
Modulation of Cancer Cell Autophagic Responses by Graphene-Based Nanomaterials: Molecular Mechanisms and Therapeutic Implications

 ,
,
Abstract
:Simple Summary
Abstract
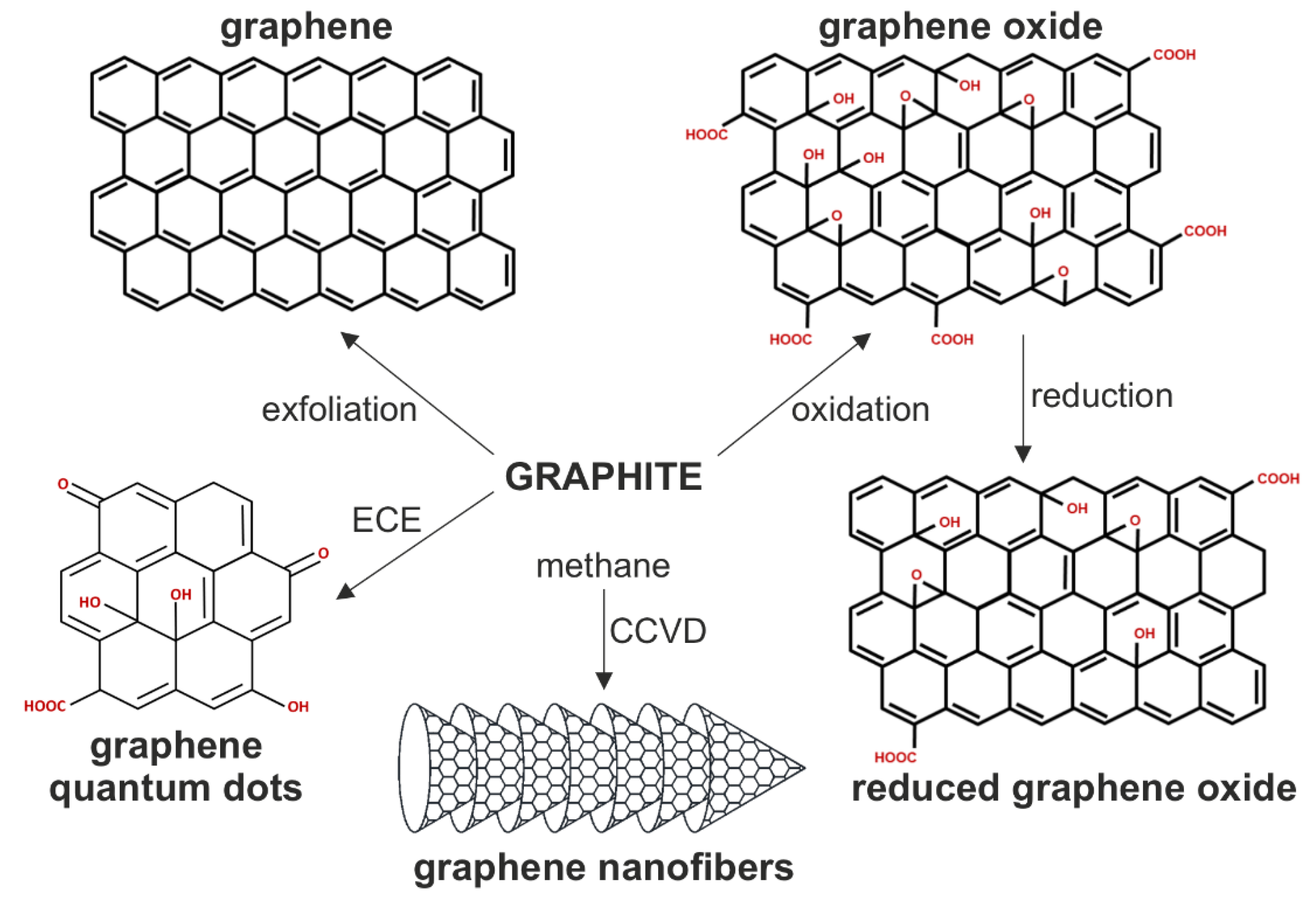
1. Introduction
2. Autophagy Regulation and Role in Cell Death
3. Autophagy Modulation by GNM in Cancer Cells
3.1. Autophagy Induction by GNM
3.2. Suppression of Autophagic Flux by GNM
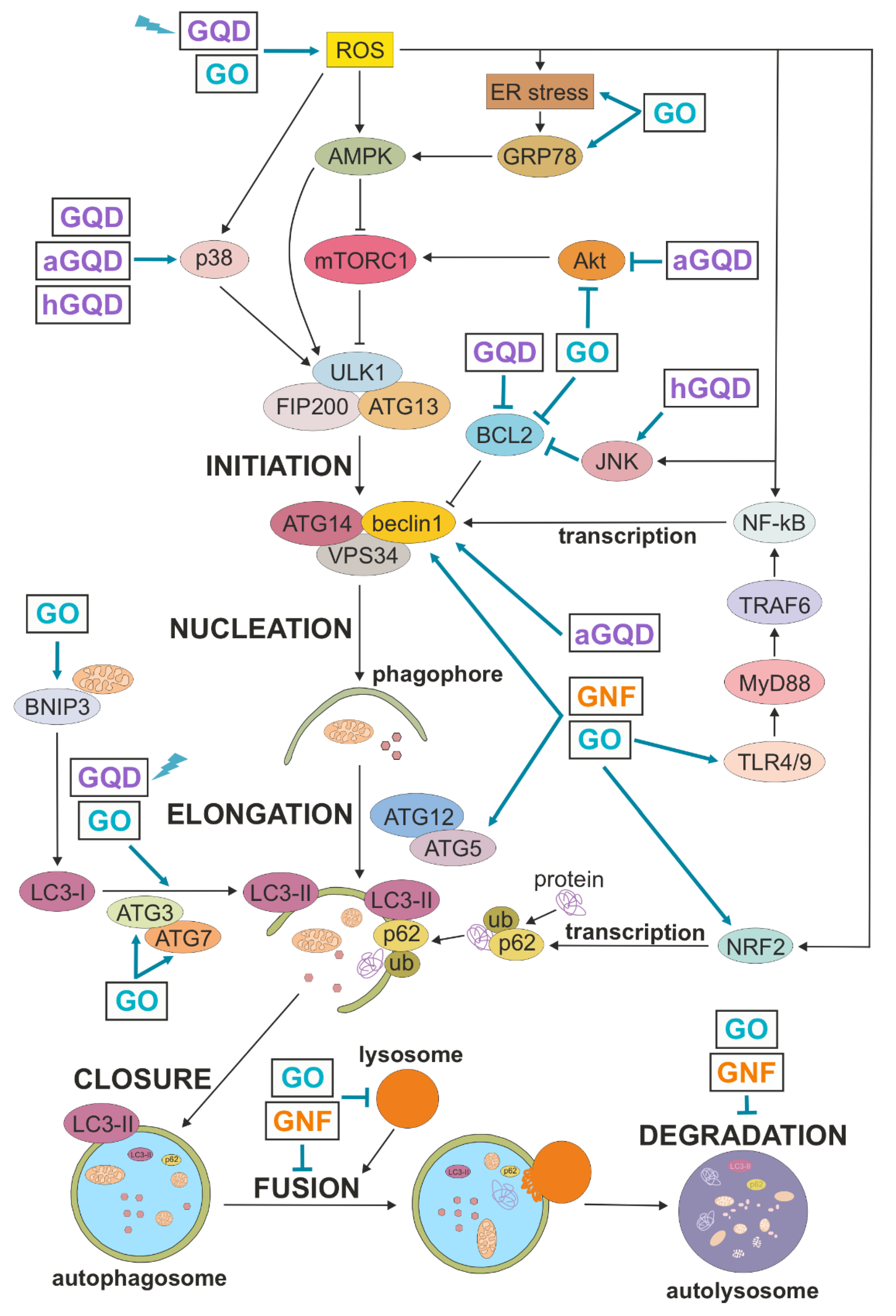
4. Mechanisms of Autophagy Induction by GNM
4.1. Transcriptional Induction of Autophagy
4.2. AMPK/AKT/mTOR Signaling
4.3. MAPK Signaling
4.4. TLR Signaling
4.5. Oxidative Stress
4.6. ER Stress
5. Mechanisms of Autophagic Flux Suppression by GNM
6. Structure–Activity Relationship of GNM as Autophagy Modulators
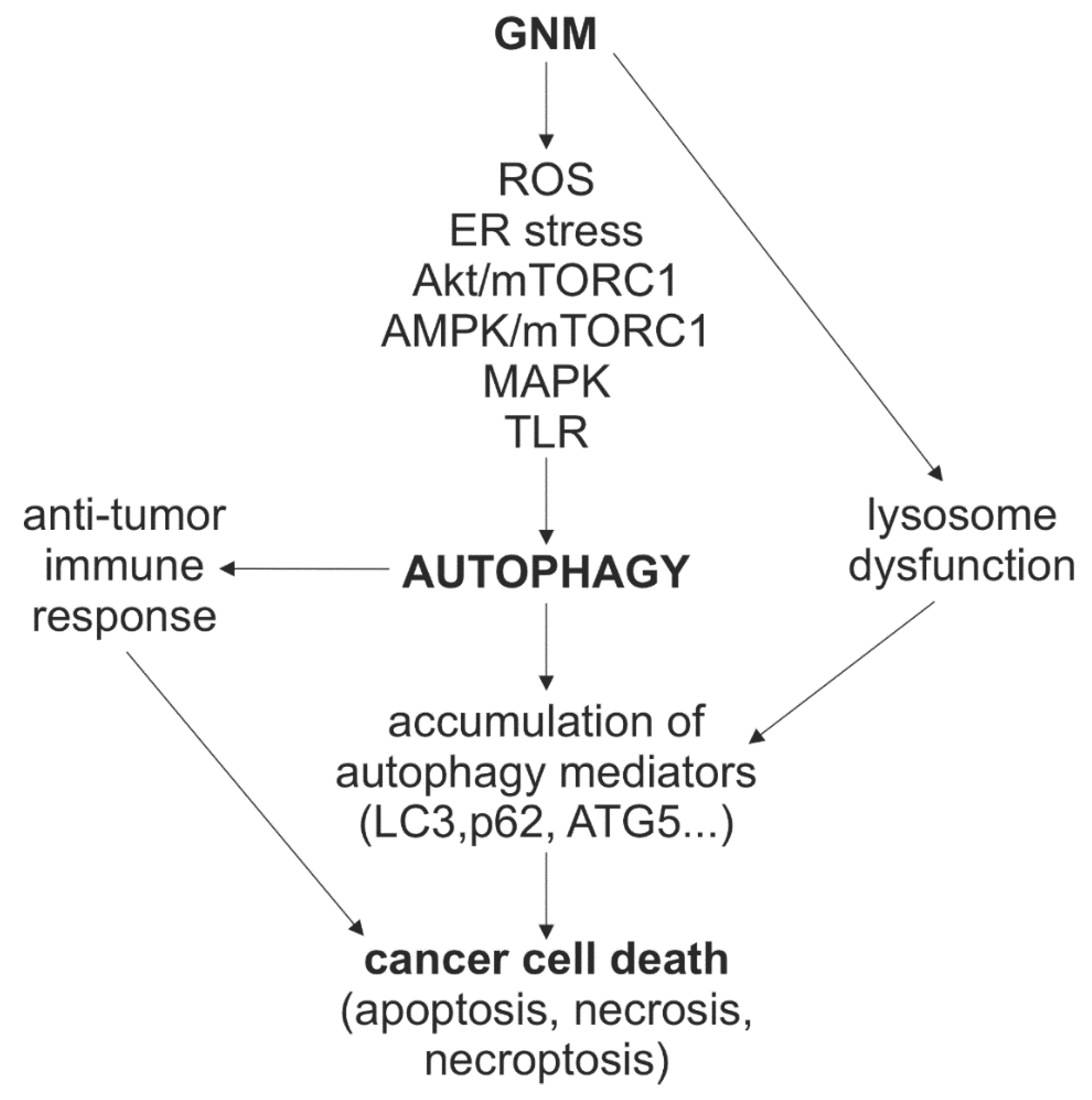
7. The Role of Autophagy Modulation in GNM-Induced Cancer Cell Death
7.1. Cytotoxic Effect of GNM-Mediated Autophagy Induction
7.2. Cytotoxic Effect of GNM-Mediated Blockade of Autophagic Flux
8. Implications of Autophagy Modulation by GNM for Anticancer Therapy
8.1. Biocompatibility and Biological Fate of GNM
8.2. Selectivity of GNM-Mediated Autophagy Modulation in Anticancer Therapy
8.3. Autophagy Modulation by GNM as Chemo/Radio-Sensitizers and Drug Delivery Systems
8.4. Immunomodulatory Effects of GNM-Induced Autophagy
9. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Luong, D.X.; Bets, K.V.; Algozeeb, W.A.; Stanford, M.G.; Kittrell, C.; Chen, W.; Salvatierra, R.V.; Ren, M.; McHugh, E.A.; Advincula, P.A.; et al. Gram-scale bottom-up flash graphene synthesis. Nature 2020, 577, 647–651. [Google Scholar] [CrossRef]
- Compton, O.C.; Nguyen, S.T. Graphene oxide, highly reduced graphene oxide, and graphene: Versatile building blocks for carbon-based materials. Small 2010, 6, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Li, X.; Ji, R.; Teng, K.S.; Tai, G.; Ye, J.; Wei, C.; Lau, S.P. Bottom-up synthesis of large-scale graphene oxide nanosheets. J. Mater. Chem. 2012, 22, 5676–5683. [Google Scholar] [CrossRef]
- Yan, Y.; Gong, J.; Chen, J.; Zeng, Z.; Huang, W.; Pu, K.; Liu, J.; Chen, P. Recent advances on graphene quantum dots: From chemistry and physics to applications. Adv. Mater. 2019, 31, e1808283. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.; Pérez-Rodríguez, S.; Sebastián, D.; Pinilla, J.L.; Lázaro, M.J.; Suelves, I. Graphene oxide nanofibers: A nanocarbon material with tuneable electrochemical properties. Appl. Surface Sci. 2020, 509, 144774. [Google Scholar] [CrossRef]
- Tibbetts, G.G.; Lake, M.L.; Strong, K.L.; Rice, B.P. A review of the fabrication and properties of vapor-grown carbon nanofiber/polymer composites. Compos. Sci. Technol. 2007, 67, 1709–1718. [Google Scholar] [CrossRef]
- Qu, Y.; He, F.; Yu, C.; Liang, X.; Liang, D.; Ma, L.; Zhang, Q.; Lv, J.; Wu, J. Advances on graphene-based nanomaterials for biomedical applications. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 90, 764–780. [Google Scholar] [CrossRef] [PubMed]
- de Melo-Diogo, D.; Lima-Sousa, R.; Alves, C.G.; Correia, I.J. Graphene family nanomaterials for application in cancer combination photothermal therapy. Biomater. Sci. 2019, 7, 3534–3551. [Google Scholar] [CrossRef]
- Li, Y.; Dong, H.; Li, Y.; Shi, D. Graphene-based nanovehicles for photodynamic medical therapy. Int. J. Nanomed. 2015, 10, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Markovic, Z.M.; Harhaji-Trajkovic, L.M.; Todorovic-Markovic, B.M.; Kepic, D.P.; Arsikin, K.M.; Jovanovic, S.P.; Pantovic, A.C.; Dramicanin, M.D.; Trajkovic, V.S. In vitro comparison of the photothermal anticancer activity of graphene nanoparticles and carbon nanotubes. Biomaterials 2011, 32, 1121–1129. [Google Scholar] [CrossRef]
