
Erlotinib Promotes Ligand-Induced EGFR Degradation in 3D but Not 2D Cultures of Pancreatic Ductal Adenocarcinoma Cells




Abstract
:Simple Summary
Abstract
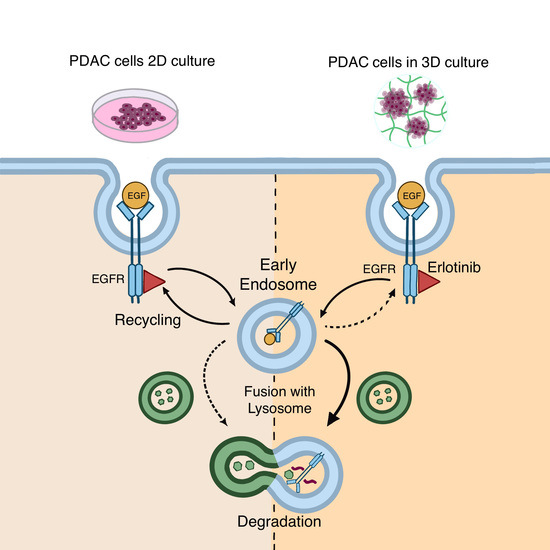
1. Introduction
2. Materials and Methods
2.1. 2D Cell Culture
2.2. 3D Cell Culture in the Self-Assembling Peptide Scaffold RAD16-I
