
TP53 in Biology and Treatment of Osteosarcoma

 ,
,  ,
,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
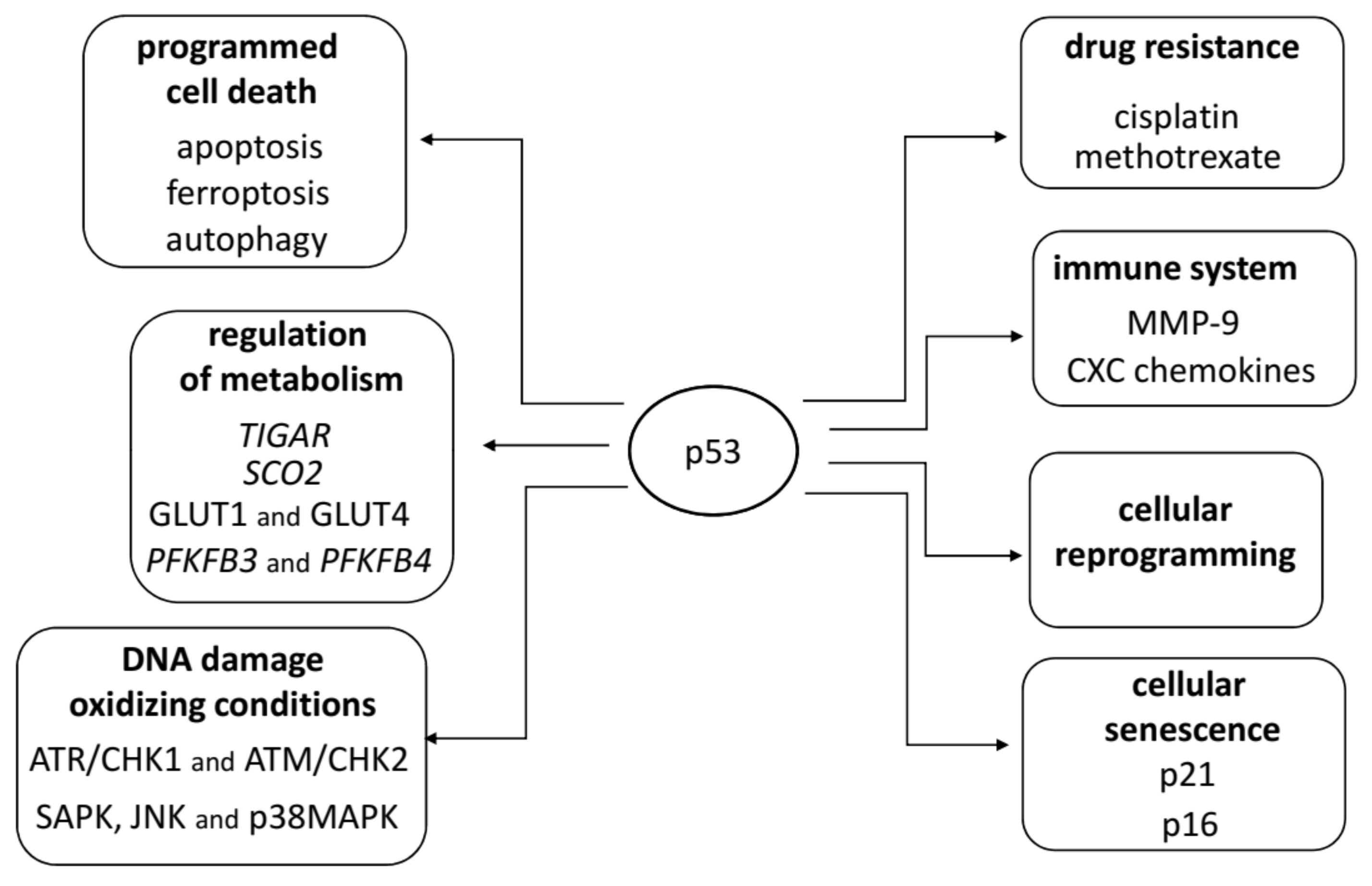
2. Structure and Functions of TP53
3. Malfunction of TP53 in Osteosarcoma
3.1. Osteosarcoma in Li-Fraumeni Syndrome
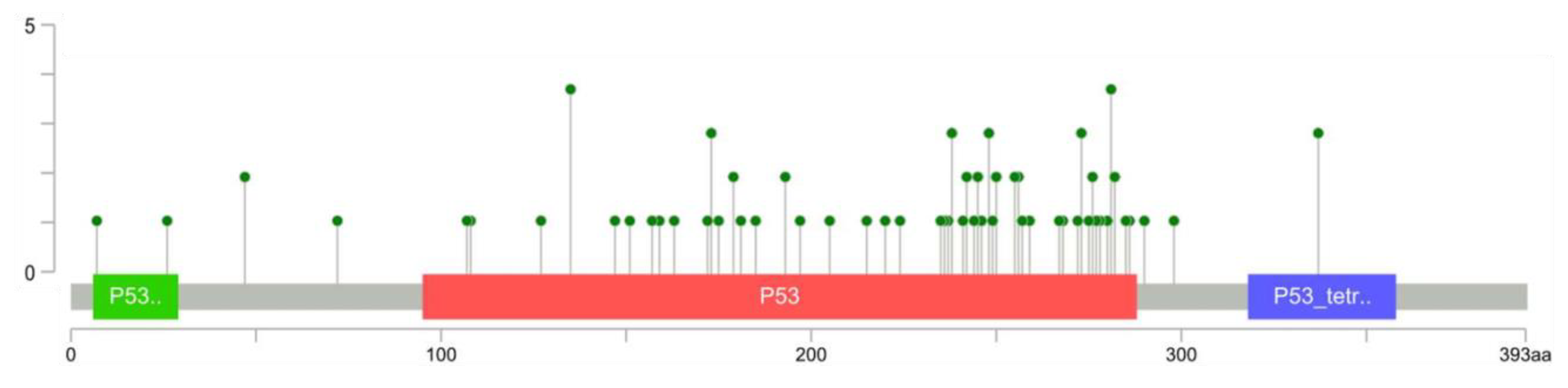
3.2. TP53 Alterations in Osteosarcoma Patient-Derived Samples
4. Animal Models
5. p53 as a Therapeutic Target in Osteosarcoma
5.1. Gene Therapy Approaches
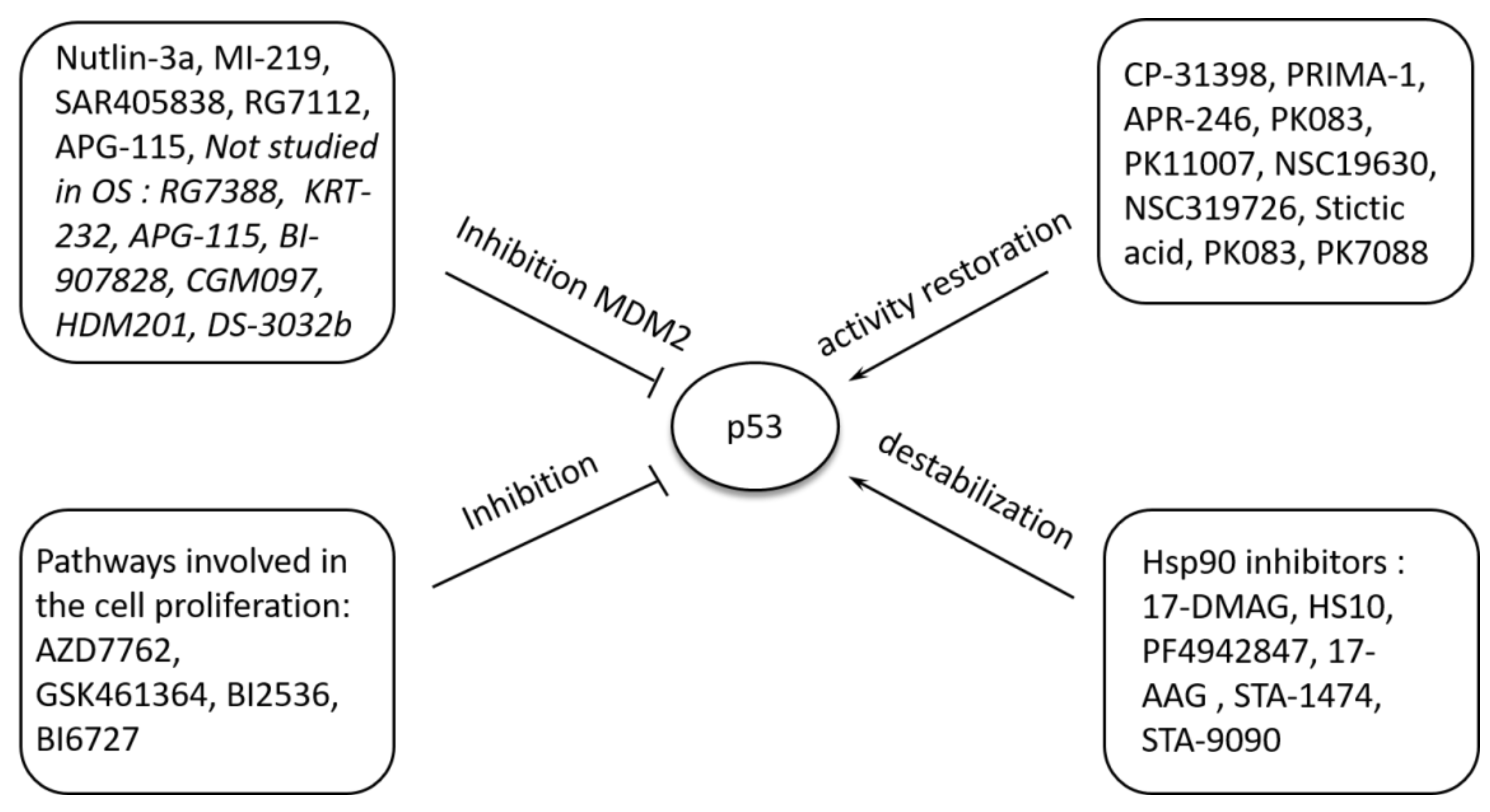
5.2. Pharmacological Modulation of P53 Function
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rutkowski, P.; Świtaj, T. Bone sarcomas. Oncol. Clin. Pract. 2018, 14, 115–128. [Google Scholar] [CrossRef]
- Wang, H.; Miao, R.; Jacobson, A.; Goldberg, S.; Harmon, D.C.; Cote, G.; Hornicek, F.J.; Raskin, K.; Nielsen, G.; DeLaney, T.F.; et al. Osteosarcoma prognostic nomograms for predicting the 10-year probability of mortality and recurrence. J. Clin. Oncol. 2017, 35, 11020. [Google Scholar] [CrossRef]
- Spalek, M.J.; Poleszczuk, J.; Czarnecka, A.M.; Dudzisz-Sledz, M.; Napieralska, A.; Matysiakiewicz, J.; Chojnacka, M.; Raciborska, A.; Sztuder, A.; Maciejczyk, A.; et al. Radiotherapy in the Management of Pediatric and Adult Osteosarcomas: A Multi-Institutional Cohort Analysis. Cells 2021, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brennan, B.; et al. Bone sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv79–iv95. [Google Scholar] [CrossRef] [PubMed]
- Picci, P. Osteosarcoma (osteogenic sarcoma). Orphanet. J. Rare Dis. 2007, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; Asaftei, S.D.; D’Ambrosio, L.; Pignochino, Y.; Nuzzo, A.; Gambarotti, M.; Marchesi, E.; Biagini, R.; et al. A nonrandomized phase II trial of sorafenib (S) and everolimus (E) in unresectable metastatic osteosarcoma (OST) patients (pts) relapsed after standard chemotherapy. J. Clin. Oncol. 2014, 32, 10533. [Google Scholar] [CrossRef]
- Xie, L.; Guo, W.; Xu, J.; Sun, X.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; Yan, T.; et al. Apatinib plus camrelizumab (SHR-1210) for unresectable high-grade osteosarcoma (APFAO) progressing after chemotherapy: A prospective, open label, phase II trial. J. Clin. Oncol. 2019, 37, 11013. [Google Scholar] [CrossRef]
- Smrke, A.; Anderson, P.M.; Gulia, A.; Gennatas, S.; Huang, P.H.; Jones, R.L. Future Directions in the Treatment of Osteosarcoma. Cells 2021, 10, 172. [Google Scholar] [CrossRef]
- Bishop, M.W.; Janeway, K.A.; Gorlick, R. Future directions in the treatment of osteosarcoma. Curr. Opin. Pediatr. 2016, 28, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Kang, N.; Wang, Y.; Guo, S.; Ou, Y.; Wang, G.; Chen, J.; Li, D.; Zhan, Q. Mutant TP53 G245C and R273H promote cellular malignancy in esophageal squamous cell carcinoma. BMC Cell Biol. 2018, 19, 16. [Google Scholar] [CrossRef]
- Doyle, B.; Morton, J.P.; Delaney, D.W.; Ridgway, R.A.; Wilkins, J.A.; Sansom, O.J. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J. Pathol. 2010, 222, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Min, L.; Seebacher, N.A.; Li, X.; Zhou, Y.; Hornicek, F.J.; Wei, Y.; Tu, C.; Duan, Z. Targeting mutant TP53 as a potential therapeutic strategy for the treatment of osteosarcoma. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2019, 37, 789–798. [Google Scholar] [CrossRef]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Wu, Q.; Gong, X.; Liu, J.; Ma, Y. Osteosarcoma: A review of current and future therapeutic approaches. Biomed. Eng. Online 2021, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Synoradzki, K.; Firlej, W.; Bartnik, E.; Sobczuk, P.; Fiedorowicz, M.; Grieb, P.; Rutkowski, P. Molecular Biology of Osteosarcoma. Cancers 2020, 12, 2130. [Google Scholar] [CrossRef] [PubMed]
- Vieler, M.; Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [Green Version]
- Courtois, S.; Verhaegh, G.; North, S.; Luciani, M.-G.; Lassus, P.; Hibner, U.; Oren, M.; Hainaut, P. ΔN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002, 21, 6722–6728. [Google Scholar] [CrossRef] [Green Version]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Bourdon, J.C. p53 and its isoforms in cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.S.; Mhoumadi, Y.; Verma, C.S. Roles of computational modelling in understanding p53 structure, biology, and its therapeutic targeting. J. Mol. Cell Biol. 2019, 11, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, N.; Attardi, L.D. The Transactivation Domains of the p53 Protein. Cold Spring Harb. Perspect. Med. 2017, 7, a026047. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, K.; Li, H.; Zhang, Y. Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol. Cell Biol. 2003, 23, 6396–6405. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Shi, T.; Polderman, P.E.; Burgering, B.M.T.; Dansen, T.B. DNA damage and oxidizing conditions activate p53 through differential upstream signaling pathways. bioRxiv 2020. [Google Scholar] [CrossRef]
