
Histone Modifying Enzymes as Targets for Therapeutic Intervention in Oesophageal Adenocarcinoma





Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Oesophageal Adenocarcinoma
1.2. Epigenetics Overview
1.3. Histone Modifications
2. Histone-Modifying Enzymes in OAC
2.1. Histone Deacetylases in OAC Pathogenesis
2.2. HDAC Inhibitors for the Treatment of OAC
2.3. Histone Methyltransferases in OAC
2.4. Potential for EZH2 Inhibition in OAC
3. Tumour Microenvironment and HMEs
3.1. Tumour Microenivironment in OAC
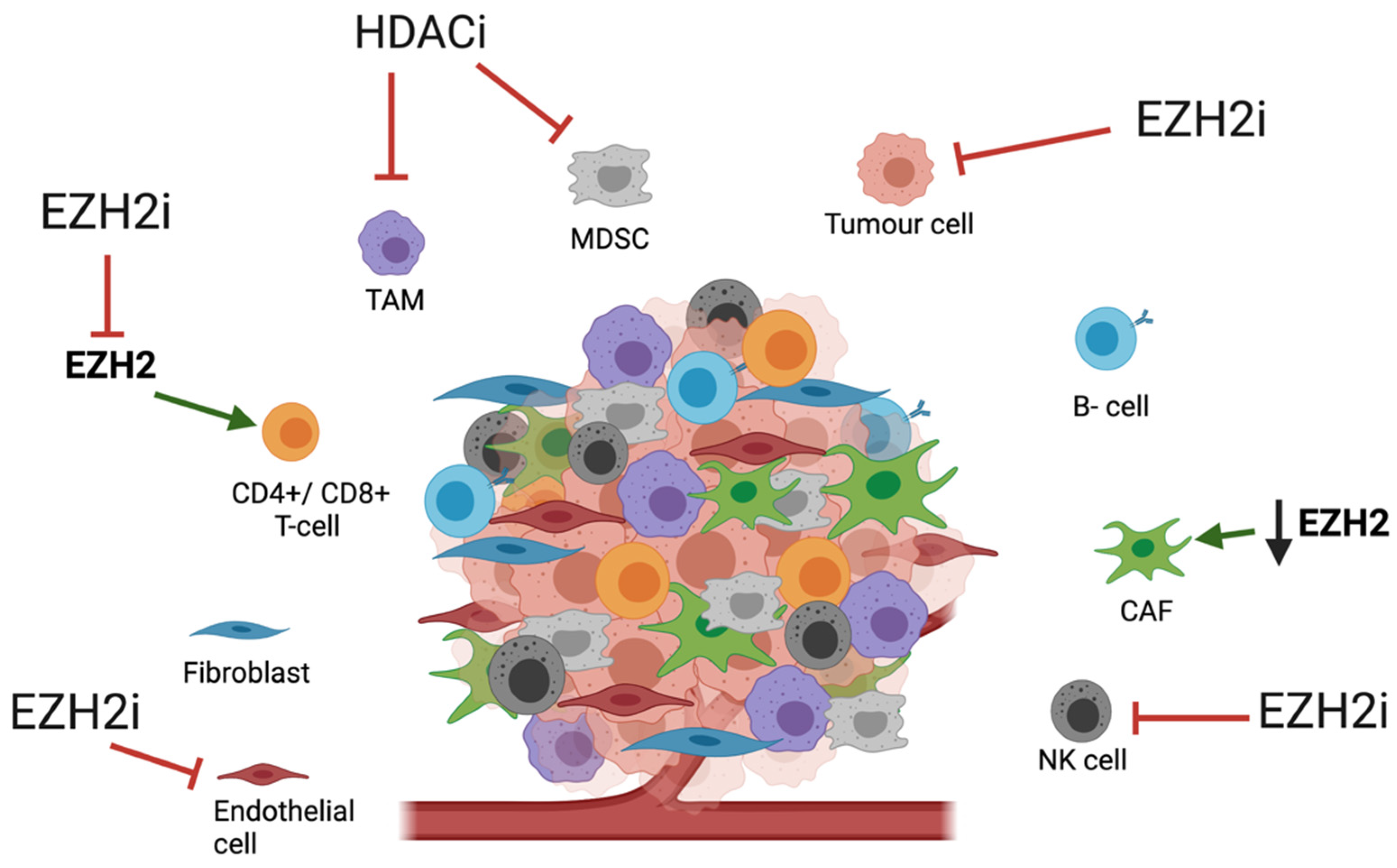
3.2. Impact of HME Therapies on the TME
4. Combination Therapies
5. Future Considerations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3D-TGA | 3 dimensional-tumour growth assay |
| ADAMTS1 | ADAM metallopeptidase with thrombospondin type 1 motif |
| ARID1A | AT-Rich Interaction Domain 1A |
| AZA | Azacytidine |
| BET | Bromodomain and Extra- Terminal motif protein |
| CAF | Cancer-associated fibroblast |
| CTCL | Primary cutaneous T-cell lymphoma |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DAC | Decitabine |
| DNMT | DNA methyltransferase |
| DNMTi | DNA methyltransferase inhibitors |
| ECF | Epiribucin, cisplatin and fluorouracil |
| EZH2 | Enhancer of zeste homolog 2 |
| EZH2i | EZH2 inhibitor |
| FDA | US Food and Drug Administration |
| FLOT | Docetaxel, oxaliplatin, leucovorin and 5-FU |
| H3K27 | Histone 3 lysine 27 |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HDACi | Histone deacetylase inhibitor |
| HDM | Histone demethylases |
| HME | Histone modifying enzyme |
| HMEi | HME inhibitors |
| HMT | Histone methyltransferases |
| MTA1 | Metastasis-associated gene 1 |
| MDSC | Myeloid-derived suppressor cells |
| NK cell | Natural killer cell |
| OAC | Oesophageal adenocarcinoma |
| OCCC | Ovarian clear cell carcinoma |
| OSCC | Oesophageal squamous cell carcinoma |
| PD-1 | Programmed cell death protein 1 |
| PDE5i | Phosphodiesterase type 5 inhibitors |
| PRC2 | Polycomb Repressive Complex 2 |
| PTCL | Peripheral T-cell lymphoma |
| qRT-PCR | Real-Time Quantitative Reverse Transcription PCR) |
| SAM | S-adenosyl-L-methionine |
| SMARCA4 | SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A, Member 4 |