- Robinson, J.T.; Tabakman, S.M.; Liang, Y.; Wang, H.; Casalongue, H.S.; Vinh, D.; Dai, H. Ultrasmall reduced graphene oxide with high near-infrared absorbance for photothermal therapy. J. Am. Chem. Soc. 2011, 133, 6825–6831. [Google Scholar] [CrossRef]
- Yang, K.; Wan, J.; Zhang, S.; Tian, B.; Zhang, Y.; Liu, Z. The influence of surface chemistry and size of nanoscale graphene oxide on photothermal therapy of cancer using ultra-low laser power. Biomaterials 2012, 33, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.Y.; Yu, X.H.; Wang, K.; Yin, Y.J.; Tang, Y.J.; Tang, Y.L.; Liang, X.H. Graphene quantum dots (GQDs)-based nanomaterials for improving photodynamic therapy in cancer treatment. Eur. J. Med. Chem. 2019, 182, 111620. [Google Scholar] [CrossRef]
- Jarosz, A.; Skoda, M.; Dudek, I.; Szukiewicz, D. Oxidative stress and mitochondrial activation as the main mechanisms underlying graphene toxicity against human cancer cells. Oxid. Med. Cell Longev. 2016, 2016, 5851035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; He, N.; Zhao, Y.; Liu, T.; Deng, Y. Autophagy modulated by inorganic nanomaterials. Theranostics 2020, 10, 3206–3222. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Duan, Y. Crosstalk between autophagy and nanomaterials: Internalization, activation, termination. Adv. Biosyst. 2019, 3, e1800259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, L.; Gao, J.; Wen, L. Pro-death or pro-survival: Contrasting paradigms on nanomaterial-induced autophagy and exploitations for cancer therapy. Acc. Chem. Res. 2019, 52, 3164–3176. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Kögel, D. Autophagy in cancer cell death. Biology 2019, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2020, 219, e201909033. [Google Scholar] [CrossRef]
- Ou, L.; Lin, S.; Song, B.; Liu, J.; Lai, R.; Shao, L. The mechanisms of graphene-based materials-induced programmed cell death: A review of apoptosis, autophagy, and programmed necrosis. Int. J. Nanomed. 2017, 12, 6633–6646. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Debnath, J. Beyond self-eating: The control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol. 2018, 217, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamenkovic, M.; Janjetovic, K.; Paunovic, V.; Ciric, D.; Kravic-Stevovic, T.; Trajkovic, V. Comparative analysis of cell death mechanisms induced by lysosomal autophagy inhibitors. Eur. J. Pharmacol. 2019, 859, 172540. [Google Scholar] [CrossRef]
- Kosic, M.; Paunovic, V.; Ristic, B.; Mircic, A.; Bosnjak, M.; Stevanovic, D.; Kravic-Stevovic, T.; Trajkovic, V.; Harhaji-Trajkovic, L. 3-Methyladenine prevents energy stress-induced necrotic death of melanoma cells through autophagy-independent mechanisms. J. Pharmacol. Sci. 2021, 147, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhao, L.; Yang, Z.; Liu, Z.; Gu, J.; Bai, B.; Liu, J.; Xu, J.; Yang, H. Mechanisms of oxidative stress, apoptosis, and autophagy involved in graphene oxide nanomaterial anti-osteosarcoma effect. Int. J. Nanomed. 2018, 13, 2907–2919. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.C.; Lin, M.W.; Hsu, M.N.; Yu-Chen, G.; Chao, Y.C.; Tuan, H.Y.; Chiang, C.S.; Hu, Y.C. Graphene oxide sensitizes cancer cells to chemotherapeutics by inducing early autophagy events, promoting nuclear trafficking and necrosis. Theranostics 2018, 8, 2477–2487. [Google Scholar] [CrossRef]
- Li, X.; Li, K.; Chu, F.; Huang, J.; Yang, Z. Graphene oxide enhances β-amyloid clearance by inducing autophagy of microglia and neurons. Chem. Biol. Interact. 2020, 325, 109126. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Meng, C.L.; Lin, K.C.; Tuan, H.Y.; Yang, H.J.; Chen, C.L.; Li, K.C.; Chiang, C.S.; Hu, Y.C. Graphene oxide as a chemosensitizer: Diverted autophagic flux, enhanced nuclear import, elevated necrosis and improved antitumor effects. Biomaterials 2015, 40, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Chen, C.L.; Tuan, H.Y.; Yuan, P.X.; Li, K.C.; Yang, H.J.; Hu, Y.C. Graphene oxide triggers toll-like receptors/autophagy responses in vitro and inhibits tumor growth in vivo. Adv. Healthc. Mater. 2014, 3, 1486–1495. [Google Scholar] [CrossRef]