- el-Deiry, W.S. Regulation of p53 downstream genes. Semin. Cancer Biol. 1998, 8, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sax, J.K.; El-Deiry, W.S. p53 downstream targets and chemosensitivity. Cell Death Differ. 2003, 10, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Hwang, P.M.; Rago, C.; Kinzler, K.W.; Vogelstein, B. Identification and classification of p53-regulated genes. Proc. Natl. Acad. Sci. USA 1999, 96, 14517–14522. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., III.; Meng, R.D.; Kao, G.; Muschel, R.; Yen, T.; El-Deiry, W.S. Induction of the TRAIL receptor KILLER/DR5 in p53-dependent apoptosis but not growth arrest. Oncogene 1999, 18, 6411–6418. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Yu, J.; Yue, W.; Wu, B.; Zhang, L. PUMA sensitizes lung cancer cells to chemotherapeutic agents and irradiation. Clin. Cancer Res. 2006, 12, 2928–2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-Only Member of the Bcl-2 Family and Candidate Mediator of p53-Induced Apoptosis. Science 2000, 288, 1053. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Michalak, E.M.; Villunger, A.; Adams, J.M.; Strasser, A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008, 15, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Wang, Y.; Chen, J.; Hu, Y.; Cao, Z.; Ren, P.; Zhang, Y. p21 overexpression sensitizes osteosarcoma U2OS cells to cisplatin via evoking caspase-3 and Bax/Bcl-2 cascade. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 3119–3123. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Yang, D.; Li, L.; Luo, G.; Li, T.; Fan, X.; Wang, Q.; Zhang, X.; Wang, Y.; Le, W. An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy 2009, 5, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar]
- Waldman, T.; Kinzler Kw Fau-Vogelstein, B.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, Y.; Zhang, G.; Huang, S.; Dong, Z.; Kong, C.; Su, D.; Du, J.; Zhu, S.; Liang, Q.; et al. DNMT3a plays a role in switches between doxorubicin-induced senescence and apoptosis of colorectal cancer cells. Int. J. Cancer 2011, 128, 551–561. [Google Scholar] [CrossRef]
- Gartel, A.L.; Tyner, A.L. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp. Cell Res. 1999, 246, 280–289. [Google Scholar] [CrossRef]
- Holzer, K.; Drucker, E.; Roessler, S.; Dauch, D.; Heinzmann, F.; Waldburger, N.; Eiteneuer, E.M.; Herpel, E.; Breuhahn, K.; Zender, L.; et al. Proteomic Analysis Reveals GMP Synthetase as p53 Repression Target in Liver Cancer. Am. J. Pathol. 2017, 187, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Yang, C.; Guo, Z.; Fu, Y.; Yu, X.; Liu, B.; Zhou, H.; Wang, J.; Li, W.; Pang, Q. P16 protein expression as a useful predictive biomarker for neoadjuvant chemotherapy response in patients with high-grade osteosarcoma: A systematic meta-analysis under guideline of PRISMA. Medicine 2017, 96, e6714. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisúa Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yin, X.; Qin, H.; Zhu, F.; Liu, H.; Yang, W.; Zhang, Q.; Xiang, C.; Hou, P.; Song, Z.; et al. Two Supporting Factors Greatly Improve the Efficiency of Human iPSC Generation. Cell Stem Cell 2008, 3, 475–479. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Lu, C.; Hu, W.; Sun, Y.; Levine, A.J. Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res. 2012, 72, 5635–5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarig, R.; Rivlin, N.; Brosh, R.; Bornstein, C.; Kamer, I.; Ezra, O.; Molchadsky, A.; Goldfinger, N.; Brenner, O.; Rotter, V. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J. Exp. Med. 2010, 207, 2127–2140. [Google Scholar] [CrossRef] [PubMed]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Fau-Karnieli, E.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros, S.; Flöter, J.; Kaymak, I.; Da Costa, C.; Houddane, A.; Dubuis, S.; Griffiths, B.; Mitter, R.; Walz, S.; Blake, S.; et al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 2017, 36, 3287–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, D.A.; He, Y.; Leslie, P.L.; Tikunov, A.P.; Fenger, N.; Macdonald, J.M.; Zhang, Y. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci. Rep. 2016, 6, 38067. [Google Scholar] [CrossRef] [Green Version]
- Matoba, S.; Kang, J.G.; Fau-Patino, W.D.; Patino Wd Fau-Wragg, A.; Wragg, A.; Fau-Boehm, M.; Boehm, M.; Fau-Gavrilova, O.; Gavrilova, O.; Fau-Hurley, P.J.; et al. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Yeudall, W.A.; Vaughan, C.A.; Miyazaki, H.; Ramamoorthy, M.; Choi, M.Y.; Chapman, C.G.; Wang, H.; Black, E.; Bulysheva, A.A.; Deb, S.P.; et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2021, 33, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Z.; Wu, H.; Xue, M.; Lin, P.; Wang, S.; Lin, N.; Huang, X.; Pan, W.; Liu, M.; et al. Epigenetic Regulation of CXCL12 Plays a Critical Role in Mediating Tumor Progression and the Immune Response In Osteosarcoma. Cancer Res. 2018, 78, 3938–3953. [Google Scholar] [CrossRef] [Green Version]
