
Chromoanagenesis Landscape in 10,000 TCGA Patients



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Machine Learning Pipeline
2.3. Statistical Analysis
2.4. Visualization
3. Results
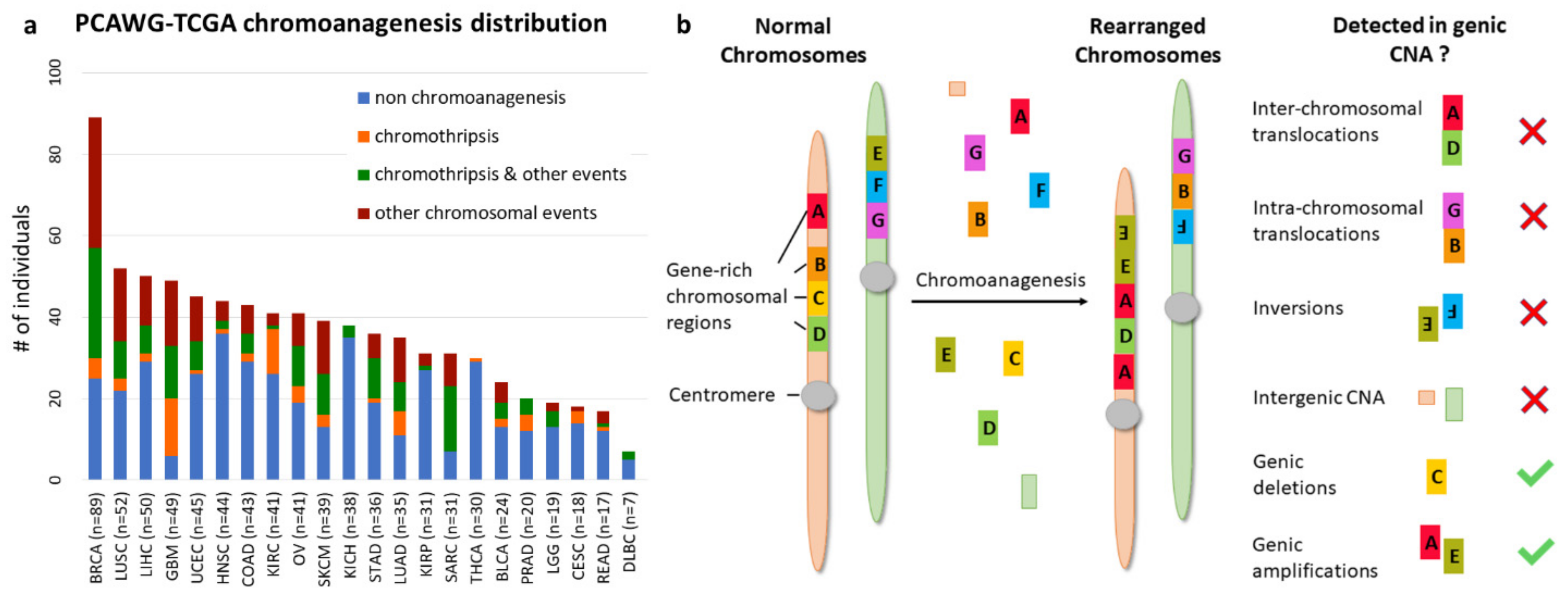
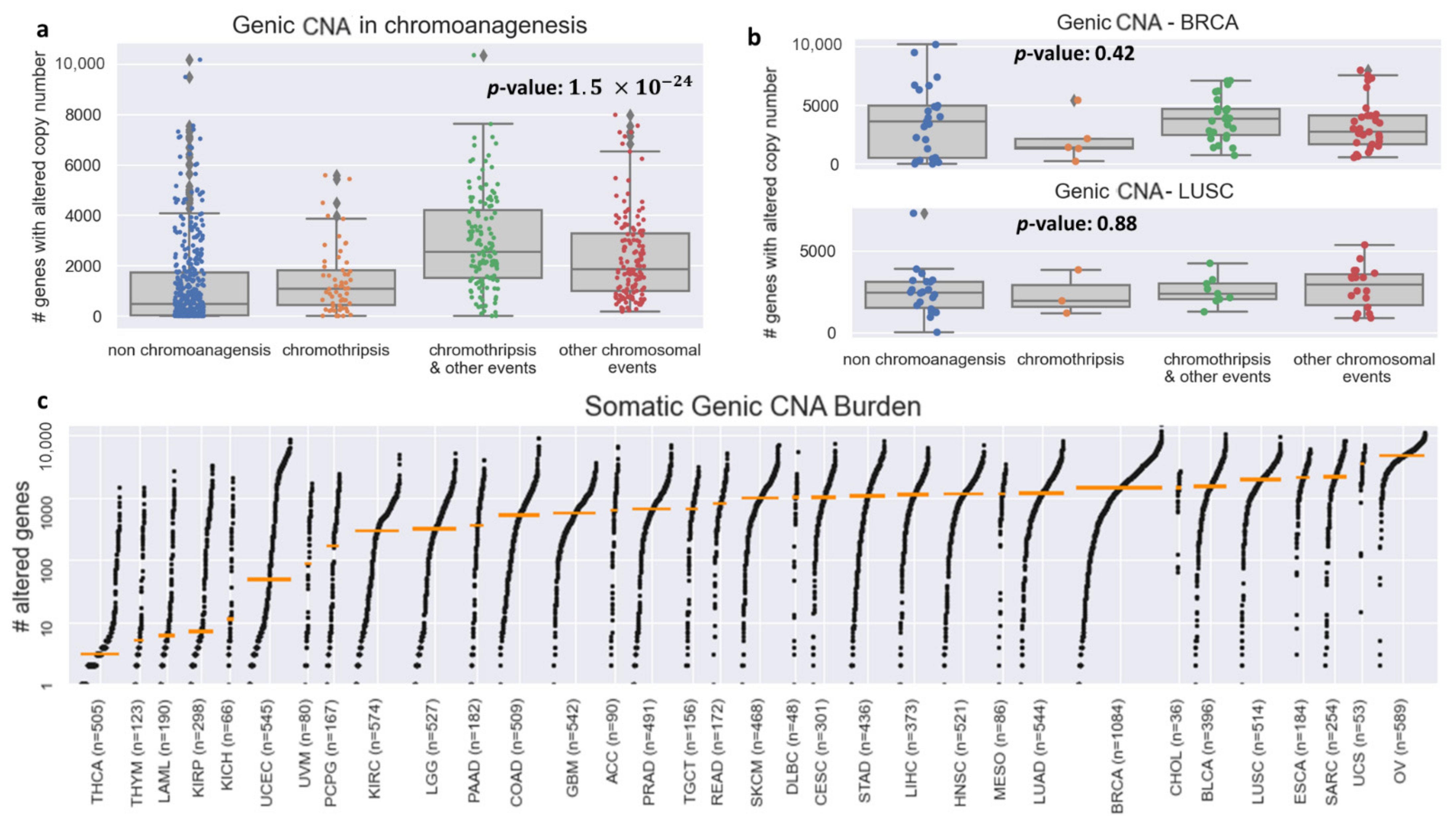
3.1. Cancer Type Impacts Genic CNA Frequency
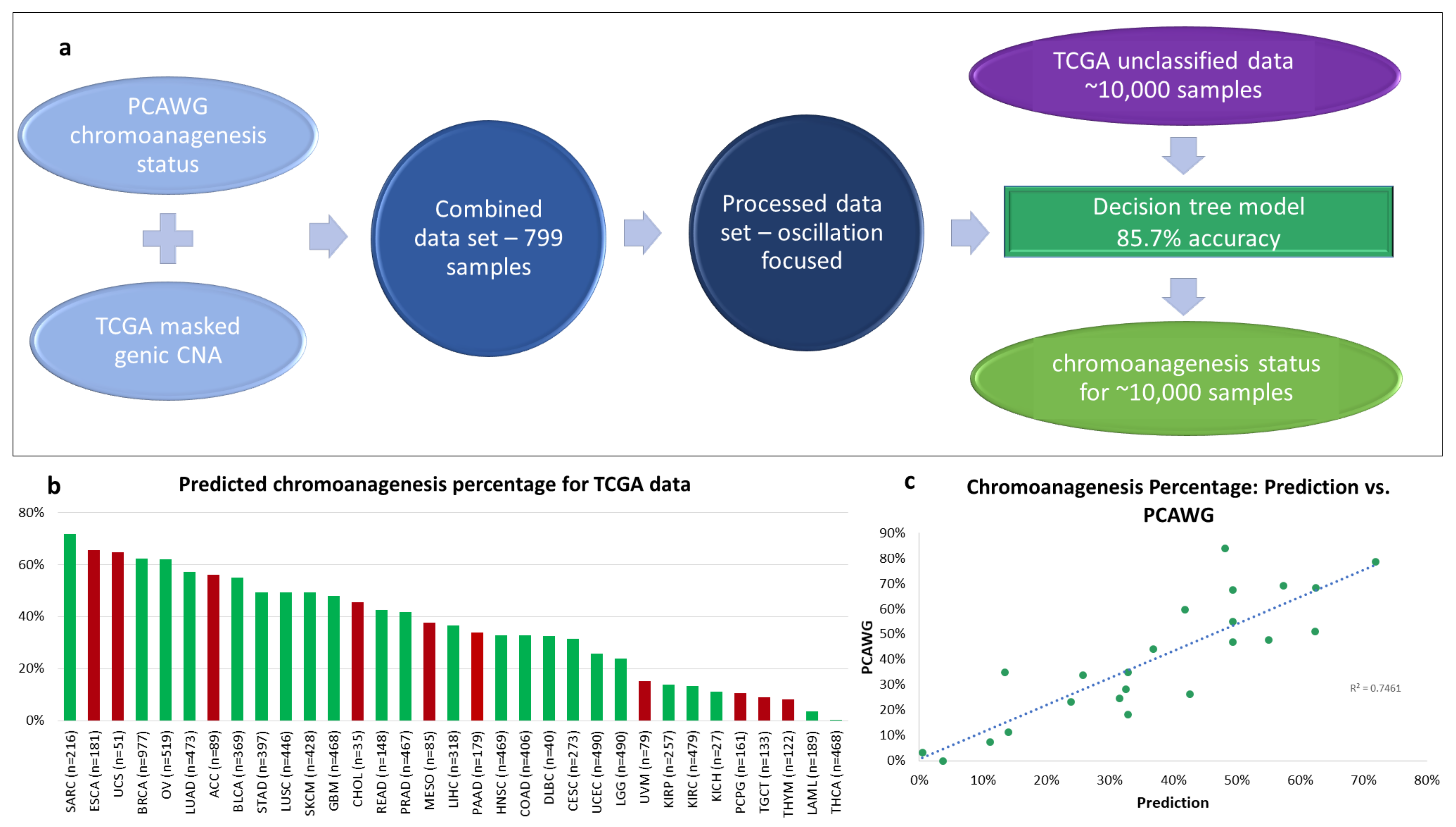
3.2. Predicting Chromoanagenesis at High Accuracy
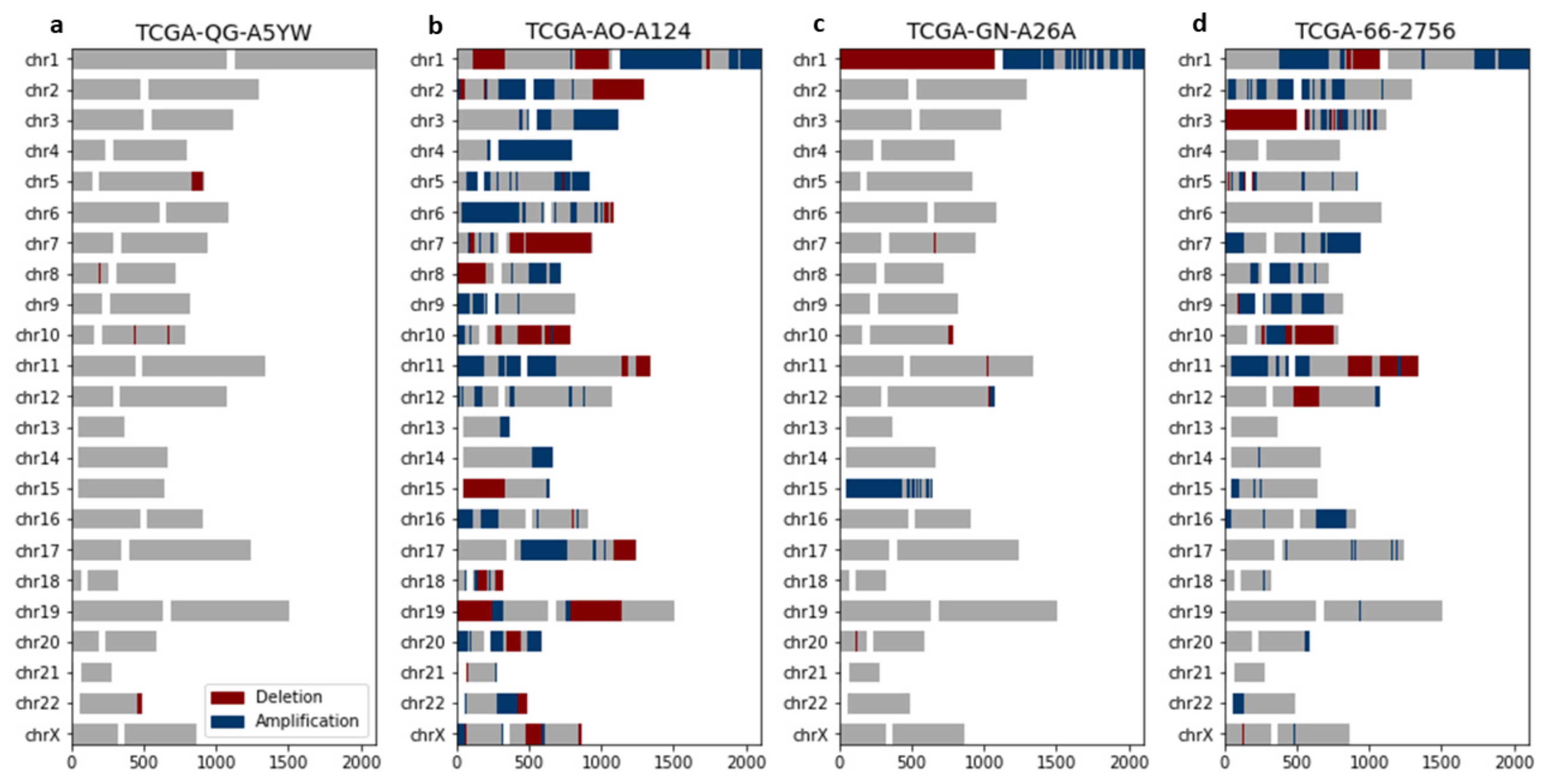
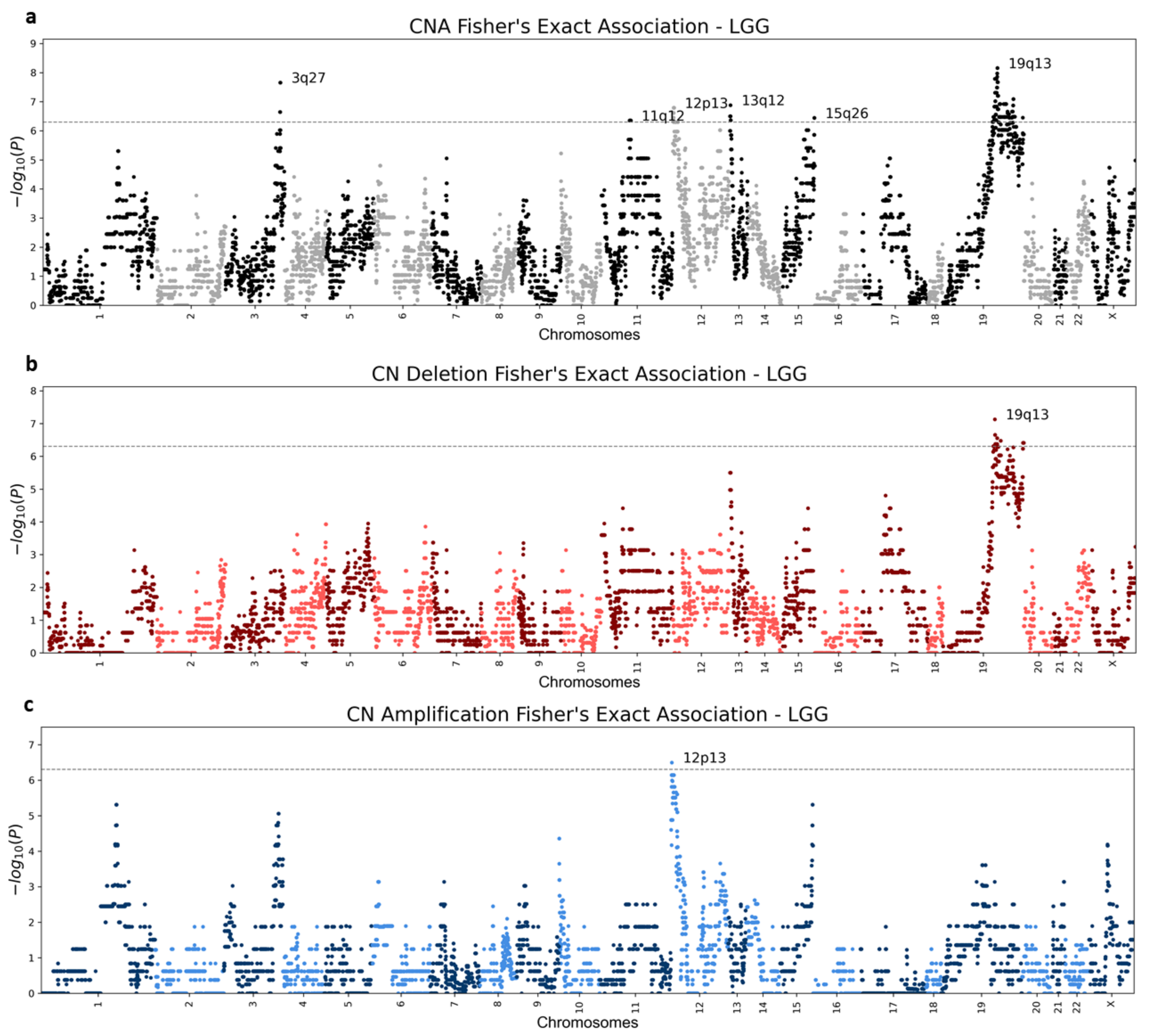
3.3. Chromoanagenesis Cancer Specific CNA Patterns
3.4. Chromoanagenesis Single Gene Focal Alterations
3.5. Chromoanagenesis CNA Pattern Overlaps with Existing Knowledge
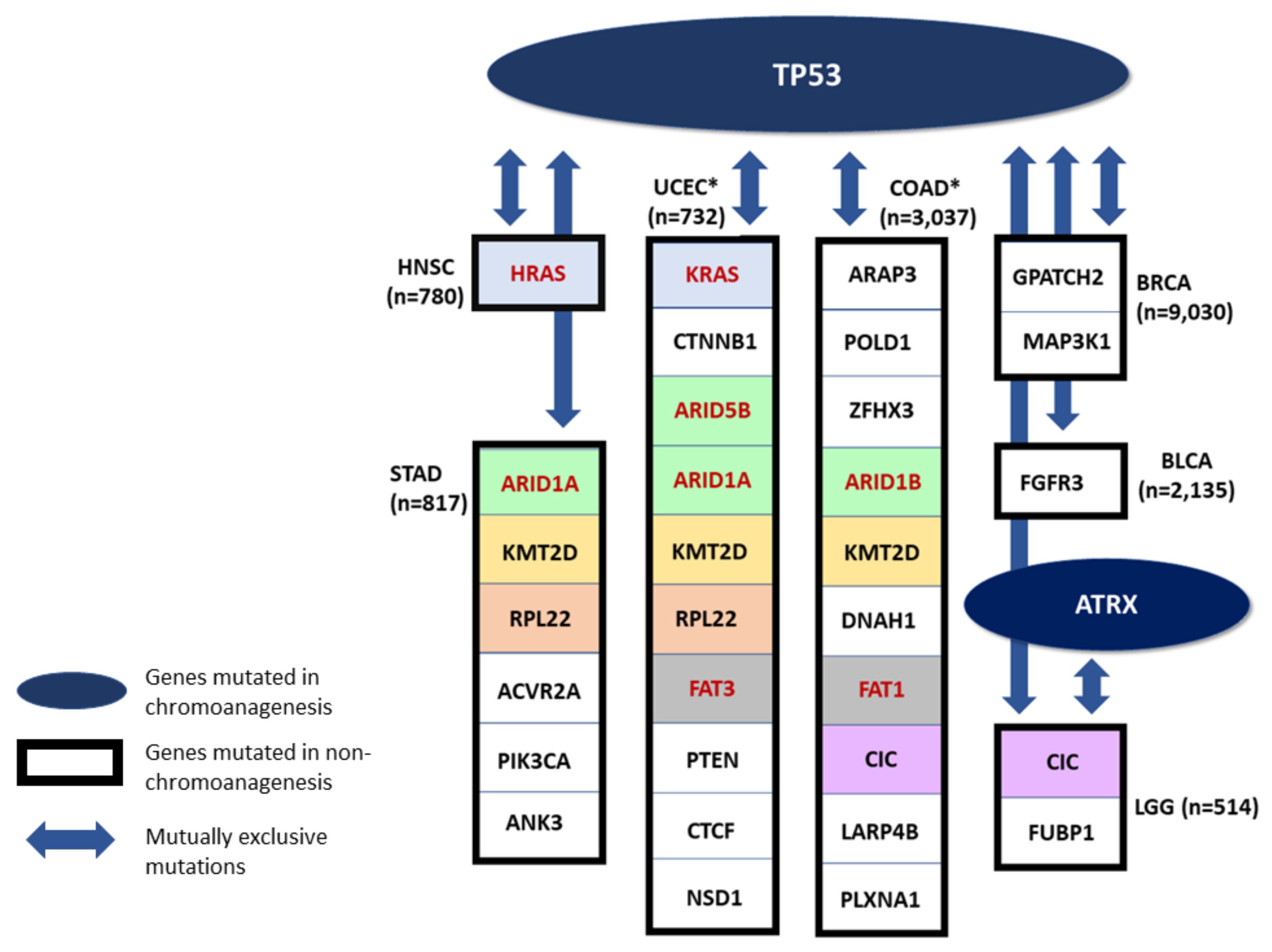
3.6. Somatic SNV Reveal Chromoanagenesis Gene Differentiation
3.7. Mutual Exclusivity Imply Distinct Tumorigenesis Pathways
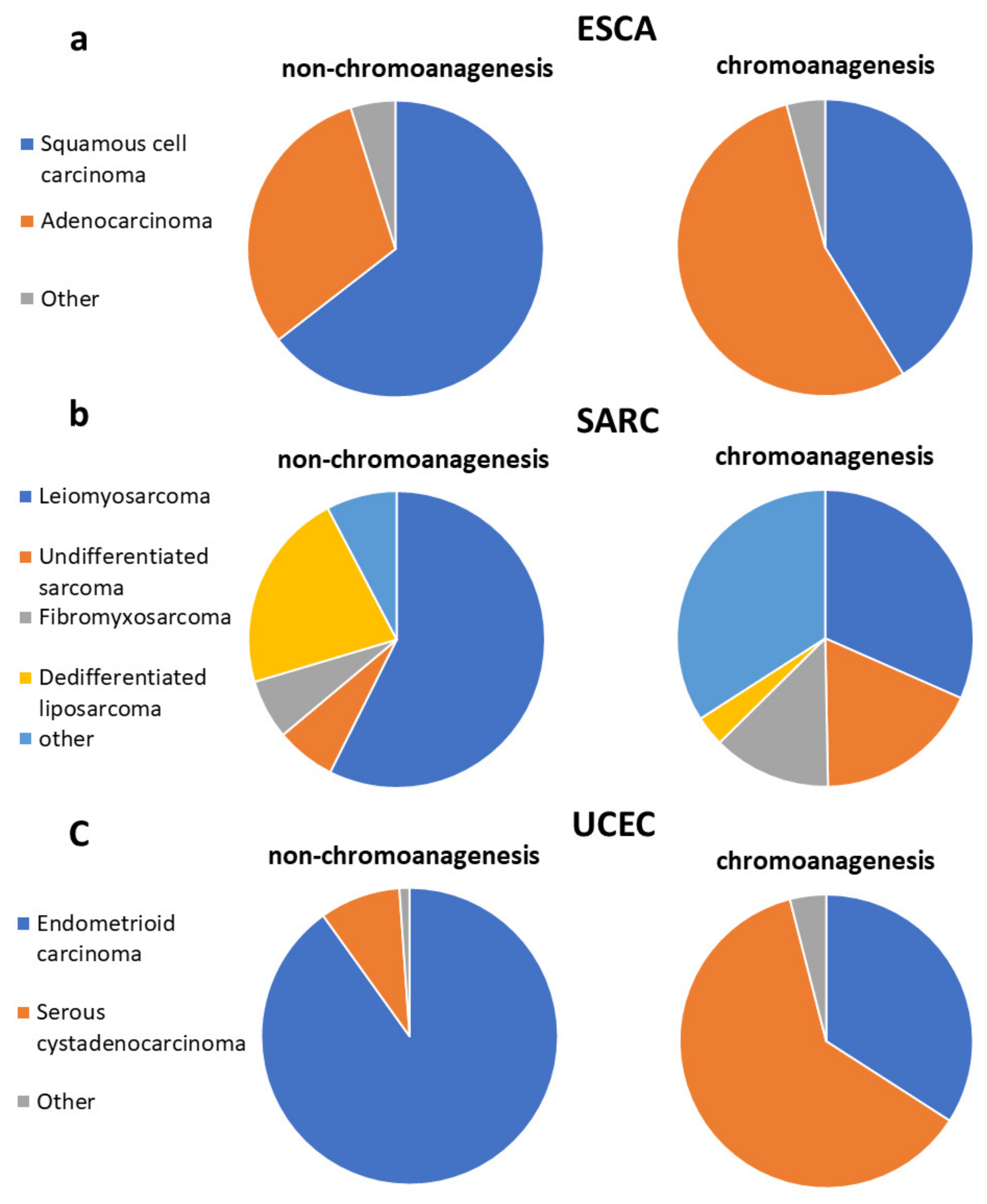
3.8. Chromoanagenesis Samples Are Mostly Not Signified by Distinct Clinical Characteristics
3.9. Chromoanagenesis Does Not Correlate with HPV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- De Pagter, M.S.; van Roosmalen, M.J.; Baas, A.F.; Renkens, I.; Duran, K.J.; van Binsbergen, E.; Tavakoli-Yaraki, M.; Hochstenbach, R.; van der Veken, L.T.; Cuppen, E.; et al. Chromothripsis in Healthy Individuals Affects Multiple Protein-Coding Genes and Can Result in Severe Congenital Abnormalities in Offspring. Am. J. Hum. Genet. 2015, 96, 651–656. [Google Scholar] [CrossRef] [Green Version]
- Pellestor, F. Chromoanagenesis: Cataclysms behind complex chromosomal rearrangements. Mol. Cytogenet. 2019, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Pellestor, F.; Gatinois, V. Chromoanagenesis: A piece of the macroevolution scenario. Mol. Cytogenet. 2020, 13, 3. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Gaillard, J.; Schneider, A.; Puechberty, J.; Gatinois, V. Chromoanagenesis, the mechanisms of a genomic chaos. Semin. Cell Dev. Biol. 2021. [Google Scholar] [CrossRef]