| SOC | Standard of care |
| SWI/SNF | SWItch/Sucrose Non-Fermentable complex |
| TAM | Tumour-associated macrophages |
| TGF-β | Transforming growth factor-β |
| TME | Tumour microenvironment |
| Tregs | Regulatory T-cells |
| TXNIP | Thioredoxin Interacting Protein |
| VEGF | Vascular endothelial growth factor |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubecz, A.; Gall, I.; Solymosi, N.; Schweigert, M.; Peters, J.H.; Feith, M.; Stein, H.J. Temporal Trends in Long-Term Survival and Cure Rates in Esophageal Cancer: A SEER Database Analysis. J. Thorac. Oncol. 2012, 7, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Gavin, A.T.; Francisci, S.; Foschi, R.; Donnelly, D.; Lemmens, V.; Brenner, H.; Anderson, L. Oesophageal cancer survival in Europe: A EUROCARE-4 study. Cancer Epidemiol. 2012, 36, 505–512. [Google Scholar] [CrossRef]
- Kauppila, J.H.; Mattsson, F.; Brusselaers, N.; Lagergren, J. Prognosis of oesophageal adenocarcinoma and squamous cell carcinoma following surgery and no surgery in a nationwide Swedish cohort study. BMJ Open 2018, 8, 21495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, Y.T.; Zheng, R.S.; Zhang, S.W.; Chen, W.Q. Analysis of esophageal cancer time trends in china, 1989–2008. Asian Pac. J. Cancer Prev. 2012, 13, 4613–4617. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.B.; Chow, W.H.; Devesa, S.S. Oesophageal cancer incidence in the United States by race, sex, and histologic type, 1977–2005. Br. J. Cancer 2009, 101, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal cancer. Nat. Rev. Dis. Prim. 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Levi, F.; Ferlay, J.; Garavello, W.; Lucchini, F.; Bertuccio, P.; Negri, E.; La Vecchia, C. Trends in oesophageal cancer incidence and mortality in Europe. Int. J. Cancer 2008, 122, 1118–1129. [Google Scholar] [CrossRef]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am. J. Gastroenterol. 2017, 112, 1247–1255. [Google Scholar] [CrossRef]
- Oesophago-Gastric Cancer Audit. Available online: https://digital.nhs.uk/data-and-information/clinical-audits-and-registries/national-oesophago-gastric-cancer-audit (accessed on 27 June 2021).