- Rahnamoun, H.; Lu, H.; Duttke, S.H.; Benner, C.; Glass, C.A.-O.; Lauberth, S.M. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, T.; Wang, W. Prognostic significance of matrix metalloproteinase 9 expression in osteosarcoma: A meta-analysis of 16 studies. Medicine 2018, 97, e13051. [Google Scholar] [CrossRef]
- Martelli, L.; Di Mario, F.; Botti, P.; Ragazzi, E.; Martelli, M.; Kelland, L. Accumulation, platinum–DNA adduct formation and cytotoxicity of cisplatin, oxaliplatin and satraplatin in sensitive and resistant human osteosarcoma cell lines, characterized by p53 wild-type status. Biochem. Pharmacol. 2007, 74, 20–27. [Google Scholar] [CrossRef]
- Goto, A.; Kanda, H.; Ishikawa, Y.; Matsumoto, S.; Kawaguchi, N.; Machinami, R.; Kato, Y.; Kitagawa, T. Association of loss of heterozygosity at the p53 locus with chemoresistance in osteosarcomas. Jpn. J. Cancer Res. 1998, 89, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, S.; Qi, H.; Wang, Z.; Chen, H.-W.; Shao, J.; Shen, J. JMJD5 interacts with p53 and negatively regulates p53 function in control of cell cycle and proliferation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2286–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midgley, C.A.; Desterro, J.M.; Saville, M.K.; Howard, S.; Sparks, A.; Hay, R.T.; Lane, D.P. An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 2000, 19, 2312–2323. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.-W.; et al. The Ink4a Tumor Suppressor Gene Product, p19Arf, Interacts with MDM2 and Neutralizes MDM2′s Inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. CR 2018, 37, 30. [Google Scholar] [CrossRef] [Green Version]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Liu, D.P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Ang, H.C.; Veprintsev, D.B.; Blair, C.M.; Fersht, A.R. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 2005, 280, 16030–16037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J. Targeting Therapies for the p53 Protein in Cancer Treatments. Ann. Rev. Cancer Biol. 2019, 3, 21–34. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef]
- Gokgoz, N.; Wunder, J.S.; Mousses, S.; Eskandarian, S.; Bell, R.S.; Andrulis, I.L. Comparison of p53 mutations in patients with localized osteosarcoma and metastatic osteosarcoma. Cancer 2001, 92, 2181–2189. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Mai, P.L.; Gastier-Foster, J.M.; Gorlick, R.; Khanna, C.; Patiño-Garcia, A.; Sierrasesúmaga, L.; Lecanda, F.; Andrulis, I.L.; et al. Germline TP53 variants and susceptibility to osteosarcoma. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Noirot, C.; Accadbled, F.; Sales de Gauzy, J.; Castex, M.P.; Brousset, P.; Gomez-Brouchet, A. Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann. Oncol. 2016, 27, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Rao, P.H.; Favis, R.; Lu, X.Y.; Elowitz, M.B.; Barany, F.; Ladanyi, M.; Gorlick, R.; Levine, A.J. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc. Natl. Acad. Sci. USA 2003, 100, 11547–11552. [Google Scholar] [CrossRef] [Green Version]
- Toguchida, J.; Yamaguchi, T.; Ritchie, B.; Beauchamp, R.L.; Dayton, S.H.; Herrera, G.E.; Yamamuro, T.; Kotoura, Y.; Sasaki, M.S.; Little, J.B.; et al. Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res. 1992, 52, 6194–6199. [Google Scholar]
- Patino-Garcia, A.; Sierrasesumaga, L. Analysis of the p16INK4 and TP53 tumor suppressor genes in bone sarcoma pediatric patients. Cancer Genet. Cytogenet. 1997, 98, 50–55. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.W.; Aslo, A.; Won, A.; Tan, M.; Lampkin, B.; Koeffler, H.P. Alterations of the p53, Rb and MDM2 genes in osteosarcoma. J. Cancer Res. Clin. Oncol. 1996, 122, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Wadayama, B.; Toguchida, J.; Yamaguchi, T.; Sasaki, M.S.; Yamamuro, T. p53 expression and its relationship to DNA alterations in bone and soft tissue sarcomas. Br. J. Cancer 1993, 68, 1134–1139. [Google Scholar] [CrossRef] [Green Version]
- Mousses, S.; McAuley, L.; Bell, R.S.; Kandel, R.; Andrulis, I.L. Molecular and immunohistochemical identification of p53 alterations in bone and soft tissue sarcomas. Mod. Pathol. 1996, 9, 1–6. [Google Scholar] [PubMed]
- Lorenz, S.; Barøy, T.; Sun, J.; Nome, T.; Vodák, D.; Bryne, J.C.; Håkelien, A.M.; Fernandez-Cuesta, L.; Möhlendick, B.; Rieder, H.; et al. Unscrambling the genomic chaos of osteosarcoma reveals extensive transcript fusion, recurrent rearrangements and frequent novel TP53 aberrations. Oncotarget 2016, 7, 5273–5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Zou, Z.; Pirollo, K.; Blattner, W.; Chang, E.H. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990, 348, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Birch, J.M.; Hartley, A.L.; Tricker, K.J.; Prosser, J.; Condie, A.; Kelsey, A.M.; Harris, M.; Jones, P.H.; Binchy, A.; Crowther, D.; et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994, 54, 1298–1304. [Google Scholar]
