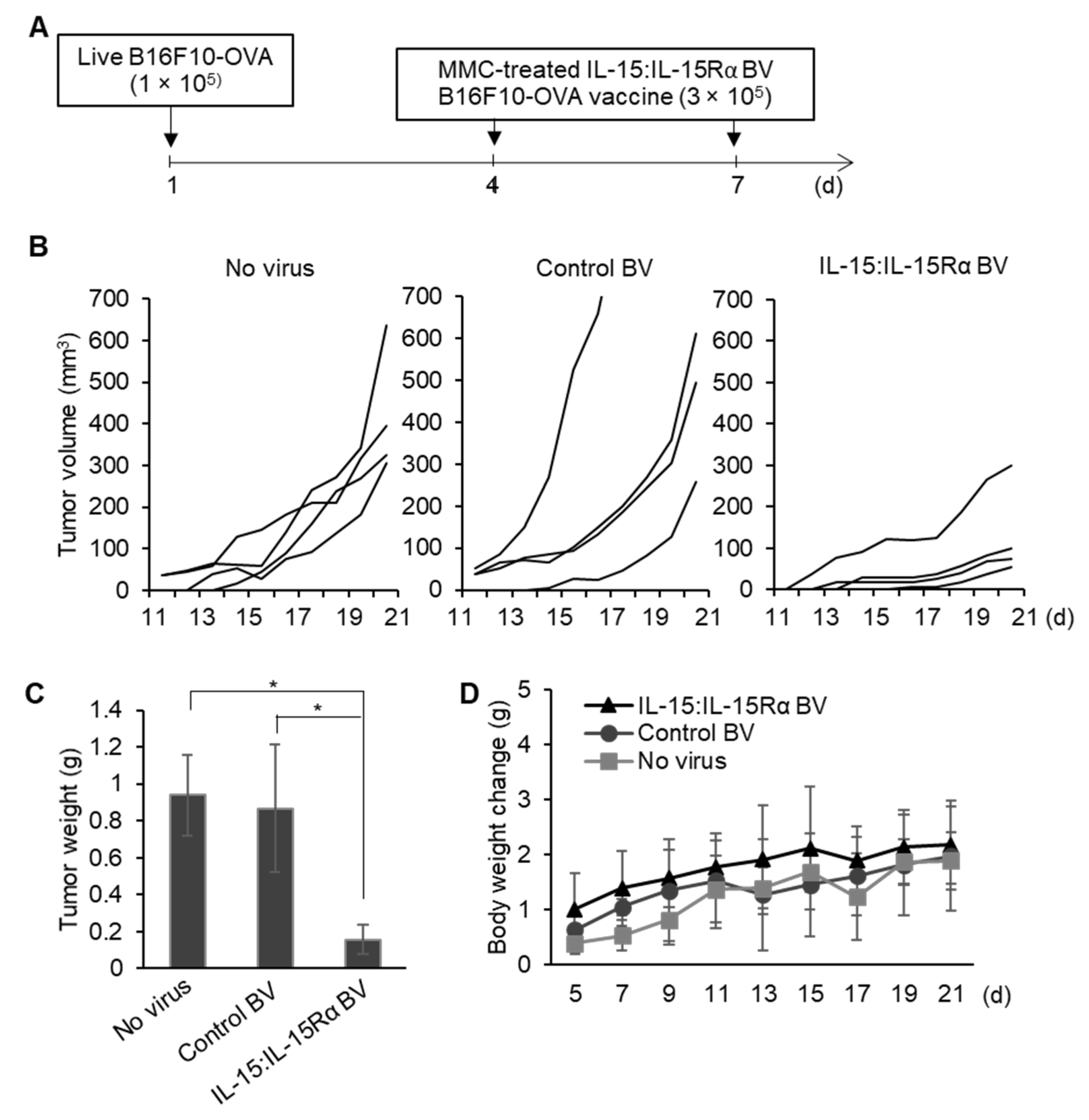
3.5. The Therapeutic Effect of Post-Treatment with IL-15:IL-15Rα-B16F10-OVA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meghnem, D.; Morisseau, S.; Frutoso, M.; Trillet, K.; Maillasson, M.; Barbieux, I.; Khaddage, S.; Leray, I.; Hildinger, M.; Quemener, A.; et al. Cutting Edge: Differential Fine-Tuning of IL-2- and IL-15-Dependent Functions by Targeting Their Common IL-2/15Rbeta/gammac Receptor. J. Immunol. 2017, 198, 4563–4568. [Google Scholar] [CrossRef] [Green Version]
- Knudson, K.M.; Hodge, J.W.; Schlom, J.; Gameiro, S.R. Rationale for IL-15 superagonists in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 705–709. [Google Scholar] [CrossRef] [Green Version]
- Thi, V.A.D.; Jeon, H.M.; Park, S.M.; Lee, H.; Kim, Y.S. Cell-Based IL-15:IL-15Ralpha Secreting Vaccine as an Effective Therapy for CT26 Colon Cancer in Mice. Mol. Cells 2019, 42, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.; Patel, H.J.; Zhang, M.; Waldmann, T.A.; Muller, J.R. Preassociation of IL-15 with IL-15R alpha-IgG1-Fc enhances its activity on proliferation of NK and CD8+/CD44high T cells and its antitumor action. J. Immunol. 2008, 180, 2099–2106. [Google Scholar] [CrossRef] [Green Version]
- Epardaud, M.; Elpek, K.G.; Rubinstein, M.P.; Yonekura, A.R.; Bellemare-Pelletier, A.; Bronson, R.; Hamerman, J.A.; Goldrath, A.W.; Turley, S.J. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. 2008, 68, 2972–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.S.; Kwilas, A.R.; Xu, W.; Alter, S.; Jeng, E.K.; Wong, H.C.; Schlom, J.; Hodge, J.W. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016, 7, 16130–16145. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Hu, Q.; Ye, X.; Qu, X.; Cui, D.; Zhang, L.; Xu, Z.; Wan, H.; Zhang, L.; Tao, W. Discovery of a novel IL-15 based protein with improved developability and efficacy for cancer immunotherapy. Sci. Rep. 2018, 8, 7675. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.; Quemener, A.; Jacques, Y. Antitumor activity of an immunocytokine composed of an anti-GD2 antibody and the IL-15 superagonist RLI. Oncoimmunology 2013, 2, e26441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochems, C.; Tritsch, S.R.; Knudson, K.M.; Gameiro, S.R.; Rumfield, C.S.; Pellom, S.T.; Morillon, Y.M.; Newman, R.; Marcus, W.; Szeto, C.; et al. The multi-functionality of N-809, a novel fusion protein encompassing anti-PD-L1 and the IL-15 superagonist fusion complex. Oncoimmunology 2019, 8, e1532764. [Google Scholar] [CrossRef] [Green Version]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Y.; Sun, X.; Muftuoglu, Y.; Wang, B.; Yu, T.; Hu, Y.; Ma, L.; Xiang, M.; Guo, G.; et al. Powerful anti-colon cancer effect of modified nanoparticle-mediated IL-15 immunogene therapy through activation of the host immune system. Theranostics 2018, 8, 3490–3503. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.; Usiskin, I.M.; Bergamaschi, C.; Hanlon, D.J.; Edelson, R.L.; Justesen, S.; Pavlakis, G.N.; Flavell, R.A.; Fahmy, T.M. Configuration-dependent Presentation of Multivalent IL-15:IL-15Ralpha Enhances the Antigen-specific T Cell Response and Anti-tumor Immunity. J. Biol. Chem. 2016, 291, 8931–8950. [Google Scholar] [CrossRef] [Green Version]
- Wege, A.K.; Weber, F.; Kroemer, A.; Ortmann, O.; Nimmerjahn, F.; Brockhoff, G. IL-15 enhances the anti-tumor activity of trastuzumab against breast cancer cells but causes fatal side effects in humanized tumor mice (HTM). Oncotarget 2017, 8, 2731–2744. [Google Scholar] [CrossRef]
- Paul, A.; Hasan, A.; Rodes, L.; Sangaralingam, M.; Prakash, S. Bioengineered baculoviruses as new class of therapeutics using micro and nanotechnologies: Principles, prospects and challenges. Adv. Drug. Deliv. Rev. 2014, 71, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.O.; Schluns, K.S. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Dubois, S.; Miljkovic, M.D.; Conlon, K.C. IL-15 in the Combination Immunotherapy of Cancer. Front Immunol. 2020, 11, 868. [Google Scholar] [CrossRef]
- Morris, J.C.; Ramlogan-Steel, C.A.; Yu, P.; Black, B.A.; Mannan, P.; Allison, J.P.; Waldmann, T.A.; Steel, J.C. Vaccination with tumor cells expressing IL-15 and IL-15Ralpha inhibits murine breast and prostate cancer. Gene. Ther. 2014, 21, 393–401. [Google Scholar] [CrossRef]
- Berger, A.; Colpitts, S.J.; Seabrook, M.S.S.; Furlonger, C.L.; Bendix, M.B.; Moreau, J.M.; McKillop, W.M.; Medin, J.A.; Paige, C.J. Interleukin-15 in cancer immunotherapy: IL-15 receptor complex versus soluble IL-15 in a cancer cell-delivered murine leukemia model. J. Immunother. Cancer 2019, 7, 355. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Liang, C.-y.; Chen, X.-w. Baculoviruses as vectors in mammalian cells. Virol. Sin. 2007, 22, 148. [Google Scholar] [CrossRef]
- Mansouri, M.; Bellon-Echeverria, I.; Rizk, A.; Ehsaei, Z.; Cianciolo Cosentino, C.; Silva, C.S.; Xie, Y.; Boyce, F.M.; Davis, M.W.; Neuhauss, S.C.; et al. Highly efficient baculovirus-mediated multigene delivery in primary cells. Nat. Commun. 2016, 7, 11529. [Google Scholar] [CrossRef] [Green Version]
- Fukui, M.; Zhu, B.T. Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radic. Biol. Med. 2010, 48, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Fornwald, J.A.; Lu, Q.; Boyce, F.M.; Ames, R.S. Gene Expression in Mammalian Cells Using BacMam, a Modified Baculovirus System. Methods Mol. Biol. 2016, 1350, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Condreay, J.P.; Watson, C.A. Pharmacological applications of baculovirus-mediated protein expression in mammalian cells. Curr. Protoc. Pharm. 