- Agarwal, A.; Polineni, R.; Hussein, Z.; Vigoda, I.; Bhagat, T.D.; Bhattacharyya, S.; Maitra, A.; Verma, A. Role of epigenetic alterations in the pathogenesis of Barrett’s esophagus and esophageal adenocarcinoma. Int. J. Clin. Exp. Pathol. 2012, 5, 382–396. [Google Scholar] [PubMed]
- Noble, F.; Lloyd, M.A.; Turkington, R.; Griffiths, E.; O’Donovan, M.; O’Neill, J.R.; Mercer, S.; Parsons, S.L.; Fitzgerald, R.C.; Underwood, T.J.; et al. Multicentre cohort study to define and validate pathological assessment of response to neoadjuvant therapy in oesophagogastric adenocarcinoma. Br. J. Surg. 2017, 104, 1816–1828. [Google Scholar] [CrossRef] [Green Version]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Trojer, P.; Reinberg, D. The key to development: Interpreting the histone code? Curr. Opin. Genet. Dev. 2005, 15, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.W.; Zhu, X.Y.; Li, Y.Y.; Meng, Z.Q. Epigenetic regulation and cancer (review). Oncol. Rep. 2014, 31, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Salta, S.; Macedo-Silva, C.; Miranda-Gonçalves, V.; Lopes, N.; Gigliano, D.; Guimarães, R.; Farinha, M.; Sousa, O.; Henrique, R.; Jerónimo, C. A DNA methylation-based test for esophageal cancer detection. Biomark Res. 2020, 8, 1–9. [Google Scholar] [CrossRef]
- Wu, D.-L.; Sui, F.-Y.; Jiang, X.-M.; Jiang, X.-H. Methylation in esophageal carcinogenesis. World J. Gastroenterol. 2006, 12, 6933–6940. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Food and Drug Administration. FDA Approves First Treatment Option Specifically for Patients with Epithelioid Sarcoma, a Rare Soft Tissue Cancer. 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-option-specifically-patients-epithelioid-sarcoma-rare-soft-tissue (accessed on 27 June 2021).
- US Food and Drug Administration. FDA Granted Accelerated Approval to Tazemetostat for Follicular Lymphoma|FDA. Available online: https://www.fda.gov/drugs/fda-granted-accelerated-approval-tazemetostat-follicular-lymphoma (accessed on 29 January 2021).