- Campbell, P.J.; Raine, K.M.; Butler, A.P.; Garrison, E.; Zamora, J.; Wedge, D.C.; Tarabichi, M.; Roberts, N.D.; Li, Y.; Alexandrov, L.B.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82. [Google Scholar]
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciampi, R.; Giordano, T.J.; Wikenheiser-Brokamp, K.; Koenig, R.J.; Nikiforov, Y.E. HOOK3-RET: A novel type of RET/PTC rearrangement in papillary thyroid carcinoma. Endocr. Relat. Cancer 2007, 14, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Mauri, F.; Song, Y.; de Cock, F.; Meeusen, B.; Swedlund, B.; Impens, F.; Van Haver, D.; Opitz, M.; Thery, M.; et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 2021, 589, 448–455. [Google Scholar] [CrossRef]
- Lamy, P.J.; Fina, F.; Bascoul-Mollevi, C.; Laberenne, A.C.; Martin, P.M.; Ouafik, L.H.; Jacot, W. Quantification and clinical relevance of gene amplification at chromosome 17q12-q21 in human epidermal growth factor receptor 2-amplified breast cancers. Breast Cancer Res. 2011, 13, R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varis, A.; Wolf, M.; Monni, O.; Vakkari, M.-L.; Kokkola, A.; Moskaluk, C.; Frierson, H.; Powell, S.M.; Knuutila, S.; Kallioniemi, A.; et al. Targets of gene amplification and overexpression at 17q in gastric cancer. Cancer Res. 2002, 62, 2625–2629. [Google Scholar]
- Sircoulomb, F.; Bekhouche, I.; Finetti, P.; Adélaïde, J.; Ben Hamida, A.; Bonansea, J.; Raynaud, S.; Innocenti, C.; Charafe-Jauffret, E.; Tarpin, C.; et al. Genome profiling of ERBB2-amplified breast cancers. BMC Cancer 2010, 10, 539. [Google Scholar] [CrossRef]