2.3. Drug Incubation
2.4. MTT Assay for Cell Viability and Proliferation
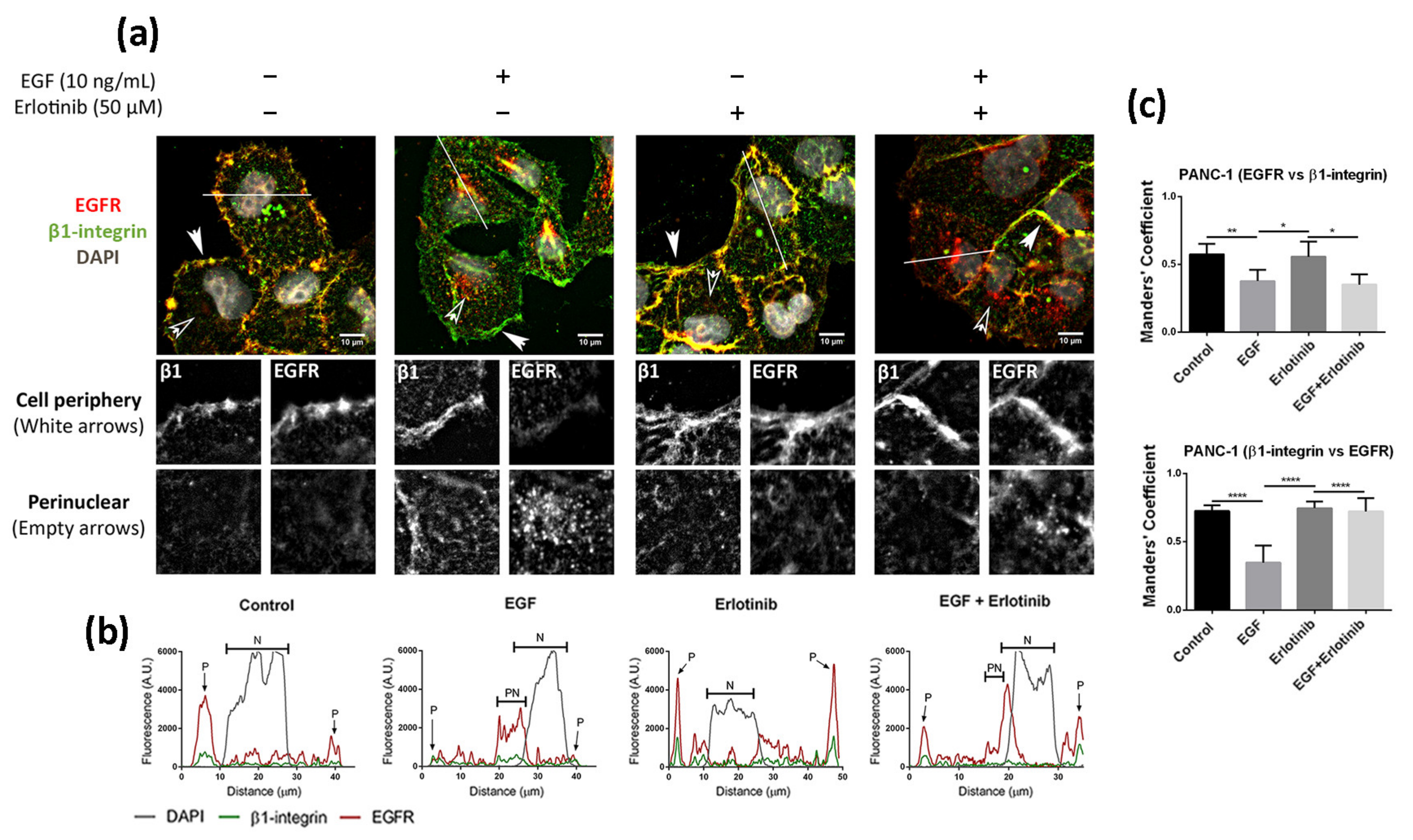
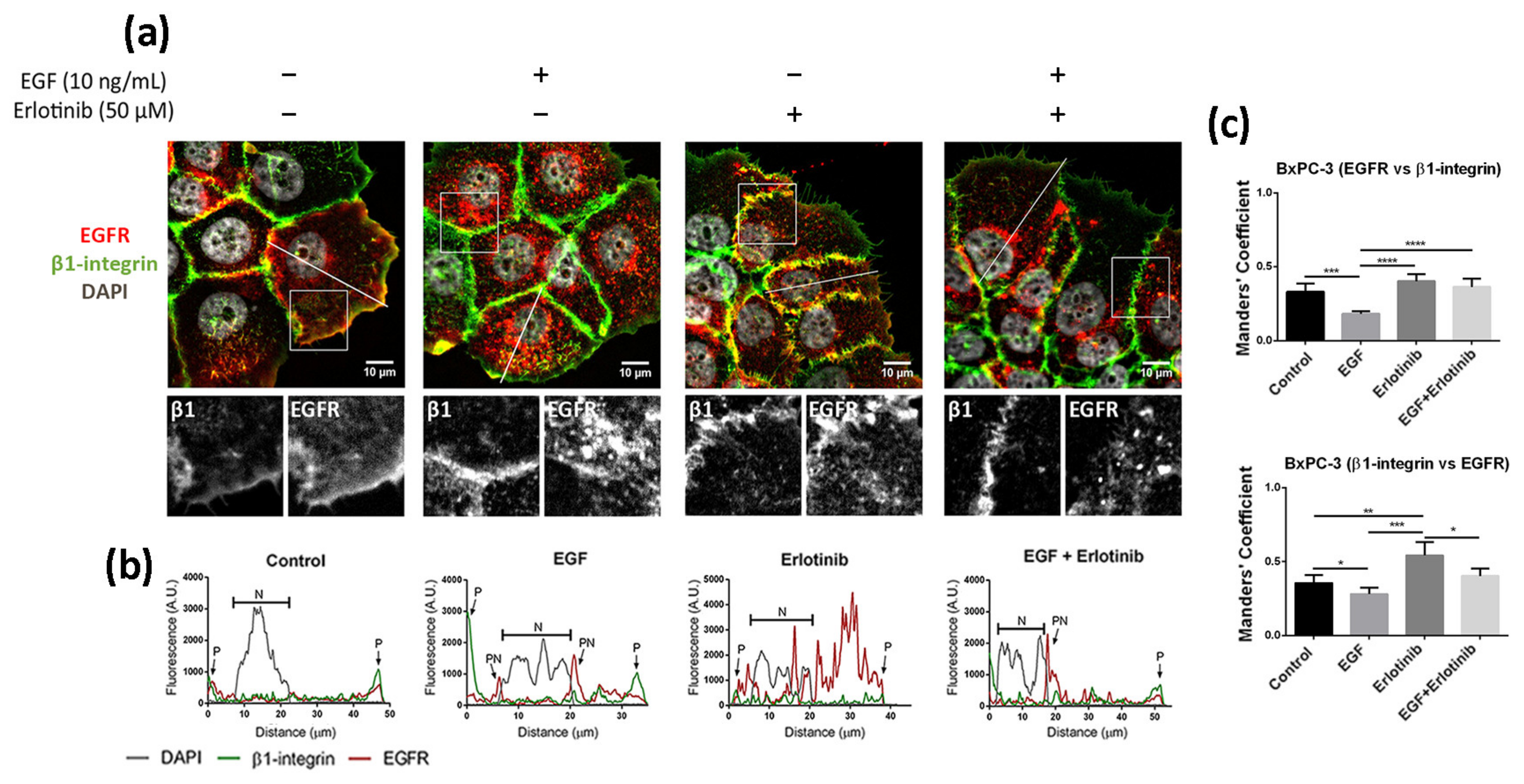