- Jeong, J.K.; Lee, Y.J.; Jeong, S.Y.; Jeong, S.; Lee, G.W.; Park, S.Y. Autophagic flux induced by graphene oxide has a neuroprotective effect against human prion protein fragments. Int. J. Nanomed. 2017, 12, 8143–8158. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Wei, P.; Zhang, Y.; Lin, J.; Sha, R.; Hu, Y.; Zhang, J.; Zhou, W.; Yao, H.; Ren, L.; et al. Autophagy-mediated clearance of ubiquitinated mutant huntingtin by graphene oxide. Nanoscale 2016, 8, 18740–18750. [Google Scholar] [CrossRef]
- Chen, G.Y.; Yang, H.J.; Lu, C.H.; Chao, Y.C.; Hwang, S.M.; Chen, C.L.; Lo, K.W.; Sung, L.Y.; Luo, W.Y.; Tuan, H.Y.; et al. Simultaneous induction of autophagy and toll-like receptor signaling pathways by graphene oxide. Biomaterials 2012, 33, 6559–6569. [Google Scholar] [CrossRef]
- Mari, E.; Mardente, S.; Morgante, E.; Tafani, M.; Lococo, E.; Fico, F.; Valentini, F.; Zicari, A. Graphene oxide nanoribbons induce autophagic vacuoles in neuroblastoma cell lines. Int. J. Mol. Sci. 2016, 17, 1995. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Yang, X.; Luo, L.H.; Ning, Z. Graphene oxide regulates endoplasmic reticulum stress: Autophagic pathways in nasopharyngeal carcinoma cells. Int. J. Clin. Exp. Pathol. 2018, 11, 5801–5808. [Google Scholar]
- Markovic, Z.M.; Ristic, B.Z.; Arsikin, K.M.; Klisic, D.G.; Harhaji-Trajkovic, L.M.; Todorovic-Markovic, B.M.; Kepic, D.P.; Kravic-Stevovic, T.K.; Jovanovic, S.P.; Milenkovic, M.M.; et al. Graphene quantum dots as autophagy-inducing photodynamic agents. Biomaterials 2012, 33, 7084–7092. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wan, B.; Yang, Y.; Cui, X.; Xin, Y.; Guo, L.H. Cytotoxicity and autophagy induction by graphene quantum dots with different functional groups. J. Environ. Sci. 2019, 77, 198–209. [Google Scholar] [CrossRef]
- Qin, Y.; Zhou, Z.W.; Pan, S.T.; He, Z.X.; Zhang, X.; Qiu, J.X.; Duan, W.; Yang, T.; Zhou, S.F. Graphene quantum dots induce apoptosis, autophagy, and inflammatory response via p38 mitogen-activated protein kinase and nuclear factor-κB mediated signaling pathways in activated THP-1 macrophages. Toxicology 2015, 327, 62–76. [Google Scholar] [CrossRef]
- Yuan, Y.G.; Wang, Y.H.; Xing, H.H.; Gurunathan, S. Quercetin-mediated synthesis of graphene oxide-silver nanoparticle nanocomposites: A suitable alternative nanotherapy for neuroblastoma. Int. J. Nanomed. 2017, 12, 5819–5839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arya, B.D.; Mittal, S.; Joshi, P.; Pandey, A.K.; Ramirez-Vick, J.E.; Singh, S.P. Graphene oxide-chloroquine nanoconjugate induce necroptotic death in A549 cancer cells through autophagy modulation. Nanomedicine 2018, 13, 2261–2282. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, L.; Guo, W.; Zhang, Y.; Lai, X.; Shao, L.; Li, Y. Graphene oxide induces p62/SQSTM-dependent apoptosis through the impairment of autophagic flux and lysosomal dysfunction in PC12 cells. Acta Biomater. 2018, 81, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Sharma, P.K.; Tiwari, R.; Rayavarapu, R.G.; Shankar, J.; Chauhan, L.K.S.; Pandey, A.K. Impaired lysosomal activity mediated autophagic flux disruption by graphite carbon nanofibers induce apoptosis in human lung epithelial cells through oxidative stress and energetic impairment. Part Fibre. Toxicol. 2017, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Feng, X.; He, L.; Zhang, Y.; Shao, L. The interrupted effect of autophagic flux and lysosomal function induced by graphene oxide in p62-dependent apoptosis of F98 cells. J. Nanobiotechnol. 2020, 18, 52. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, Y.; Lai, W.; Xiang, Z.; Tu, B.; Li, D.; Nan, X.; Chen, C.; Hu, Z.; Fang, Q. Proteomic profiling of RAW264.7 macrophage cells exposed to graphene oxide: Insights into acute cellular responses. Nanotoxicology 2019, 13, 35–49. [Google Scholar] [CrossRef]