- Borg, A.; Baldetorp, B.; Fernö, M.; Killander, D.; Olsson, H.; Sigurdsson, H. ERBB2 amplification in breast cancer with a high rate of proliferation. Oncogene 1991, 6, 137–143. [Google Scholar]
- Cowin, P.A.; George, J.; Fereday, S.; Loehrer, E.; Van Loo, P.; Cullinane, C.; Etemadmoghadam, D.; Ftouni, S.; Galletta, L.; Anglesio, M.S.; et al. LRP1B deletion in high-grade serous ovarian cancers is associated with acquired chemotherapy resistance to liposomal doxorubicin. Cancer Res. 2012, 72, 4060–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.I.; Zhu, Y.; McAvoy, S.; Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, J.; Takahashi, Y.; Goto, J.; Tomiyama, H.; Ishikawa, S.; Yoshino, H.; Minami, N.; Smith, D.I.; Lesage, S.; Aburatani, H.; et al. Mechanisms of genomic instabilities underlying two common fragile-site-associated Loci, PARK2 and DMD, in germ cell and cancer cell lines. Am. J. Hum. Genet. 2010, 87, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smida, J.; Xu, H.-E.; Zhang, Y.; Baumhoer, D.; Ribi, S.; Kovac, M.; Von Luettichau, I.; Bielack, S.; O’Leary, V.B.; Leib-Mösch, C.; et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int. J. Cancer 2017, 141, 816–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Oeggerli, M.; Tomovska, S.; Schraml, P.; Calvano-Forte, D.; Schafroth, S.; Simon, R.; Gasser, T.; Mihatsch, M.J.; Sauter, G. E2F3 amplification and overexpression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Oncogene 2004, 23, 5616–5623. [Google Scholar] [CrossRef] [Green Version]