- Li, F.P.; Fraumeni, J.F., Jr.; Mulvihill, J.J.; Blattner, W.A.; Dreyfus, M.G.; Tucker, M.A.; Miller, R.W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48, 5358–5362. [Google Scholar]
- Chompret, A.; Abel, A.; Stoppa-Lyonnet, D.; Brugieres, L.; Pagès, S.; Feunteun, J.; Bonaïti-Pellié, C.A. Sensitivity and predictive value of criteria for p53 germline mutation screening. J. Med. Genet. 2001, 38, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Bougeard, G.; Sesboüé, R.; Baert-Desurmont, S.; Vasseur, S.; Martin, C.; Tinat, J.; Brugières, L.; Chompret, A.; Paillerets, B.B.-D.; Stoppa-Lyonnet, D.; et al. Molecular basis of the Li–Fraumeni syndrome: An update from the French LFS families. J. Med. Genet. 2008, 45, 535. [Google Scholar] [CrossRef] [PubMed]
- Jennis, M.; Kung, C.P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Chun, S.M.; Kim, K.R.; Sohn, I.; Sung, C.O. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS ONE 2013, 8, e72609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.-P.; Ho, F.Y.F.; Fung, K.-P.; Kong, S.-K.; Kwok, T.-T. p53-R175H mutant gains new function in regulation of doxorubicin-induced apoptosis. Int. J. Cancer 2005, 114, 331–336. [Google Scholar] [CrossRef]
- Wong, R.P.C.; Tsang, W.P.; Chau, P.Y.; Co, N.N.; Tsang, T.Y.; Kwok, T.T. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol. Cancer Therapeut. 2007, 6, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hu, Q.; Li, G.; Li, L.; Liang, S.; Zhang, Y.; Liu, J.; Fan, Z.; Li, L.; Zhou, B.; et al. ONZIN Upregulation by Mutant p53 Contributes to Osteosarcoma Metastasis Through the CXCL5-MAPK Signaling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 1099–1111. [Google Scholar] [CrossRef]
- Xiong, S.; Tu, H.; Kollareddy, M.; Pant, V.; Li, Q.; Zhang, Y.; Jackson, J.G.; Suh, Y.A.; Elizondo-Fraire, A.C.; Yang, P.; et al. Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc. Natl. Acad. Sci. USA 2014, 111, 11145–11150. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Pourebrahim, R.; Zhang, Y.; Liu, B.; Gao, R.; Xiong, S.; Lin, P.P.; McArthur, M.J.; Ostrowski, M.C.; Lozano, G. Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes Dev. 2017, 31, 1847–1857. [Google Scholar] [CrossRef]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056. [Google Scholar] [CrossRef] [Green Version]
- Jordan, J.J.; Inga, A.; Conway, K.; Edmiston, S.; Carey, L.A.; Wu, L.; Resnick, M.A. Altered-function p53 missense mutations identified in breast cancers can have subtle effects on transactivation. Mol. Cancer Res. 2010, 8, 701–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Zhao, M.; Tang, L.; Patel, A.A.; Xi, Q.; Van, H.T.; Takahashi, H.; Osman, A.A.; Zhang, J.; Wang, J.; et al. Gain-of-function mutant p53 promotes the oncogenic potential of head and neck squamous cell carcinoma cells by targeting the transcription factors FOXO3a and FOXM1. Oncogene 2018, 37, 1279–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neskey, D.M.; Osman, A.A.; Ow, T.J.; Katsonis, P.; McDonald, T.; Hicks, S.C.; Hsu, T.K.; Pickering, C.R.; Ward, A.; Patel, A.; et al. Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer Res. 2015, 75, 1527–1536. [Google Scholar] [CrossRef] [Green Version]
- Pintus, S.S.; Ivanisenko, N.V.; Demenkov, P.S.; Ivanisenko, T.V.; Ramachandran, S.; Kolchanov, N.A.; Ivanisenko, V.A. The substitutions G245C and G245D in the Zn(2+)-binding pocket of the p53 protein result in differences of conformational flexibility of the DNA-binding domain. J. Biomol. Struct. Dyn. 2013, 31, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, N.; Lam, S.W.; Cleton-Jansen, A.M.; Bovée, J. What’s new in bone forming tumours of the skeleton? Virchows Arch. Int. J. Pathol. 2020, 476, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Guo, J.; Zhang, K.; Guo, Y. TP53 Mutations and Survival in Osteosarcoma Patients: A Meta-Analysis of Published Data. Dis. Mark. 2016, 2016, 4639575. [Google Scholar] [CrossRef] [Green Version]
- Simpson, D.A.; Livanos, E.; Heffernan, T.P.; Kaufmann, W.K. Telomerase expression is sufficient for chromosomal integrity in cells lacking p53 dependent G1 checkpoint function. J. Carcinog. 2005, 4, 18. [Google Scholar] [CrossRef]
- Slováčkova, J.; Grochova, D.; Navratilova, J.; Smarda, J.; Smardova, J. Transactivation by temperature-dependent p53 mutants in yeast and human cells. Cell Cycle 2010, 9, 2141–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grochova, D.; Vankova, J.; Damborsky, J.; Ravcukova, B.; Smarda, J.; Vojtesek, B.; Smardova, J. Analysis of transactivation capability and conformation of p53 temperature-dependent mutants and their reactivation by amifostine in yeast. Oncogene 2008, 27, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Schärer, E.; Iggo, R. Mammalian p53 can function as a transcription factor in yeast. Nucleic Acids Res. 1992, 20, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424. [Google Scholar] [CrossRef] [Green Version]