- Huang, J.; Lam, G.Y.; Brumell, J.H. Autophagy signaling through reactive oxygen species. Antioxid. Redox. Signal. 2011, 14, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Hurley, J.H.; Schulman, B.A. Atomistic autophagy: The structures of cellular self-digestion. Cell 2014, 157, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Chourasia, A.H.; Macleod, K.F. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 2015, 11, 1937–1938. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [Green Version]
- Hulea, L.; Markovic, Z.; Topisirovic, I.; Simmet, T.; Trajkovic, V. Biomedical potential of mTOR modulation by nanoparticles. Trends Biotechnol. 2016, 34, 349–353. [Google Scholar] [CrossRef]
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, S.; Jain, K.; Basu, A. Regulation of autophagy by kinases. Cancers 2011, 3, 2630–2654. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slobodnyuk, K.; Radic, N.; Ivanova, S.; Llado, A.; Trempolec, N.; Zorzano, A.; Nebreda, A.R. Autophagy-induced senescence is regulated by p38α signaling. Cell Death Dis. 2019, 10, 376. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The role of TLRs in anti-cancer immunity and tumor rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Jang, Y.J.; Kim, J.H.; Byun, S. Modulation of autophagy for controlling immunity. Cells 2019, 8, 138. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Hyttinen, J.M.; Kauppinen, A.; Kaarniranta, K. Context-dependent regulation of autophagy by IKK-NF-κB signaling: Impact on the aging process. Int. J. Cell. Biol. 2012, 2012, 849541. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Paunovic, V.; Kosic, M.; Misirkic-Marjanovic, M.; Trajkovic, V.; Harhaji-Trajkovic, L. Dual targeting of tumor cell energy metabolism and lysosomes as an anticancer strategy. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118944. [Google Scholar] [CrossRef]
- Chong, Y.; Ge, C.; Fang, G.; Tian, X.; Ma, X.; Wen, T.; Wamer, W.G.; Chen, C.; Chai, Z.; Yin, J.J. Crossover between anti- and pro-oxidant activities of graphene quantum dots in the absence or presence of light. ACS Nano 2016, 10, 8690–8699. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Lee, A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.L.; Shajahan, A.N.; Wärri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012, 72, 3337–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heberle, A.M.; Prentzell, M.T.; van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular mechanisms of mTOR regulation by stress. Mol. Cell. Oncol. 2015, 2, e970489. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Zhao, W.; Cao, L.; Huang, J. Involvement of the actin machinery in programmed cell death. Front. Cell Dev. Biol. 2020, 8, 634849. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The cytoskeleton-autophagy connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef] [Green Version]
- Mendes, R.G.; Koch, B.; Bachmatiuk, A.; Ma, X.; Sanchez, S.; Damm, C.; Schmidt, O.G.; Gemming, T.; Eckert, J.; Rümmeli, M.H. A size dependent evaluation of the cytotoxicity and uptake of nanographene oxide. J. Mater. Chem. B 2015, 3, 2522–2529. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Lv, M.; Xiu, P.; Huynh, T.; Zhang, M.; Castelli, M.; Liu, Z.; Huang, Q.; Fan, C.; Fang, H.; et al. Destructive extraction of phospholipids from Escherichia coli membranes by graphene nanosheets. Nat. Nanotechnol. 2013, 8, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Duan, G.; Yang, Z.; Weber, J.K.; Liu, X.; Lu, S.; Meng, X.; Xu, J. Particle size-dependent antibacterial activity and murine cell cytotoxicity induced by graphene oxide nanomaterials. J. Nanomater. 2016, 2016, 6709764. [Google Scholar] [CrossRef] [Green Version]