- López, V.; González-Peramato, P.; Suela, J.; Serrano, A.; Algaba, F.; Cigudosa, J.C.; Vidal, A.; Bellmunt, J.; Heredero, O.; Sánchez-Carbayo, M. Identification of prefoldin amplification (1q23.3-q24.1) in bladder cancer using comparative genomic hybridization (CGH) arrays of urinary DNA. J. Transl. Med. 2013, 11, 182. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.; Richter, J.; Wagner, U.; Fijan, A.; Bruderer, J.; Schmid, U.; Ackermann, D.; Maurer, R.; Alund, G.; Knönagel, H.; et al. High-throughput tissue microarray analysis of 3p25 (RAF1) and 8p12 (FGFR1) copy number alterations in urinary bladder cancer. Cancer Res. 2001, 61, 4514–4519. [Google Scholar]
- Uysal, D.; Kowalewski, K.-F.; Kriegmair, M.C.; Wirtz, R.; Popovic, Z.V.; Erben, P. A comprehensive molecular characterization of the 8q22.2 region reveals the prognostic relevance of OSR2 mRNA in muscle invasive bladder cancer. PLoS ONE 2021, 16, e0248342. [Google Scholar] [CrossRef]
- Letessier, A.; Sircoulomb, F.; Ginestier, C.; Cervera, N.; Monville, F.; Gelsi-Boyer, V.; Esterni, B.; Geneix, J.; Finetti, P.; Zemmour, C.; et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer 2006, 6, 245. [Google Scholar] [CrossRef] [Green Version]
- Escudero-Esparza, A.; Bartoschek, M.; Gialeli, C.; Okroj, M.; Owen, S.; Jirström, K.; Orimo, A.; Jiang, W.G.; Pietras, K.; Blom, A.M. Complement inhibitor CSMD1 acts as tumor suppressor in human breast cancer. Oncotarget 2016, 7, 76920–76933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, M.; Shaaban, A.; Zhang, L.; Walker, C.; Gray, S.L.; Thakker, N.; Toomes, C.; Speirs, V.; Bell, S.M. Loss of CSMD1 expression is associated with high tumour grade and poor survival in invasive ductal breast carcinoma. Breast Cancer Res. Treat. 2009, 121, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Collins, V.P.; Schmidt, E.E.; Collins, V.P. Amplification and Overexpression of the MDM2 Gene in a Subset of Human Malignant Gliomas without p53 Mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar]