2010, 49, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Condreay, J.P.; Witherspoon, S.M.; Clay, W.C.; Kost, T.A. Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc. Natl. Acad. Sci. USA 1999, 96, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.; Jardin, B.A.; Kulamarva, A.; Malhotra, M.; Elias, C.B.; Prakash, S. Recombinant baculovirus as a highly potent vector for gene therapy of human colorectal carcinoma: Molecular cloning, expression, and in vitro characterization. Mol. Biotechnol. 2010, 45, 129–139. [Google Scholar] [CrossRef]
- Hitchman, E.; Hitchman, R.B.; King, L.A. BacMam Delivery of a Protective Gene to Reduce Renal Ischemia-Reperfusion Injury. Hum Gene Ther. 2017, 28, 747–756. [Google Scholar] [CrossRef]
- Sprick, G.; Weidner, T.; Salzig, D.; Czermak, P. Baculovirus-induced recombinant protein expression in human mesenchymal stromal stem cells: A promoter study. New Biotechnol. 2017, 39, 161–166. [Google Scholar] [CrossRef]
- Shukla, S.; Schwartz, C.; Kapoor, K.; Kouanda, A.; Ambudkar, S.V. Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug. Metab. Dispos. 2012, 40, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Li, F.; Yang, Y.; Guo, H.Y.; Wu, C.X.; Wang, S. Recombinant baculovirus containing the diphtheria toxin A gene for malignant glioma therapy. Cancer Res. 2006, 66, 5798–5806. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Fang, L.; Fan, H.; Luo, R.; Zhao, Q.; Chen, H.; Xiao, S. Antitumor effects of a recombinant pseudotype baculovirus expressing Apoptin in vitro and in vivo. Int. J. Cancer 2010, 126, 2741–2751. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.Y.; Shih, Y.S.; Lo, W.H.; Chen, H.R.; Wang, S.C.; Wang, C.H.; Chien, C.H.; Chiang, C.S.; Chuang, Y.J.; Hu, Y.C. Baculovirus vectors for antiangiogenesis-based cancer gene therapy. Cancer Gene. Ther. 2011, 18, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, M.; Takaku, H. Intradermal immunization with combined baculovirus and tumor cell lysate induces effective antitumor immunity in mice. Int. J. Oncol. 2013, 43, 2023–2030. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Chung, Y.-C.; Hu, Y.-C. Update on baculovirus as an expression and/or delivery vehicle for vaccine antigens. Expert Rev. Vaccines 2014, 13, 1501–1521. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sanderson, N.S.; Wawrowsky, K.; Puntel, M.; Castro, M.G.; Lowenstein, P.R. Kupfer-type immunological synapse characteristics do not predict anti-brain tumor cytolytic T-cell function in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4716–4721. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Shin, S.J.; Lee, M.H.; Lee, M.G.; Kang, T.H.; Park, W.S.; Soh, B.Y.; Park, J.H.; Shin, Y.K.; Kim, H.W.; et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS ONE 2014, 9, e104351. [Google Scholar] [CrossRef]