- Koner, A.L.; Krndija, D.; Hou, Q.; Sherratt, D.J.; Howarth, M. Hydroxy-terminated conjugated polymer nanoparticles have near-unity bright fraction and reveal cholesterol-dependence of IGF1R nanodomains. ACS Nano 2013, 7, 1137–1144. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, C.; Wu, L.; Bai, X.; Zhai, S. Cytotoxicity-related bioeffects induced by nanoparticles: The role of surface chemistry. Front. Bioeng. Biotechnol. 2019, 7, 414. [Google Scholar] [CrossRef] [Green Version]
- Lunova, M.; Prokhorov, A.; Jirsa, M.; Hof, M.; Olżyńska, A.; Jurkiewicz, P.; Kubinová, Š.; Lunov, O.; Dejneka, A. Nanoparticle core stability and surface functionalization drive the mTOR signaling pathway in hepatocellular cell lines. Sci. Rep. 2017, 7, 16049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzapfel, V.; Musyanovych, A.; Landfester, K.; Lorenz, M.R.; Mailänder, V. Preparation of fluorescent carboxyl and amino functionalized polystyrene particles by miniemulsion polymerization as markers for cells. Macromol. Chem. Phys. 2005, 206, 2440–2449. [Google Scholar] [CrossRef]
- Ozbek, O.; Ulgen, K.O.; Ileri Ercan, N. The toxicity of polystyrene-based nanoparticles in Saccharomyces cerevisiae is associated with nanoparticle charge and uptake mechanism. Chem. Res. Toxicol. 2021, 34, 1055–1068. [Google Scholar] [CrossRef]
- Kessel, D.; Reiners, J.J. Photodynamic therapy: Autophagy and mitophagy, apoptosis and paraptosis. Autophagy 2020, 16, 2098–2101. [Google Scholar] [CrossRef]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [Green Version]
- Halcrow, P.W.; Geiger, J.D.; Chen, X. Overcoming chemoresistance: Altering pH of cellular compartments by chloroquine and hydroxychloroquine. Front. Cell Dev. Biol. 2021, 9, 627639. [Google Scholar] [CrossRef]
- Liao, C.; Li, Y.; Tjong, S.C. Graphene nanomaterials: Synthesis, biocompatibility, and cytotoxicity. Int. J. Mol. Sci. 2018, 19, 3564. [Google Scholar] [CrossRef] [Green Version]
- Tabish, T.A.; Scotton, C.J.; Ferguson, D.C.J.; Lin, L.; der Veen, A.V.; Lowry, S.; Ali, M.; Jabeen, F.; Ali, M.; Winyard, P.G.; et al. Biocompatibility and toxicity of graphene quantum dots for potential application in photodynamic therapy. Nanomedicine 2018, 13, 1923–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Hu, X.; Guan, P.; Hou, T.; Chen, P.; Wan, D.; Zhang, X.; Wang, J.; Wang, C. Highly biocompatible graphene quantum dots: Green synthesis, toxicity comparison and fluorescence imaging. J. Mater. Sci. 2020, 55, 1198–1215. [Google Scholar] [CrossRef]
- Martín, C.; Jun, G.; Schurhammer, R.; Reina, G.; Chen, P.; Bianco, A.; Ménard-Moyon, C. Enzymatic degradation of graphene quantum dots by human peroxidases. Small 2019, 15, e1905405. [Google Scholar] [CrossRef] [PubMed]
- Kurapati, R.; Mukherjee, S.P.; Martín, C.; Bepete, G.; Vázquez, E.; Pénicaud, A.; Fadeel, B.; Bianco, A. Degradation of single-layer and few-layer graphene by neutrophil myeloperoxidase. Angew. Chem. Int. Ed. Engl. 2018, 57, 11722–11727. [Google Scholar] [CrossRef]
- Shannahan, J. The biocorona: A challenge for the biomedical application of nanoparticles. Nanotechnol. Rev. 2017, 6, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, G.; Kang, S.G.; Tian, X.; Garate, J.A.; Zhao, L.; Ge, C.; Zhou, R. Protein corona mitigates the cytotoxicity of graphene oxide by reducing its physical interaction with cell membrane. Nanoscale 2015, 7, 15214–15224. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Chen, W.; Laurent, S.; Thirifays, C.; Burtea, C.; Rezaee, F.; Mahmoudi, M. Hard corona composition and cellular toxicities of the graphene sheets. Colloids Surf. B Biointerfaces 2013, 109, 212–218. [Google Scholar] [CrossRef]