- Furgason, J.M.; Koncar, R.F.; Michelhaugh, S.K.; Sarkar, F.H.; Mittal, S.; Sloan, A.E.; Barnholtz-Sloan, J.; Bahassi, E.M. Whole genome sequence analysis links chromothripsis to EGFR, MDM2, MDM4, and CDK4 amplification in glioblastoma. Oncoscience 2015, 2, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Flørenes, V.A.; Mælandsmo, G.M.; Forus, A.; Andreassen, Å.; Myklebost, O.; Fodstad, Ø. MDM2 gene amplification and transcript levels in human sarcomas: Relationship to TP53 gene status. J. Natl. Cancer Inst. 1994, 86, 1297–1302. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Voronina, N.; Wong, J.K.L.; Hübschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat. Commun. 2020, 11, 3572. [Google Scholar] [CrossRef]
- Jiao, Y.; Killela, P.J.; Reitman, Z.; Rasheed, B.A.; Heaphy, C.M.; De Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Shinjo, S.; Marie, S.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, J.-Y.; Lee, J.-C.; Tsai, J.-H.; Yang, C.-Y.; Liu, T.-L.; Ke, Z.-L.; Hsu, H.-H.; Jeng, Y.-M. Comprehensive screening of alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas. Mod. Pathol. 2015, 28, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, L.; Parsons, R. PTEN: Life as a tumor suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, H.; Li, M.; Jiang, Z.; Liu, Z.; Wang, X. Correlate the TP53 Mutation and the HRAS Mutation with Immune Signatures in Head and Neck Squamous Cell Cancer. Comput. Struct. Biotechnol. J. 2019, 17, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Baldo, O.; Hamden, P.; Knowles, M.A. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J. Pathol. 2007, 213, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Song, W.; Bi, X.; Zhao, J.; Huang, Z.; Li, Z.; Zhou, J.; Cai, J.; Zhao, H. Recent advances in the ARID family: Focusing on roles in human cancer. OncoTargets Ther. 2014, 7, 315–324. [Google Scholar]
- Wu, S.; Fatkhutdinov, N.; Zhang, R. Harnessing mutual exclusivity between TP53 and ARID1 A mutations. Cell Cycle 2017, 16, 2313–2314. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Wang, T.L.; Shih, I.M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef] [Green Version]
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Cherniack, A.D.; Pashtan, I.; Saksena, G.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169. [Google Scholar]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Li, S.C.; Tachiki, L.M.L.; Kabeer, M.H.; Dethlefs, B.A.; Anthony, M.J.; Loudon, W.G. Cancer genomic research at the crossroads: Realizing the changing genetic landscape as intratumoral spatial and temporal heterogeneity becomes a confounding factor. Cancer Cell Int. 2014, 14, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wen, Q.; Stucky, A.; Zeng, Y.; Gao, S.; Loudon, W.G.; Ho, H.W.; Kabeer, M.H.; Li, S.C.; Zhang, X.; et al. Relapse pathway of glioblastoma revealed by single-cell molecular analysis. Carcinogenesis 2018, 39, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Mendoza, C.J.; Morton, C.C. The Iceberg under Water: Unexplored Complexity of Chromoanagenesis in Congenital Disorders. Am. J. Hum. Genet. 2019, 104, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Zanardo, É.A.; Piazzon, F.B.; Dutra, R.L.; Dias, A.T.; Montenegro, M.M.; Novo-Filho, G.M.; Costa, T.V.M.M.; Nascimento, A.M.; Kim, C.A.; Kulikowski, L.D. Complex structural rearrangement features suggesting chromoanagenesis mechanism in a case of 1p36 deletion syndrome. Mol. Genet. Genom. 2014, 289, 1037–1043. [Google Scholar] [CrossRef]