- Achatz, M.I.; Olivier, M.; Le Calvez, F.; Martel-Planche, G.; Lopes, A.; Rossi, B.M.; Ashton-Prolla, P.; Giugliani, R.; Palmero, E.I.; Vargas, F.R.; et al. The TP53 mutation, R337H, is associated with Li-Fraumeni and Li-Fraumeni-like syndromes in Brazilian families. Cancer Lett. 2007, 245, 96–102. [Google Scholar] [CrossRef]
- Giacomazzi, J.; Graudenz, M.S.; Osorio, C.A.B.T.; Koehler-Santos, P.; Palmero, E.I.; Zagonel-Oliveira, M.; Michelli, R.A.D.; Neto, C.S.; Fernandes, G.C.; Achatz, M.I.W.S.; et al. Prevalence of the TP53 p.R337H Mutation in Breast Cancer Patients in Brazil. PLoS ONE 2014, 9, e99893. [Google Scholar] [CrossRef]
- Seidinger, A.L.; Mastellaro, M.J.; Paschoal Fortes, F.; Godoy Assumpção, J.; Aparecida Cardinalli, I.; Aparecida Ganazza, M.; Correa Ribeiro, R.; Brandalise, S.R.; Dos Santos Aguiar, S.; Yunes, J.A. Association of the highly prevalent TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in southeast Brazil. Cancer 2011, 117, 2228–2235. [Google Scholar] [CrossRef]
- Herrmann, L.J.; Heinze, B.; Fassnacht, M.; Willenberg, H.S.; Quinkler, M.; Reisch, N.; Zink, M.; Allolio, B.; Hahner, S. TP53 germline mutations in adult patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E476–E485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol. 2020, 6, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Akouchekian, M.; Hemati, S.; Jafari, D.; Jalilian, N.; Dehghan Manshadi, M. Does PTEN gene mutation play any role in Li-Fraumeni syndrome. Med. J. Islam. Repub. Iran 2016, 30, 378. [Google Scholar]
- Saba, K.H.; Cornmark, L.; Kovac, M.; Magnusson, L.; Nilsson, J.; van den Bos, H.; Spierings, D.C.J.; Bidgoli, M.; Jonson, T.; Sumathi, V.P.; et al. Oncogenes hijack a constitutively active TP53promoter in osteosarcoma. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, R.; Marcatti, M.; Drago-Ferrante, R.; D’Anneo, A.; Giuliano, M.; Carlisi, D.; De Blasio, A.; Querques, F.; Pastore, L.; Tesoriere, G.; et al. Mutant p53 gain of function can be at the root of dedifferentiation of human osteosarcoma MG63 cells into 3AB-OS cancer stem cells. Bone 2014, 60, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Xu, A.; Gingold, J.; Strong, L.C.; Zhao, R.; Lee, D.F. Li-Fraumeni Syndrome Disease Model: A Platform to Develop Precision Cancer Therapy Targeting Oncogenic p53. Trends Pharm. Sci. 2017, 38, 908–927. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A.; Donehower, L.A. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat. Genet. 1993, 5, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Curtis, S.J.; Roy, D.M.; Flesken-Nikitin, A.; Nikitin, A.Y. Local mesenchymal stem/progenitor cells are a preferential target for initiation of adult soft tissue sarcomas associated with p53 and Rb deficiency. Am. J. Pathol. 2010, 177, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G. Mouse models of p53 functions. Cold Spring Harb. Perspect. Biol. 2010, 2, a001115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef]
- van Boxtel, R.; Kuiper, R.V.; Toonen, P.W.; van Heesch, S.; Hermsen, R.; de Bruin, A.; Cuppen, E. Homozygous and Heterozygous p53 Knockout Rats Develop Metastasizing Sarcomas with High Frequency. Am. J. Pathol. 2011, 179, 1616–1622. [Google Scholar] [CrossRef] [Green Version]
- Tong, C.; Li, P.; Wu, N.L.; Yan, Y.; Ying, Q.L. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 2010, 467, 211–213. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.X.; Wu, H.P.; Ashton, C.; Tong, C.; Ying, Q.L. Rats deficient for p53 are susceptible to spontaneous and carcinogen-induced tumorigenesis. Carcinogenesis 2012, 33, 2001–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, S.A.; Hart, M.L.; Busi, S.; Parker, T.; Goerndt, A.; Jones, K.; Amos-Landgraf, J.M.; Bryda, E.C. Fischer-344 Tp53-knockout rats exhibit a high rate of bone and brain neoplasia with frequent metastasis. Dis. Model. Mech. 2016, 9, 1139–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyczyńska, U.; Strzemecki, D.; Czarnecka, A.M.; Fendler, W.; Fiedorowicz, M.; Wełniak-Kamińska, M.; Guzowska, M.; Synoradzki, K.; Cheda, Ł.; Rogulski, Z.; et al. TP53-Deficient Angiosarcoma Expression Profiling in Rat Model. Cancers 2020, 12, 1525. [Google Scholar] [CrossRef] [PubMed]