- Di Santo, R.; Digiacomo, L.; Quagliarini, E.; Capriotti, A.L.; Laganà, A.; Zenezini Chiozzi, R.; Caputo, D.; Cascone, C.; Coppola, R.; Pozzi, D.; et al. Personalized graphene oxide-protein corona in the human plasma of pancreatic cancer patients. Front. Bioeng. Biotechnol. 2020, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Molinaro, R.; Tabatabaei, M.; Farokhzad, O.C.; Mahmoudi, M. Personalized protein corona on nanoparticles and its clinical implications. Biomater. Sci. 2017, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Dong, C.; Ren, T.; Li, Y.; Shi, D. Surface-engineered graphene-based nanomaterials for drug delivery. J. Biomed. Nanotechnol. 2014, 10, 2086–2106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wei, C.; Li, Y.; Li, Y.; Chen, G.; He, Y.; Yi, C.; Wang, C.; Yu, D. Dose-dependent cytotoxicity induced by pristine graphene oxide nanosheets for potential bone tissue regeneration. J. Biomed. Mater. Res. A 2020, 108, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Fu, Z.; Wang, C.; Zheng, W.; Li, S.; Le, W. Graphene oxide nanocolloids induce autophagy-lysosome dysfunction in mouse embryonic stem cells. J. Biomed. Nanotechnol. 2019, 15, 340–351. [Google Scholar] [CrossRef]
- Lim, M.H.; Jeung, I.C.; Jeong, J.; Yoon, S.J.; Lee, S.H.; Park, J.; Kang, Y.S.; Lee, H.; Park, Y.J.; Lee, H.G.; et al. Graphene oxide induces apoptotic cell death in endothelial cells by activating autophagy via calcium-dependent phosphorylation of c-Jun N-terminal kinases. Acta Biomater. 2016, 46, 191–203. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, A.; Shen, Q.; Xie, Y.; Liu, S.; Wang, X. Graphene oxide aggravated dextran sulfate sodium-induced colitis through intestinal epithelial cells autophagy dysfunction. J. Toxicol. Sci. 2021, 46, 43–55. [Google Scholar] [CrossRef]
- Wan, B.; Wang, Z.X.; Lv, Q.Y.; Dong, P.X.; Zhao, L.X.; Yang, Y.; Guo, L.H. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicol. Lett. 2013, 221, 118–127. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Wang, J.; Nie, Y.; Du, H.; Dai, H.; Wang, J.; Wang, M.; Chen, S.; Hei, T.K.; et al. Graphene oxide attenuates the cytotoxicity and mutagenicity of PCB 52 via activation of genuine autophagy. Environ. Sci. Technol. 2016, 50, 3154–3164. [Google Scholar] [CrossRef]
- Tomic, S.; Janjetovic, K.; Mihajlovic, D.; Milenkovic, M.; Kravic-Stevovic, T.; Markovic, Z.; Todorovic-Markovic, B.; Spitalsky, Z.; Micusik, M.; Vucevic, D.; et al. Graphene quantum dots suppress proinflammatory T cell responses via autophagy-dependent induction of tolerogenic dendritic cells. Biomaterials 2017, 146, 13–28. [Google Scholar] [CrossRef]
- Bramini, M.; Sacchetti, S.; Armirotti, A.; Rocchi, A.; Vázquez, E.; León Castellanos, V.; Bandiera, T.; Cesca, F.; Benfenati, F. Graphene oxide nanosheets disrupt lipid composition, Ca2+ homeostasis, and synaptic transmission in primary cortical neurons. ACS Nano 2016, 10, 7154–7171. [Google Scholar] [CrossRef] [Green Version]
- Malanagahalli, S.; Murera, D.; Martín, C.; Lin, H.; Wadier, N.; Dumortier, H.; Vázquez, E.; Bianco, A. Few layer graphene does not affect cellular homeostasis of mouse macrophages. Nanomaterials 2020, 10, 228. [Google Scholar] [CrossRef] [Green Version]
- Di Cristo, L.; Grimaldi, B.; Catelani, T.; Vázquez, E.; Pompa, P.P.; Sabella, S. Repeated exposure to aerosolized graphene oxide mediates autophagy inhibition and inflammation in a three-dimensional human airway model. Mater. Today Bio. 2020, 6, 100050. [Google Scholar] [CrossRef]
- Li, X.; Liu, H.; Yu, Y.; Ma, L.; Liu, C.; Miao, L. Graphene oxide quantum dots-induced mineralization via the reactive oxygen species-dependent autophagy pathway in dental pulp stem cells. J. Biomed. Nanotechnol. 2020, 16, 965–974. [Google Scholar] [CrossRef]