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- McDermott, J.; Jimeno, A. Belinostat for the treatment of peripheral T-cell lymphomas. Drugs Today 2014, 50, 337–345. [Google Scholar] [CrossRef]
- Richardson, P.G.; Laubach, J.P.; Lonial, S.; Moreau, P.; Yoon, S.-S.; Hungria, V.T.; Dimopoulos, M.; Beksac, M.; Alsina, M.; San-Miguel, J.F. Panobinostat: A novel pan-deacetylase inhibitor for the treatment of relapsed or relapsed and refractory multiple myeloma. Expert Rev. Anticancer. Ther. 2015, 15, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.; Myers, M.; Axelrod, K.C.; Ness, E.A.; Piekarz, R.L.; Bates, S.E.; Booher, S. Romidepsin: A new drug for the treatment of cutaneous T-cell lymphoma. Clin. J. Oncol. Nurs. 2012, 16, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Vaquero, A.; Loyola, A.; Reinberg, D. The constantly changing face of chromatin. Sci. Aging Knowl. Environ. 2003, 2003, 4. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Almouzni, G. Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett. 2005, 220, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Al-Ali, H.; Volmar, C.-H.; Trotta, A.D.A.; Brothers, S.P.; Capobianco, A.J.; Wahlestedt, C. Ex vivo drug sensitivity testing as a means for drug repurposing in esophageal adenocarcinoma. PLoS ONE 2018, 13, e0203173. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.H.; Onion, D.; Collier, P.; Dorrington, M.S.; Argent, R.H.H.; Clarke, P.; Reece-Smith, A.M.; Parsons, S.L.; Grabowska, A.M. Individual patient oesophageal cancer 3D models for tailored treatment. Oncotarget 2017, 8, 24224–24236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrens, T.D.; Timme, S.; Hoeppner, J.; Ostendorp, J.; Hembach, S.; Follo, M.; Hopt, U.T.; Werner, M.; Busch, H.; Boerries, M.; et al. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics 2015, 10, 431–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, P.L.; Surman, D.R.; Brown, K.; Xu, Y.; McDuffie, L.A.; Shukla, V.; Reardon, E.S.; Crooks, D.R.; Trepel, J.B.; Lee, S.; et al. Induction of Thioredoxin-Interacting Protein by a Histone Deacetylase Inhibitor, Entinostat, Is Associated with DNA Damage and Apoptosis in Esophageal Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 2013–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofonikolas, K.; Frankell, A.M.; Smyth, E.C.; Fitzgerald, R.C. Examining the effects of BET, HDAC2, and EZH2 inhibition on esophageal adenocarcinoma (EAC) cell line proliferation. J. Clin. Oncol. 2019, 37 (Suppl. S4), 68. [Google Scholar] [CrossRef]
- Miwa, K.; Segawa, M.; Takano, Y.; Matsumoto, H.; Sahara, H.; Yagi, M.; Miyazaki, I.; Hattori, T. Induction of oesophageal and forestomach carcinomas in rats by reflux of duodenal contents. Br. J. Cancer 1994, 70, 185–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miwa, K.; Sahara, H.; Segawa, M.; Kinami, S.; Sato, T.; Miyazaki, I.; Hattori, T. Reflux of duodenal or gastro-duodenal contents induces esophageal carcinoma in rats. Int J. Cancer 1996, 67, 269–274. [Google Scholar] [CrossRef]
- Miyashita, T.; Ohta, T.; Fujimura, T.; Ninomiya, I.; Fushida, S.; Hattori, T.; Miwa, K. Duodenal juice stimulates oesophageal stem cells to induce Barrett’s oesophagus and oesophageal adenocarcinoma in rats. Oncol. Rep. 2006, 15, 1469–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, T.; Tajima, H.; Munemoto, M.; Shah, F.A.; Harmon, J.W.; Watanabe, T.; Shoji, M.; Okamoto, K.; Nakanuma, S.; Sakai, S.; et al. Impact of histone deacetylase 1 and metastasis-associated gene 1 expression in esophageal carcinogenesis. Oncol. Lett. 2014, 8, 758–764. [Google Scholar] [CrossRef]
- Nicolson, G.L.; Nawa, A.; Toh, Y.; Taniguchi, S.; Nishimori, K.; Moustafa, A. Tumor metastasis-associated human MTA1 gene and its MTA1 protein product: Role in epithelial cancer cell invasion, proliferation and nuclear regulation. Clin. Exp. Metastasis 2003, 20, 19–24. [Google Scholar] [CrossRef]