- Arya, P.; Hodge, J.C.; Matlock, P.A.; Vance, G.H.; Breman, A.M. Two Patients with Complex Rearrangements Suggestive of Germline Chromoanagenesis. Cytogenet. Genome Res. 2020, 160, 671–679. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Fukami, M.; Shima, H.; Suzuki, E.; Ogata, T.; Matsubara, K.; Kamimaki, T. Catastrophic cellular events leading to complex chromosomal rearrangements in the germline. Clin. Genet. 2016, 91, 653–660. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | # Amplified Regions | # Deleted Regions | # Additional Altered Regions | |
|---|---|---|---|---|
| BLCA | Bladder urothelial carcinoma | 4 | 3 | |
| BRCA | Breast invasive carcinoma | 20 | 3 | 14 |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma | 2 | ||
| COAD | Colon adenocarcinoma | 1 | 1 | |
| GBM | Glioblastoma multiforme | 1 | 1 | |
| HNSC | Head and neck squamous cell carcinoma | 5 | ||
| LGG | Brain lower grade glioma | 1 | 5 | |
| LUAD | Lung adenocarcinoma | 1 | 6 | |
| LUSC | Lung squamous cell carcinoma | 1 | ||
| OV | Ovarian serous cystadenocarcinoma | 2 | ||
| PRAD | Prostate adenocarcinoma | 1 | ||
| SARC | Sarcoma | 1 | ||
| SKCM | Skin cutaneous melanoma | 3 | 3 | |
| STAD | Stomach adenocarcinoma | 16 | 4 | 38 |
| UCEC | Uterine corpus endometrial carcinoma | 166 | 57 | 74 |
| Gene | Gene Full Name | Amplified in | Deleted in | Altered in | Is Driver |
|---|---|---|---|---|---|
| ANKS1B | Ankyrin Repeat And Sterile Alpha Motif Domain Containing 1B | UCEC | − | ||
| CSMD1 | CUB And Sushi Multiple Domains 1 | BRCA | − | ||