- Jiao, D.; Wang, J.; Yu, W.; Zhang, N.; Zhang, K.; Bai, Y. Gelatin reduced graphene oxide nanosheets as kartogenin nanocarrier induces rat ADSCs chondrogenic differentiation combining with autophagy modification. Materials 2021, 14, 1053. [Google Scholar] [CrossRef]
- Rastogi, S.K.; Raghavan, G.; Yang, G.; Cohen-Karni, T. Effect of graphene on nonneuronal and neuronal cell viability and stress. Nano Lett. 2017, 17, 3297–3301. [Google Scholar] [CrossRef]
- Murera, D.; Malaganahalli, S.; Martín, C.; Reina, G.; Fauny, J.D.; Dumortier, H.; Vázquez, E.; Bianco, A. Few layer graphene does not affect the function and the autophagic activity of primary lymphocytes. Nanoscale 2019, 11, 10493–10503. [Google Scholar] [CrossRef]
- Yuan, Y.G.; Gurunathan, S. Combination of graphene oxide-silver nanoparticle nanocomposites and cisplatin enhances apoptosis and autophagy in human cervical cancer cells. Int. J. Nanomed. 2017, 12, 6537–6558. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Miao, H.; Luo, Y.; Sun, Y.; Tian, X.; Wang, F.; You, C.; Peng, S.; Tang, G.; Yang, C.; et al. FePt/GO nanosheets suppress proliferation, enhance radiosensitization and induce autophagy of human non-small cell lung cancer cells. Int. J. Biol. Sci. 2019, 15, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Guan, W.; Qing, X.; Yang, W.; Que, Y.; Tan, L.; Liang, H.; Zhang, Z.; Wang, B.; Liu, X.; et al. Ultrafast low-temperature photothermal therapy activates autophagy and recovers immunity for efficient antitumor treatment. ACS Appl. Mater. Interfaces 2020, 12, 4265–4275. [Google Scholar] [CrossRef]
- Huang, X.; Chen, J.; Wu, W.; Yang, W.; Zhong, B.; Qing, X.; Shao, Z. Delivery of MutT homolog 1 inhibitor by functionalized graphene oxide nanoparticles for enhanced chemo-photodynamic therapy triggers cell death in osteosarcoma. Acta Biomater. 2020, 109, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Qiu, Y.; Dinesh, P.; Gong, W.; Jiang, L.; Feng, X.; Li, J.; Jiang, Y.; Lei, Y.L.; Chen, Q. The functions of autophagy at the tumour-immune interface. J. Cell Mol. Med. 2021, 25, 2333–2341. [Google Scholar] [CrossRef]
- Wallace, P.R. The band theory of graphite. Phys. Rev. 1947, 71, 622–634. [Google Scholar] [CrossRef]
- Boehm, H.P.; Setton, R.; Stumpp, E. Nomenclature and terminology of graphite intercalation compounds. Carbon 1986, 24, 241–245. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Hoseini-Ghahfarokhi, M.; Mirkiani, S.; Mozaffari, N.; Abdolahi Sadatlu, M.A.; Ghasemi, A.; Abbaspour, S.; Akbarian, M.; Farjadian, F.; Karimi, M. Applications of graphene and graphene oxide in smart drug/gene delivery: Is the world still flat? Int. J. Nanomed. 2020, 15, 9469–9496. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GNM | Cell Type | LC3-II, LC3 Puncta | AV (TEM) | Autophagic Flux | Mechanisms | Ref. |
|---|---|---|---|---|---|---|
| GO | MG-63 human osteosarcoma | ↑ | n.a. | ↑ (↑LC3-II—flux assay) | ↑ROS, ↑ATG3, ↑ATG5, ↑ATG7, ↑NRF2, ↓BCL2 | [29] |
| GO | CT26 mouse colon carcinoma, Skov-3 human ovarian carcinoma | ↑ | n.a. | ↑ (AP-LY fusion) | n.a. | [30] |
| GO | SH-SY5Y human neuroblastoma | ↑ | n.a. | ↑ (↓p62) | ↑AMPK, ↓mTOR, ↑beclin | [31] |
| GO | CT26 mouse colon carcinoma | ↑ | n.a. | ↑ (AP-LY fusion) | n.a. | [32] |
| GO | CT26 mouse colon carcinoma | ↑ | ↑ | ↑ (AP-LY fusion) | ↑TLR4/TLR9, ↑MyD88, ↑TRAF6, ↑NF-κB, ↑beclin | [33] |
| GO | SK-N-SH human neuroblastoma | ↑ | ↑ | ↑ (↓p62) | n.a. | [34] |
| GO | PC12 rat pheochromocytoma, HeLa cervical carcinoma | ↑ | ↑ | ↑ (↑LC3-II—flux assay) | ↑ERK | [35] |
| GO | RAW264.7 mouse monocytic leukemia | ↑ | ↑ | n.a. | ↑TLR4/TLR9, ↑MyD88, ↑TRAF6, ↑NF-κB, ↑beclin | [36] |
| GO | SK-N-BE(2) and SH-SY5Y human neuroblastoma | ↑ | ↑ | n.a. | ↑ROS, ↑BNIP3, ↑beclin | [37] |
| GO | HONE1 human nasopharingeal carcinoma | ↑ | ↑ | n.a. | ↑LC3 mRNA, ↑ER stress, ↑GRP78 | [38] |