- Moon, H.-E.; Cheon, H.; Lee, M.-S. Metastasis-associated protein 1 inhibits p53-induced apoptosis. Oncol. Rep. 2007, 18, 1311–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Frankell, A.M.; Jammula, S.; Li, X.; Contino, G.; Killcoyne, S.; Abbas, S.; Perner, J.; Perner, J.; Bower, L.; Devonshire, G.; et al. The landscape of selection in 551 esophageal adenocarcinomas defines genomic biomarkers for the clinic. Nat. Genet. 2019, 51, 506–516. [Google Scholar] [CrossRef]
- Langer, R.; Mutze, K.; Becker, K.; Feith, M.; Ott, K.; Höfler, H.; Keller, G. Expression of class I histone deacetylases (HDAC1 and HDAC2) in oesophageal adenocarcinomas: An immunohistochemical study. J. Clin. Pathol. 2010, 63, 994–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Chen, Y.-Y.; Scott, G.K.; Devries, S.; Chin, K.; Benz, C.C.; Waldman, F.M.; Hwang, E.S. Protein Acetylation and Histone Deacetylase Expression Associated with Malignant Breast Cancer Progression. Clin. Cancer Res. 2009, 15, 3163–3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, Y.; Yamamoto, M.; Endo, K.; Ikeda, Y.; Baba, H.; Kohnoe, S.; Yonemasu, H.; Hachitanda, Y.; Okamura, T.; Sugimachi, K. Histone H4 acetylation and histone deacetylase 1 expression in esophageal squamous cell carcinoma. Oncol. Rep. 2003, 10, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Elahi, A.; Ajidahun, A.; Clark, W.; Hernandez, J.; Achille, A.; Hao, J.-H.; Seto, E.; Shibata, D. The interplay between histone deacetylases and c-Myc in the transcriptional suppression of HPP1 in colon cancer. Cancer Biol. Ther. 2014, 15, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.-P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Raetz, E.A.; Kim, M.K.H.; Moos, P.; Carlson, M.; Bruggers, C.; Hooper, D.K.; Foot, L.; Liu, T.; Seeger, R.; Carroll, W.L. Identification of genes that are regulated transcriptionally by Myc in childhood tumors. Cancer 2003, 98, 841–853. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Abbondanza, C.; Martens, J.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res. 2017, 23, 2542–2555. [Google Scholar] [CrossRef] [Green Version]
- Von Rahden, B.H.A.; Stein, H.J.; Pühringer-Oppermann, F.; Sarbia, M. c-myc Amplification Is Frequent in Esophageal Adenocarcinoma and Correlated with the Upregulation of VEGF-A Expression. Neoplasia 2006, 8, 702–707. [Google Scholar] [CrossRef] [Green Version]
- Tselepis, C.; Morris, C.D.; Wakelin, D.; Hardy, R.; Perry, I.; Luong, Q.T.; Harper, E.; Harrison, R.; Attwood, S.E.A.; Jankowski, J. Upregulation of the oncogene c-myc in Barrett’s adenocarcinoma: Induction of c-myc by acidified bile acid in vitro. Gut 2003, 52, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.; Suzuki, R.; Mazitschek, R.; Gorgun, G.; Ghosh, B.; Cirstea, D.; Hu, Y.; Mimura, N.; Ohguchi, H.; Cottini, F.; et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2013, 28, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stronach, E.A.; Alfraidi, A.; Rama, N.; Datler, C.; Studd, J.B.; Agarwal, R.; Guney, T.G.; Gourley, C.; Hennessy, B.T.; Mills, G.B.; et al. HDAC4-Regulated STAT1 Activation Mediates Platinum Resistance in Ovarian Cancer. Cancer Res. 2011, 71, 4412–4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone Deacetylase (HDAC) Inhibitors with a Novel Connecting Unit Linker Region Reveal a Selectivity Profile for HDAC4 and HDAC5 with Improved Activity against Chemoresistant Cancer Cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Kaadige, M.R.; Looper, R.E.; Kamalanaadhan, S.; Ayer, D.E. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proc. Natl. Acad. Sci. USA 2009, 106, 14878–14883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Herz, H.