- Strzemecki, D.; Guzowska, M.; Grieb, P. Survival rates of homozygotic Tp53 knockout rats as a tool for preclinical assessment of cancer prevention and treatment. Cell. Mol. Biol. Lett. 2017, 22, 9. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.; Besch-Williford, C.L.; Franklin, C.L.; Weinstein, E.J.; Cui, X. Creation and preliminary characterization of a Tp53 knockout rat. Dis. Model. Mech. 2013, 6, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Parant, J.M.; George, S.A.; Holden, J.A.; Yost, H.J. Genetic modeling of Li-Fraumeni syndrome in zebrafish. Dis. Model. Mech. 2010, 3, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Ignatius, M.S.; Hayes, M.N.; Moore, F.E.; Tang, Q.; Garcia, S.P.; Blackburn, P.R.; Baxi, K.; Wang, L.; Jin, A.; Ramakrishnan, A.; et al. tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. eLife 2018, 7, e37202. [Google Scholar] [CrossRef]
- Regan, D.; Garcia, K.; Thamm, D. Clinical, Pathological, and Ethical Considerations for the Conduct of Clinical Trials in Dogs with Naturally Occurring Cancer: A Comparative Approach to Accelerate Translational Drug Development. ILAR J. 2018, 59, 99–110. [Google Scholar] [CrossRef]
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Kirpensteijn, J.; Kik, M.; Teske, E.; Rutteman, G.R. TP53 gene mutations in canine osteosarcoma. Vet. Surg. VS 2008, 37, 454–460. [Google Scholar] [CrossRef]
- Mori, M.; Hitora, T.; Nakamura, O.; Yamagami, Y.; Horie, R.; Nishimura, H.; Yamamoto, T. Hsp90 inhibitor induces autophagy and apoptosis in osteosarcoma cells. Int. J. Oncol. 2015, 46, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCleese, J.K.; Bear, M.D.; Fossey, S.L.; Mihalek, R.M.; Foley, K.P.; Ying, W.; Barsoum, J.; London, C.A. The novel HSP90 inhibitor STA-1474 exhibits biologic activity against osteosarcoma cell lines. Int. J. Cancer 2009, 125, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.D.; Lizardo, M.M.; Reed, D.R.; Hingorani, P.; Glover, J.; Allen-Rhoades, W.; Fan, T.; Khanna, C.; Sweet-Cordero, E.A.; Cash, T.; et al. Provocative questions in osteosarcoma basic and translational biology: A report from the Children’s Oncology Group. Cancer 2019, 125, 3514–3525. [Google Scholar] [CrossRef]
- Samsa, W.E.; Mamidi, M.K.; Bashur, L.A.; Elliott, R.; Miron, A.; Chen, Y.; Lee, B.; Greenfield, E.M.; Chan, R.; Danielpour, D.; et al. The crucial p53-dependent oncogenic role of JAB1 in osteosarcoma in vivo. Oncogene 2020, 39, 4581–4591. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2015, 108, djv303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corney, D.C.; Flesken-Nikitin, A.; Godwin, A.K.; Wang, W.; Nikitin, A.Y. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007, 67, 8433–8438. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Slabáková, E.; Culig, Z.; Remšík, J.; Souček, K. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis. 2017, 8, e3100. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, C.; Jiang, K.; He, J.; Cao, X.; Wu, S. MicroRNA-503 suppresses cell proliferation and invasion in osteosarcoma via targeting insulin-like growth factor 1 receptor. Exp. Ther. Med. 2017, 14, 1547–1553. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Zhao, Y.-k.; Liu, Y.; Zhao, J.-f. MicroRNA-34a inhibits tumor invasion and metastasis in osteosarcoma partly by effecting C-IAP2 and Bcl-2. Tumor. Biol. 2017, 39, 1010428317705761. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pan, Y.; Xie, C.; Zhang, Y. miR-34a exerts as a key regulator in the dedifferentiation of osteosarcoma via PAI-1-Sox2 axis. Cell Death Dis. 2018, 9, 777. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Xu, C.; Wang, W.; Li, X. The DNMT1/miR-34a Axis Is Involved in the Stemness of Human Osteosarcoma Cells and Derived Stem-Like Cells. Stem Cells Int. 2019, 2019, 7028901. [Google Scholar] [CrossRef]
- Li, Q.-C.; Xu, H.; Wang, X.; Wang, T.; Wu, J. miR-34a increases cisplatin sensitivity of osteosarcoma cells in vitro through up-regulation of c-Myc and Bim signal. Cancer Biomark. 2018, 21, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Ganjavi, H.; Gee, M.; Narendran, A.; Parkinson, N.; Krishnamoorthy, M.; Freedman, M.H.; Malkin, D. Adenovirus-mediated p53 gene therapy in osteosarcoma cell lines: Sensitization to cisplatin and doxorubicin. Cancer Gene Ther. 2006, 13, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. p53 as a target for the treatment of cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Xia, Y.; Li, X.; Sun, W. Applications of Recombinant Adenovirus-p53 Gene Therapy for Cancers in the Clinic in China. Curr. Gene Ther. 2020, 20, 127–141. [Google Scholar] [CrossRef]
- Witlox, M.A.; Lamfers, M.L.; Wuisman, P.I.; Curiel, D.T.; Siegal, G.P. Evolving gene therapy approaches for osteosarcoma using viral vectors: Review. Bone 2007, 40, 797–812. [Google Scholar] [CrossRef] [Green Version]
- Nakase, M.; Inui, M.; Okumura, K.; Kamei, T.; Nakamura, S.; Tagawa, T. p53 gene therapy of human osteosarcoma using a transferrin-modified cationic liposome. Mol. Cancer Ther. 2005, 4, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Fang, L.; Zhao, H.; Xiang, T.; Wang, D. MDM2 inhibitor Nutlin-3a suppresses proliferation and promotes apoptosis in osteosarcoma cells. Acta Biochim. Biophys. Sin. 2012, 44, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Pishas, K.I.; Neuhaus, S.J.; Clayer, M.T.; Schreiber, A.W.; Lawrence, D.M.; Perugini, M.; Whitfield, R.J.; Farshid, G.; Manavis, J.; Chryssidis, S.; et al. Nutlin-3a efficacy in sarcoma predicted by transcriptomic and epigenetic profiling. Cancer Res. 2014, 74, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [Green Version]