| blue light-excited GQD | U251 human glioma | ↑ | ↑ | ↑ (↓p62) | ↑ROS | [39] |
| hydroxy-GQD | A549 human lung carcinoma | ↑ | ↑ | ? (→p62) | ↑p38MAPK, ↑JNK | [40] |
| amino-GQD | A549 human lung carcinoma | ↑ | ↑ | ? (→p62) | ↑p38MAPK, ↓Akt, ↑beclin | [40] |
| GQD | THP-1 human monocytic leukemia | ↑ | n.a. | n.a. | ↑ROS, ↑p38MAPK, ↑NF-κB, ↑beclin, ↓BCL2 | [41] |
| GO, GO-Ag composite | SH-SY5Y human neuroblastoma | n.a. | ↑ | n.a. | ↑ROS, ↓BCL2 mRNA | [42] |
| GO-CQ conjugate | A549 human lung carcinoma | ↑ | ↑ | ↓ (↑p62) | ↑ROS, ↑ATG5 | [43] |
| GO | PC12 rat pheochromocytoma | ↑ | ↑ | ↓ (↓AP-LY fusion, ↑p62) | ↓PI3K, ↓Akt, ↓mTOR, ↑ATG5, ↓BCL2, ↓LY acidity, ↓ACP, ↓CTSB, ↑LMP | [44] |
| GNF | A549 human lung adenocarcinoma | ↑ | ↑ | ↓ (↑p62) | ↑ROS, ↓mTOR, ↑ATG5, ↓BCL2, ↑beclin, ↑beclin/LC3/p62 mRNA, ↓LY acidity, ↑LMP, ↓actin cytoskeleton | [45] |
| GO | F98 rat glioma | ↑ | ↑ | ↓ (↑p62) | ↓PI3K, ↓Akt, ↓mTOR, ↓LY acidity, ↓CTSB | [46] |
| GO | RAW264.7 mouse monocytic leukemia | ↑ | n.a. | ↓ (↑p62) | ↑ROS, ↑NRF2, ↓CTSB, ↓CTSD | [47] |
| GNM | Cell Type | Type of Cell Death | Autophagy Involvement | Cell Death Modulation | Cell Death Mechanisms | Ref. |
|---|---|---|---|---|---|---|
| GO + cisplatin | Skov-3 human ovarian carcinoma | necrosis | cytotoxic autophagy induction | ↓ by ULK1 RNAi ↓ by ATG7 RNAi | nuclear import of LC3 | [30] |
| GO + cisplatin | CT26 mouse colon carcinoma | necrosis | cytotoxic autophagy induction | ↓ by 3-MA ↓ by BAF A1 | nuclear import of LC3 | [32] |
| blue light-excited GQD | U251 human glioma | apoptosis | cytotoxic autophagy induction | ↓ by LC3B RNAi | ↑ROS, ↑caspases | [39] |
| hydroxy-GQD | A549 human lung carcinoma | apoptosis | cytoprotective autophagy induction | ↑ by 3-MA | n.a. | [40] |
| amino-GQD | A549 human lung carcinoma | apoptosis upon autophagy inhibition | cytoprotective autophagy induction | ↑ by 3-MA | n.a. | [40] |
| GO-CQ conjugate | A549 human lung carcinoma | necroptosis | cytotoxic block of autophagic flux | ↓ by p62 RNAi | ↑ROS, ↑RIPK1, ↑RIPK3, ↑MLKL, p62 and ATG5 as scaffolds for necrosome assembly | [43] |
| GO | PC12 rat pheochromocytoma | apoptosis | cytotoxic block of autophagic flux | ↓ by rapamycin ↓ by p62 RNAi | ↑p62, ↑LMP, ↓ΔΨ, ↑Bax/Bcl-2, ↑caspase-3, ↑caspase-9 | [44] |
| GNF | A549 human lung carcinoma | apoptosis | cytotoxic block of autophagic flux | ↓ by 3-MA ↓ by LC3 RNAi | ↑ROS, ↑LMP, ↓ΔΨ, ↑Bax/Bcl-2, ↓ATP, ↑caspase-3, ↓actin cytoskeleton | [45] |
| GO | F98 rat glioma | apoptosis | cytotoxic block of autophagic flux | ↓ by rapamycin | ↑p62, ↑Bax/Bcl-2, ↓ΔΨ, ↑caspase-3 | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ristic, B.; Harhaji-Trajkovic, L.; Bosnjak, M.; Dakic, I.; Mijatovic, S.; Trajkovic, V. Modulation of Cancer Cell Autophagic Responses by Graphene-Based Nanomaterials: Molecular Mechanisms and Therapeutic Implications. Cancers 2021, 13, 4145. https://doi.org/10.3390/cancers13164145
Ristic B, Harhaji-Trajkovic L, Bosnjak M, Dakic I, Mijatovic S, Trajkovic V. Modulation of Cancer Cell Autophagic Responses by Graphene-Based Nanomaterials: Molecular Mechanisms and Therapeutic Implications. Cancers. 2021; 13(16):4145. https://doi.org/10.3390/cancers13164145
Chicago/Turabian StyleRistic, Biljana, Ljubica Harhaji-Trajkovic, Mihajlo Bosnjak, Ivana Dakic, Srdjan Mijatovic, and Vladimir Trajkovic. 2021. "Modulation of Cancer Cell Autophagic Responses by Graphene-Based Nanomaterials: Molecular Mechanisms and Therapeutic Implications" Cancers 13, no. 16: 4145. https://doi.org/10.3390/cancers13164145
APA StyleRistic, B., Harhaji-Trajkovic, L., Bosnjak, M., Dakic, I., Mijatovic, S., & Trajkovic, V. (2021). Modulation of Cancer Cell Autophagic Responses by Graphene-Based Nanomaterials: Molecular Mechanisms and Therapeutic Implications. Cancers, 13(16), 4145. https://doi.org/10.3390/cancers13164145