-M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.; Sherriff, J.; Bernstein, B.E.; Emre, T.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-Mediated Histone H3 Lysine 9 Methylation Directs DNA Methylation to Major Satellite Repeats at Pericentric Heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Nogales, E.; Ciferri, C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog. Biophys. Mol. Biol. 2010, 102, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Eich, M.-L.; Athar, M.; Ferguson, J.E.; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.-M.; Conejo-Garcia, J.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugo, H.S.; Jacobs, I.; Sharma, S.; Scappaticci, F.; Paul, T.A.; Jensen-Pergakes, K.; Malouf, G.G. The Promise for Histone Methyltransferase Inhibitors for Epigenetic Therapy in Clinical Oncology: A Narrative Review. Adv. Ther. 2020, 37, 3059–3082. [Google Scholar] [CrossRef] [PubMed]
- Zauderer, M.G.; Szlosarek, P.; Le Moulec, S.; Popat, S.; Taylor, P.; Planchard, D.; Scherpereel, A.; Jahan, T.; Koczywas, M.; Forster, M.; et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with relapsed or refractory (R/R) malignant mesothelioma (MM) with BAP1 inactivation. J. Clin. Oncol. 2018, 36 (Suppl. S15), 8515. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; Phillips, T.; Ribrag, V.; Campbell, P.; Laurent, D.G.; Jurczak, W.; McKay, P.; Opat, S.; et al. Interim Update From A Phase 2 Multicenter Study Of Tazemetostat, an Ezh2 Inhibitor, In Patients with Relapsed or Refractory Follicular Lymphoma. Hematol. Oncol. 2019, 37, 154–156. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Johnson, P.W.M.; Winter, J.; Leonard, J.; Giulino-Roth, L.; Horner, T.; Radswillas, K.; Carver, J.; Dhar, A. A phase I, open-label study of GSK2816126, an enhancer of zeste homolog 2 (EZH2) inhibitor, in patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL), transformed follicular lymphoma (tFL), other non-Hodgkin’s lymphomas (NHL), multiple myeloma (MM) and solid tumor. J. Clin. Oncol. 2016, 34 (Suppl. S15), TPS2595. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Iii, A.D.P.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Fatkhutdinov, N.; Fukumoto, T.; Bitler, B.; Park, P.H.; Kossenkov, A.V.; Trizzino, M.; Tang, H.-Y.; Zhang, L.; Gardini, A.; et al. SWI/SNF catalytic subunits’ switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells. Nat. Commun. 2018, 9, 4116. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, K.; Dvorak, B. Role of interleukin-6 in Barrett’s esophagus pathogenesis. World J. Gastroenterol. 2013, 19, 2307–2312. [Google Scholar] [CrossRef]
- Dvorak, K.; Chavarria, M.; Payne, C.M.; Ramsey, L.; Crowley-Weber, C.; Dvorakova, B.; Dvorak, B.; Bernstein, H.; Holubec, H.; Sampliner, R.E.; et al. Activation of the Interleukin-6/STAT3 Antiapoptotic Pathway in Esophageal Cells by Bile Acids and Low pH: Relevance to Barrett’s Esophagus. Clin. Cancer Res. 2007, 13 Pt 1, 5305–5313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.W.; Karakasheva, T.A.; Hicks, P.D.; Bass, A.J.; Rustgi, A.K. The tumor microenvironment in esophageal cancer. Oncogene 2016, 35, 5337–5349. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef]
- Ichihara, F.; Kono, K.; Takahashi, A.; Kawaida, H.; Sugai, H.; Fujii, H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin. Cancer Res. 2003, 9, 4404–4408. [Google Scholar] [PubMed]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-Expressed Inducible Nitric Oxide Synthase Controls Induction of Functional Myeloid-Derived Suppressor Cells through Modulation of Vascular Endothelial Growth Factor Release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovello, P.; Segal, D.M. Myeloid Suppressor Lines Inhibit T Cell Responses by an NO-Dependent Mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.; Zhang, H.-G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef] [PubMed]