- Bill, K.L.; Garnett, J.; Meaux, I.; Ma, X.; Creighton, C.J.; Bolshakov, S.; Barriere, C.; Debussche, L.; Lazar, A.J.; Prudner, B.C.; et al. SAR405838: A Novel and Potent Inhibitor of the MDM2:p53 Axis for the Treatment of Dedifferentiated Liposarcoma. Clin. Cancer Res. 2016, 22, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- de Jonge, M.; de Weger, V.A.; Dickson, M.A.; Langenberg, M.; Le Cesne, A.; Wagner, A.J.; Hsu, K.; Zheng, W.; Mace, S.; Tuffal, G.; et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer 2017, 76, 144–151. [Google Scholar] [CrossRef]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wen, X.; Chen, G.; Zeng, S.; Men, L.; Wang, H.; He, S.; Ma, Y.; Pan, Q.; Zhang, Y.; et al. Phase I study results of APG-115, a MDM2-p53 antagonist in Chinese patients with advanced liposarcoma and other solid tumors. J. Clin. Oncol. 2020, 38, 11542. [Google Scholar] [CrossRef]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Wassman, C.D.; Baronio, R.; Demir, O.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zache, N.; Lambert, J.M.R.; Rokaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2017, 11, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ 2008, 15, 718–729. [Google Scholar] [CrossRef]
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.V.; Parrales, A.; Begani, P.; Narkar, A.; Adhikari, A.S.; Martinez, L.A.; Iwakuma, T. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget 2016, 7, 5401–5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Wang, C.; Chen, J. Phosphorylation and hsp90 binding mediate heat shock stabilization of p53. J. Biol. Chem. 2003, 278, 2066–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekki, H.; Kohashi, K.; Maekawa, A.; Yamada, Y.; Yamamoto, H.; Harimaya, K.; Hakozaki, M.; Nabeshima, K.; Iwamoto, Y.; Oda, Y. Elevated expression of HSP90 and the antitumor effect of an HSP90 inhibitor via inactivation of the Akt/mTOR pathway in undifferentiated pleomorphic sarcoma. BMC Cancer 2015, 15, 804. [Google Scholar] [CrossRef] [Green Version]
- Ory, B.; Baud’huin, M.; Verrecchia, F.; Royer, B.B.; Quillard, T.; Amiaud, J.; Battaglia, S.; Heymann, D.; Redini, F.; Lamoureux, F. Blocking HSP90 Addiction Inhibits Tumor Cell Proliferation, Metastasis Development, and Synergistically Acts with Zoledronic Acid to Delay Osteosarcoma Progression. Clin. Cancer Res. 2016, 22, 2520–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozgur, A. Investigation of anticancer activities of STA-9090 (ganetespib) as a second generation HSP90 inhibitor in Saos-2 osteosarcoma cells. J. Chemother. 2021, 1–10. [Google Scholar] [CrossRef]
- Shimamura, T.; Perera, S.A.; Foley, K.P.; Sang, J.; Rodig, S.J.; Inoue, T.; Chen, L.; Li, D.; Carretero, J.; Li, Y.C.; et al. Ganetespib (STA-9090), a nongeldanamycin HSP90 inhibitor, has potent antitumor activity in in vitro and in vivo models of non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 4973–4985. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, L.; You, Q.D.; Xu, X.L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef]
- Thoenen, E.; Curl, A.; Iwakuma, T. TP53 in bone and soft tissue sarcomas. Pharmacology 2019, 202, 149–164. [Google Scholar] [CrossRef]
- Zhu, J.; Zou, H.; Yu, W.; Huang, Y.; Liu, B.; Li, T.; Liang, C.; Tao, H. Checkpoint kinase inhibitor AZD7762 enhance cisplatin-induced apoptosis in osteosarcoma cells. Cancer Cell Int. 2019, 19, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, Y.S.; Yen, C.C.; Chen, W.M.; Lin, Y.C.; Wen, Y.S.; Ke, W.T.; Wang, J.Y.; Liu, C.Y.; Yang, M.H.; Chen, T.H.; et al. Cytotoxic mechanism of PLK1 inhibitor GSK461364 against osteosarcoma: Mitotic arrest, apoptosis, cellular senescence, and synergistic effect with paclitaxel. Int. J. Oncol. 2016, 48, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Mo, H.; He, J.; Yuan, Z.; Wu, Z.; Liu, B.; Lin, X.; Guan, J. PLK1 contributes to autophagy by regulating MYC stabilization in osteosarcoma cells. OncoTargets Ther. 2019, 12, 7527–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PosthumaDeBoer, J.; Wurdinger, T.; Graat, H.C.; van Beusechem, V.W.; Helder, M.N.; van Royen, B.J.; Kaspers, G.J. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer 2011, 11, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreahling, J.M.; Foroutan, P.; Reed, D.; Martinez, G.; Razabdouski, T.; Bui, M.M.; Raghavan, M.; Letson, D.; Gillies, R.J.; Altiok, S. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS ONE 2013, 8, e57523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghelli Luserna di Rora, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, L.; Hong, B.; van den Heuvel, A.P.