- Tagaya, Y.; Kurys, G.; Thies, T.A.; Losi, J.M.; Azimi, N.; Hanover, J.A.; Bamford, R.N.; Waldmann, T.A. Generation of secretable and nonsecretable interleukin 15 isoforms through alternate usage of signal peptides. Proc. Natl. Acad. Sci. USA 1997, 94, 14444–14449. [Google Scholar] [CrossRef] [Green Version]
- Bamford, R.N.; DeFilippis, A.P.; Azimi, N.; Kurys, G.; Waldmann, T.A. The 5’ untranslated region, signal peptide, and the coding sequence of the carboxyl terminus of IL-15 participate in its multifaceted translational control. J. Immunol. 1998, 160, 4418–4426. [Google Scholar]
- Kurys, G.; Tagaya, Y.; Bamford, R.; Hanover, J.A.; Waldmann, T.A. The long signal peptide isoform and its alternative processing direct the intracellular trafficking of interleukin-15. J. Biol. Chem. 2000, 275, 30653–30659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goehring, A.; Lee, C.H.; Wang, K.H.; Michel, J.C.; Claxton, D.P.; Baconguis, I.; Althoff, T.; Fischer, S.; Garcia, K.C.; Gouaux, E. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 2014, 9, 2574–2585. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, R.; Esposito, D. A rapid method for titrating baculovirus stocks using the Sf-9 Easy Titer cell line. Biotechniques 2009, 47, 785–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaco, B.; Pires, M.J.; Colaco, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab. Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef]
- Pulle, G.; Vidric, M.; Watts, T.H. IL-15-dependent induction of 4-1BB promotes antigen-independent CD8 memory T cell survival. J. Immunol. 2006, 176, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Ochyl, L.J.; Moon, J.J. Dendritic Cell Membrane Vesicles for Activation and Maintenance of Antigen-Specific T Cells. Adv. Healthc. Mater. 2019, 8, e1801091. [Google Scholar] [CrossRef]
- Paz, M.M.; Zhang, X.; Lu, J.; Holmgren, A. A new mechanism of action for the anticancer drug mitomycin C: Mechanism-based inhibition of thioredoxin reductase. Chem. Res. Toxicol. 2012, 25, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Yamamoto, W.; Park, J.S.; Hanaoka, H.; Okamoto, R.; Kirihara, Y.; Yorishima, T.; Okamura, T.; Kumazaki, T.; Nishiyama, M. Regulatory network of mitomycin C action in human colon cancer cells. Jpn. J. Cancer Res. 1999, 90, 571–577. [Google Scholar] [CrossRef]
- Guo, Y.; Luan, L.; Rabacal, W.; Bohannon, J.K.; Fensterheim, B.A.; Hernandez, A.; Sherwood, E.R. IL-15 Superagonist-Mediated Immunotoxicity: Role of NK Cells and IFN-γ. J. Immunol. 2015, 195, 2353–2364. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.; Berger, M.; Hackman, R.C.; Gough, M.; Elliott, C.; Jensen, M.C.; Riddell, S.R. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood 2009, 114, 2417–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiore, P.F.; Di Matteo, S.; Tumino, N.; Mariotti, F.R.; Pietra, G.; Ottonello, S.; Negrini, S.; Bottazzi, B.; Moretta, L.; Mortier, E.; et al. Interleukin-15 and cancer: Some solved and many unsolved questions. J. Immunother. Cancer 2020, 8, e001428. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2021, 11, 5240. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, L.; Chowdhary, A.; Sosman, J.A.; Chandra, S. Combining Tumor Vaccination and Oncolytic Viral Approaches with Checkpoint Inhibitors: Rationale, Pre-Clinical Experience, and Current Clinical Trials in Malignant Melanoma. Am. J. Clin. Dermatol. 2018, 19, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Palucka, K. Immunotherapy: Cancer vaccines on the move. Nat. Rev. Clin. Oncol. 2018, 15, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Aldous, A.R.; Dong, J.Z. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorg. Med. Chem. 2018, 26, 2842–2849. [Google Scholar] [CrossRef]
- Alard, E.; Butnariu, A.-B.; Grillo, M.; Kirkham, C.; Zinovkin, D.A.; Newnham, L.; Macciochi, J.; Pranjol, M.Z.I. Advances in Anti-Cancer Immunotherapy: Car-T Cell, Checkpoint Inhibitors, Dendritic Cell Vaccines, and Oncolytic Viruses, and Emerging Cellular and Molecular Targets. Cancers 2020, 12, 1826. [Google Scholar] [CrossRef]
- Nandi, D.; Pathak, S.; Verma, T.; Singh, M.; Chattopadhyay, A.; Thakur, S.; Raghavan, A.; Gokhroo, A.; Vijayamahantesh. T cell costimulation, checkpoint inhibitors and anti-tumor therapy. J. Biosci. 2020, 45, 50. [Google Scholar] [CrossRef]
- McNeel, D.G. Therapeutic Cancer Vaccines: How Much Closer Are We? BioDrugs 2018, 32, 1–7. [Google Scholar] [CrossRef]
- Ye, Z.; Qian, Q.; Jin, H.; Qian, Q. Cancer vaccine: Learning lessons from immune checkpoint inhibitors. J. Cancer 2018, 9, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do-Thi, V.A.; Lee, H.; Jeong, H.J.; Lee, J.-O.; Kim, Y.S. Protective and Therapeutic Effects of an IL-15:IL-15Rα-Secreting Cell-Based Cancer Vaccine Using a Baculovirus System. Cancers 2021, 13, 4039. https://doi.org/10.3390/cancers13164039
Do-Thi VA, Lee H, Jeong HJ, Lee J-O, Kim YS. Protective and Therapeutic Effects of an IL-15:IL-15Rα-Secreting Cell-Based Cancer Vaccine Using a Baculovirus System. Cancers. 2021; 13(16):4039. https://doi.org/10.3390/cancers13164039
Chicago/Turabian StyleDo-Thi, Van Anh, Hayyoung Lee, Hye Jin Jeong, Jie-Oh Lee, and Young Sang Kim. 2021. "Protective and Therapeutic Effects of an IL-15:IL-15Rα-Secreting Cell-Based Cancer Vaccine Using a Baculovirus System" Cancers 13, no. 16: 4039. https://doi.org/10.3390/cancers13164039
APA StyleDo-Thi, V. A., Lee, H., Jeong, H. J., Lee, J. -O., & Kim, Y. S. (2021). Protective and Therapeutic Effects of an IL-15:IL-15Rα-Secreting Cell-Based Cancer Vaccine Using a Baculovirus System. Cancers, 13(16), 4039. https://doi.org/10.3390/cancers13164039