| DLG2 | Discs Large MAGUK Scaffold Protein 2 | UCEC | − | ||
| DMD | Dystrophin | UCEC, ESCA *, STAD * | − | ||
| ELAVL1 | ELAV Like RNA Binding Protein 1 | UCEC | − | ||
| ESR1 | Estrogen Receptor 1 | UCEC | + | ||
| FGF14 | Fibroblast Growth Factor 14 | PRAD | − | ||
| KAZN | Kazrin, Periplakin Interacting Protein | UCEC | − | ||
| LRP1B | LDL Receptor Related Protein 1B | UCEC, OV * | + | ||
| LSAMP | Limbic System Associated Membrane Protein | UCEC, STAD * | − | ||
| MACROD2 | Mono-ADP Ribosylhydrolase 2 | STAD | − | ||
| MECOM | MDS1 And EVI1 Complex Locus | UCEC | + | ||
| PARK2 | Parkin RBR E3 Ubiquitin Protein Ligase | COAD | − | ||
| PDE4D | Phosphodiesterase 4D | STAD, UCEC, ESCA * | BLCA | − | |
| PGM5 | Phosphoglucomutase-Related Protein | UCEC | − | ||
| RAD51B | RAD51 Paralog B | UCEC | + | ||
| SKAP1 | Src Kinase Associated Phosphoprotein 1 | UCEC | - | ||
| THSD4 | Thrombospondin Type 1 Domain Containing 4 | UCEC | − | ||
| WWOX | WW Domain Containing Oxidoreductase | UCEC | − | ||
| ZMAT4 | Zinc Finger Matrin-Type 4 | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasnic, R.; Linial, M. Chromoanagenesis Landscape in 10,000 TCGA Patients. Cancers 2021, 13, 4197. https://doi.org/10.3390/cancers13164197
Rasnic R, Linial M. Chromoanagenesis Landscape in 10,000 TCGA Patients. Cancers. 2021; 13(16):4197. https://doi.org/10.3390/cancers13164197
Chicago/Turabian StyleRasnic, Roni, and Michal Linial. 2021. "Chromoanagenesis Landscape in 10,000 TCGA Patients" Cancers 13, no. 16: 4197. https://doi.org/10.3390/cancers13164197
APA StyleRasnic, R., & Linial, M. (2021). Chromoanagenesis Landscape in 10,000 TCGA Patients. Cancers, 13(16), 4197. https://doi.org/10.3390/cancers13164197