- Butters, O.; Young, K.; Cunningham, D.; Chau, I.; Starling, N. Targeting Vascular Endothelial Growth Factor in Oesophagogastric Cancer: A Review of Progress to Date and Immunotherapy Combination Strategies. Front. Oncol. 2019, 9, 618. [Google Scholar] [CrossRef]
- Kelly, R.J.; Ajani, J.A.; Kuzdzal, J.; Zander, T.; Van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lièvre, A.; et al. Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. N. Engl. J. Med. 2021, 384, 1191–1203. [Google Scholar] [CrossRef]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.-C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Leavenworth, J.W.; Li, Y.; Luo, Q.; Xie, H.; Liu, X.; Huang, S.; Yan, H.; Fu, Z.; Zhang, L.Y.; et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc. Natl. Acad. Sci. USA 2015, 112, 15988–15993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyan, S.-W.; Hsu, C.-H.; Peng, K.-L.; Chen, C.-C.; Kuo, W.-H.; Lee, E.Y.-H.P.; Shew, J.-Y.; Chang, K.-J.; Juan, L.-J.; Lee, W.-H. Breast Cancer Cells Induce Stromal Fibroblasts to Secrete ADAMTS1 for Cancer Invasion through an Epigenetic Change. PLoS ONE 2012, 7, e35128. [Google Scholar] [CrossRef]
- Li, X.; Su, X.; Liu, R.; Pan, Y.; Fang, J.; Cao, L.; Feng, C.; Shang, Q.; Chen, Y.; Shao, C.; et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 2021, 40, 1836–1850. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; De Palma, R.; Altucci, L. HDAC inhibitors as epigenetic regulators for cancer immunotherapy. Int. J. Biochem. Cell Biol. 2018, 98, 65–74. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Johnstone, R.W.; Smyth, M.J. From cancer immunosurveillance to cancer immunotherapy. Immunol. Rev. 2007, 220, 82–101. [Google Scholar] [CrossRef]
- Sodji, Q.H.; Kornacki, J.R.; McDonald, J.F.; Mrksich, M.; Oyelere, A.K. Design and structure activity relationship of tumor-homing histone deacetylase inhibitors conjugated to folic and pteroic acids. Eur. J. Med. Chem. 2015, 96, 340–359. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. Tumor Biology and Immunology EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Johnston, L.D.; Klaus, C.R.; Wigle, T.J.; Iwanowicz, D.; Littlefield, B.A.; Porter-Scott, M.; Smith, J.J.; Moyer, M.P.; et al. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS ONE 2014, 9, e111840. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Leithner, K.; Wohlkoenig, C.; Quehenberger, F.; Bertsch, A.; Olschewski, A.; Olschewski, H.; Hrzenjak, A. Panobinostat reduces hypoxia-induced cisplatin resistance of non-small cell lung carcinoma cells via HIF-1α destabilization. Mol. Cancer 2015, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Bose, K.; Franck, C.; Müller, M.N.; Canbay, A.; Link, A.; Venerito, M. Perioperative Therapy of Oesophagogastric Adenocarcinoma: Mainstay and Future Directions. Gastroenterol. Res. Pract. 2017, 2017, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Wang, W.; Li, B.; Cheng, B.; Lin, K.; Bai, J.; Li, H.; Yang, G. Targeting Ezh2 could overcome docetaxel resistance in prostate cancer cells 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. BMC Cancer 2019, 19, 27. [Google Scholar] [CrossRef]