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Choy, E.; Harmon, D.; Yang, C.; Ryu, K.; Schwab, J.; Mankin, H.; Hornicek, F.J. Insulin-like growth factor-I receptor tyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines. Mol. Cancer Ther. 2009, 8, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- Viereck, V.; Siggelkow, H.; Pannem, R.; Braulke, T.; Scharf, J.G.; Kübler, B. Alteration of the insulin-like growth factor axis during in vitro differentiation of the human osteosarcoma cell line HOS 58. J. Cell. Biochem. 2007, 102, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.C.; Carney, S.C.; Ahlmann, E.R.; Hendifar, A.; Chawla, S.; Fedenko, A.; Angeles, C.; Menendez, L.R. A Meta-Analysis of Osteosarcoma Outcomes in the Modern Medical Era. Sarcoma 2012, 2012, 704872. [Google Scholar] [CrossRef] [Green Version]
- Marina, N.M.; Smeland, S.; Bielack, S.S.; Bernstein, M.; Jovic, G.; Krailo, M.D.; Hook, J.M.; Arndt, C.; van den Berg, H.; Brennan, B.; et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): An open-label, international, randomised controlled trial. Lancet Oncol. 2016, 17, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, N.; Occean, B.V.; Pacquement, H.; Bompas, E.; Bouvier, C.; Brisse, H.J.; Castex, M.P.; Cheurfa, N.; Corradini, N.; Delaye, J.; et al. Results of methotrexate-etoposide-ifosfamide based regimen (M-EI) in osteosarcoma patients included in the French OS2006/sarcome-09 study. Eur. J. Cancer 2018, 88, 57–66. [Google Scholar] [CrossRef]
- Brázda, V.; Fojta, M. The Rich World of p53 DNA Binding Targets: The Role of DNA Structure. Int. J. Mol. Sci. 2019, 20, 5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, A.K.; Leu, J.I.J.; Zhou, Y.; Devarajan, K.; Nedelko, T.; Klein-Szanto, A.; Hollstein, M.; Murphy, M.E. The Codon 72 Polymorphism of p53 Regulates Interaction with NF-κB and Transactivation of Genes Involved in Immunity and Inflammation. Mol. Cell Biol. 2011, 31, 1201. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Species | Mutations | Malignancies | References |
|---|---|---|---|
| Mouse | Trp53 knockout | Spindle cell sarcomas and pleomorphic sarcomas develop if Trp53 is inactivated by Cre-loxP-mediated recombination | [135] |
| Mouse | p53 R172H and p53 R270H mutations | The spectrum of tumors that develop in mice depends on the various mutant alleles in the model. p. R270H (p. R273H in humans) results in a high frequency of carcinomas, whereas the ‘structural’ mutant p. R172H (p. R175H in humans) predominantly results in OSs. | [137] |
| Rat (Dark Agouti) | Tp53 knockout | Homozygotes develop angiosarcomas and lymphomas, and their lifespan is limited to 6 months. Heterozygotes live longer (up to 12 months) and develop a broader spectrum of tumors; however, angiosarcomas and lymphomas are also the most common, and OSs are rare | [140,141] |
| Rat (Wistar) | Tp53C273X/C273X mutation. | Homozygotes develop mostly angiosarcomas, while heterozygotes develop OSs and less frequently angiosarcomas and B-cell lymphomas | [139,145]. |
| Rat (Sprague Dawley) | Tp53 knockout | Mostly sarcomas. The spectrum of tumors seems to be generally broader than in the Wistar knockout rats. OS is the most frequently observed type of tumor in homozygotes, but still, it constitutes only 18% of all tumors | [145] |
| Zebrafish (Danio rerio) | mutation p53 I166T | Both heterozygous and mutant fish developed soft tissue sarcomas with high penetrance but most frequently, spindle cell tumors | [146]. |
| Zebrafish (Danio rerio) | Tp53 knockout | Wide range of tumors, including angiosarcomas, malignant peripheral nerve sheath tumors, germ cell tumors, or leukemias | [147] |
| Pet dogs | Spontaneous Tp53 mutations | Tp53 mutations were found in 40% of OSs | [151,152] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Synoradzki, K.J.; Bartnik, E.; Czarnecka, A.M.; Fiedorowicz, M.; Firlej, W.; Brodziak, A.; Stasinska, A.; Rutkowski, P.; Grieb, P. TP53 in Biology and Treatment of Osteosarcoma. Cancers 2021, 13, 4284. https://doi.org/10.3390/cancers13174284
Synoradzki KJ, Bartnik E, Czarnecka AM, Fiedorowicz M, Firlej W, Brodziak A, Stasinska A, Rutkowski P, Grieb P. TP53 in Biology and Treatment of Osteosarcoma. Cancers. 2021; 13(17):4284. https://doi.org/10.3390/cancers13174284
Chicago/Turabian StyleSynoradzki, Kamil Jozef, Ewa Bartnik, Anna M. Czarnecka, Michał Fiedorowicz, Wiktoria Firlej, Anna Brodziak, Agnieszka Stasinska, Piotr Rutkowski, and Paweł Grieb. 2021. "TP53 in Biology and Treatment of Osteosarcoma" Cancers 13, no. 17: 4284. https://doi.org/10.3390/cancers13174284
APA StyleSynoradzki, K. J., Bartnik, E., Czarnecka, A. M., Fiedorowicz, M., Firlej, W., Brodziak, A., Stasinska, A., Rutkowski, P., & Grieb, P. (2021). TP53 in Biology and Treatment of Osteosarcoma. Cancers, 13(17), 4284. https://doi.org/10.3390/cancers13174284