- Dou, D.; Ge, X.; Wang, X.; Xu, X.; Zhang, Z.; Seng, J.; Cao, Z.; Gu, Y.; Han, M. EZH2 Contributes To Cisplatin Resistance In Breast Cancer By Epigenetically Suppressing miR-381 Expression. OncoTargets Ther. 2019, 12, 9627–9637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Li, X.; Zhang, J.; Ge, Z.; Chen, H.; Hu, J. EZH2 contributes to 5-FU resistance in gastric cancer by epigenetically suppressing FBXO32 expression. OncoTargets Ther. 2018, 11, 7853–7864. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhu, F.; Wang, Q.; Ding, Y.; Rongbiao, Y.; Zeng, L. Inhibition of EZH2 and EGFR produces a synergistic effect on cell apoptosis by increasing autophagy in gastric cancer cells. OncoTargets Ther. 2018, 11, 8455–8463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katona, B.W.; Liu, Y.; Ma, A.; Jin, J.; Hua, X. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol. Ther. 2014, 15, 1677–1687. [Google Scholar] [CrossRef] [Green Version]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint Blockade in Cancer Immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollmann, D.; Ignatova, D.; Jedamzik, J.; Chang, Y.-T.; Jomrich, G.; Baierl, A.; Kazakov, D.; Michal, M.; French, L.E.; Hoetzenecker, W.; et al. PD-L1 expression is an independent predictor of favorable outcome in patients with localized esophageal adenocarcinoma. Oncol. Immunol. 2018, 7, e1435226. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Thudium, K.B.; Han, M.; Wang, X.-T.; Huang, H.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In Vitro Characterization of the Anti-PD-1 Antibody Nivolumab, BMS-936558, and In Vivo Toxicology in Non-Human Primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, A.V.; Morel, K.L.; Burkhart, D.L.; Baca, S.C.; Labbé, D.P.; Roehle, K.; Heckler, M.; Calagua, C.; Ye, H.; Galbo, P.; et al. Targeting EZH2 Increases Therapeutic Efficacy of Check-Point Blockade in Models of Prostate Cancer. BioRxiv 2019, 730135. [Google Scholar] [CrossRef]
- Gray, J.E.; Saltos, A.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.; Antonia, S.J.; Shafique, M.; Zheng, H.; Dai, W.; Saller, J.J.; et al. Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6623–6632. [Google Scholar] [CrossRef] [PubMed]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-Z.; Chang, H.-Y. Recent advances in three-dimensional multicellular spheroid culture for biomedical research. Biotechnol. J. 2008, 3, 1172–1184. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author | Epigenetic Target | Drug | Model | Main Findings |
|---|---|---|---|---|
| Lohse et al., 2018 [33] | Epigenetic drug panel | 163 compounds | OAC cell lines | HDACi displayed significant anti-tumour activity. |
| Saunders et al., 2017 [34] | HDAC | Pabinostat | OAC 3D-TGA | Pabinostat and standard of care agents improved chemosensitivity of the 3D cell models. |
| Ahrens et al., 2015 [35] | HDAC DNMT | Vorinostat, MS-275, FK228 AZA, DAC | OAC cell lines | MS-275, FK228 and AZA showed anti-proliferative effects on OAC cell lines. |
| Feingold et al., 2018 [36] | HDAC | Entinostat | OAC cell lines | Combination treatment with entinostat and cisplatin led to cancer cell apoptosis. |
| Kofonikolas et al., 2019 * [37] | HDAC EZH2 BET | Vorinostat Tazemetostat JQ1 | OAC cell lines | Myc-amplified OAC cells showed increased sensitivity to BET inhibition. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pickering, O.J.; Breininger, S.P.; Underwood, T.J.; Walters, Z.S. Histone Modifying Enzymes as Targets for Therapeutic Intervention in Oesophageal Adenocarcinoma. Cancers 2021, 13, 4084. https://doi.org/10.3390/cancers13164084
Pickering OJ, Breininger SP, Underwood TJ, Walters ZS. Histone Modifying Enzymes as Targets for Therapeutic Intervention in Oesophageal Adenocarcinoma. Cancers. 2021; 13(16):4084. https://doi.org/10.3390/cancers13164084
Chicago/Turabian StylePickering, Oliver J., Stella P. Breininger, Timothy J. Underwood, and Zoë S. Walters. 2021. "Histone Modifying Enzymes as Targets for Therapeutic Intervention in Oesophageal Adenocarcinoma" Cancers 13, no. 16: 4084. https://doi.org/10.3390/cancers13164084
APA StylePickering, O. J., Breininger, S. P., Underwood, T. J., & Walters, Z. S. (2021). Histone Modifying Enzymes as Targets for Therapeutic Intervention in Oesophageal Adenocarcinoma. Cancers, 13(16), 4084. https://doi.org/10.3390/cancers13164084