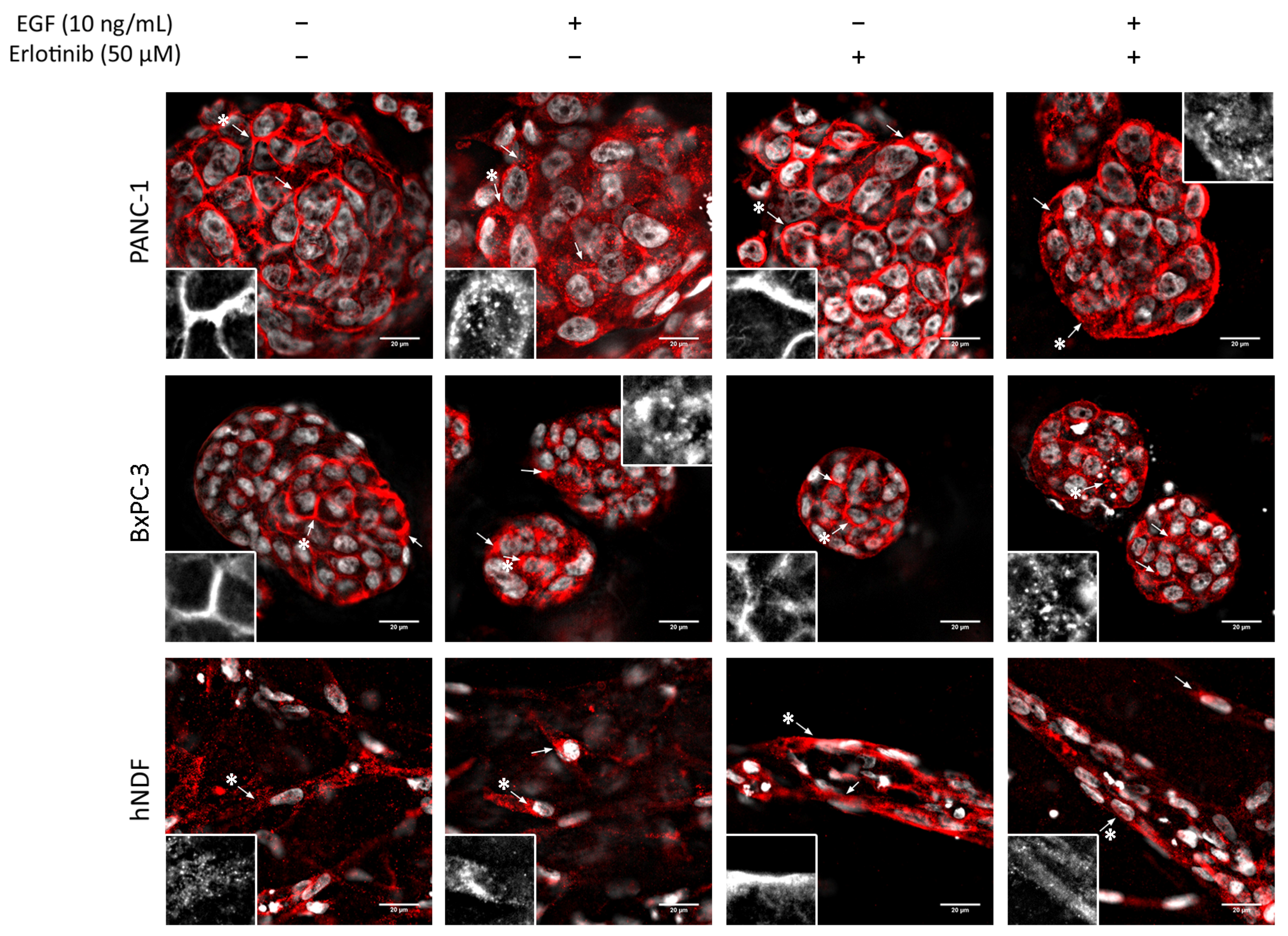
2.5. Immunofluorescence
2.6. Image Analysis
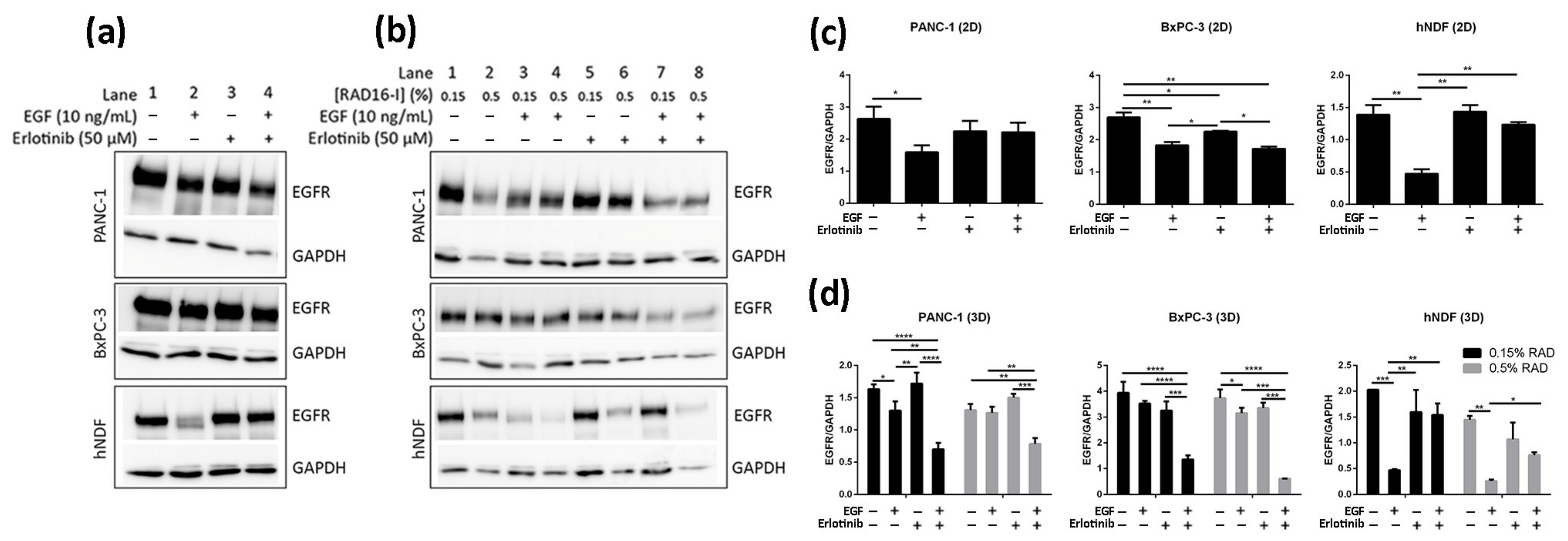
2.7. Western Blot
2.8. Statistics
3. Results
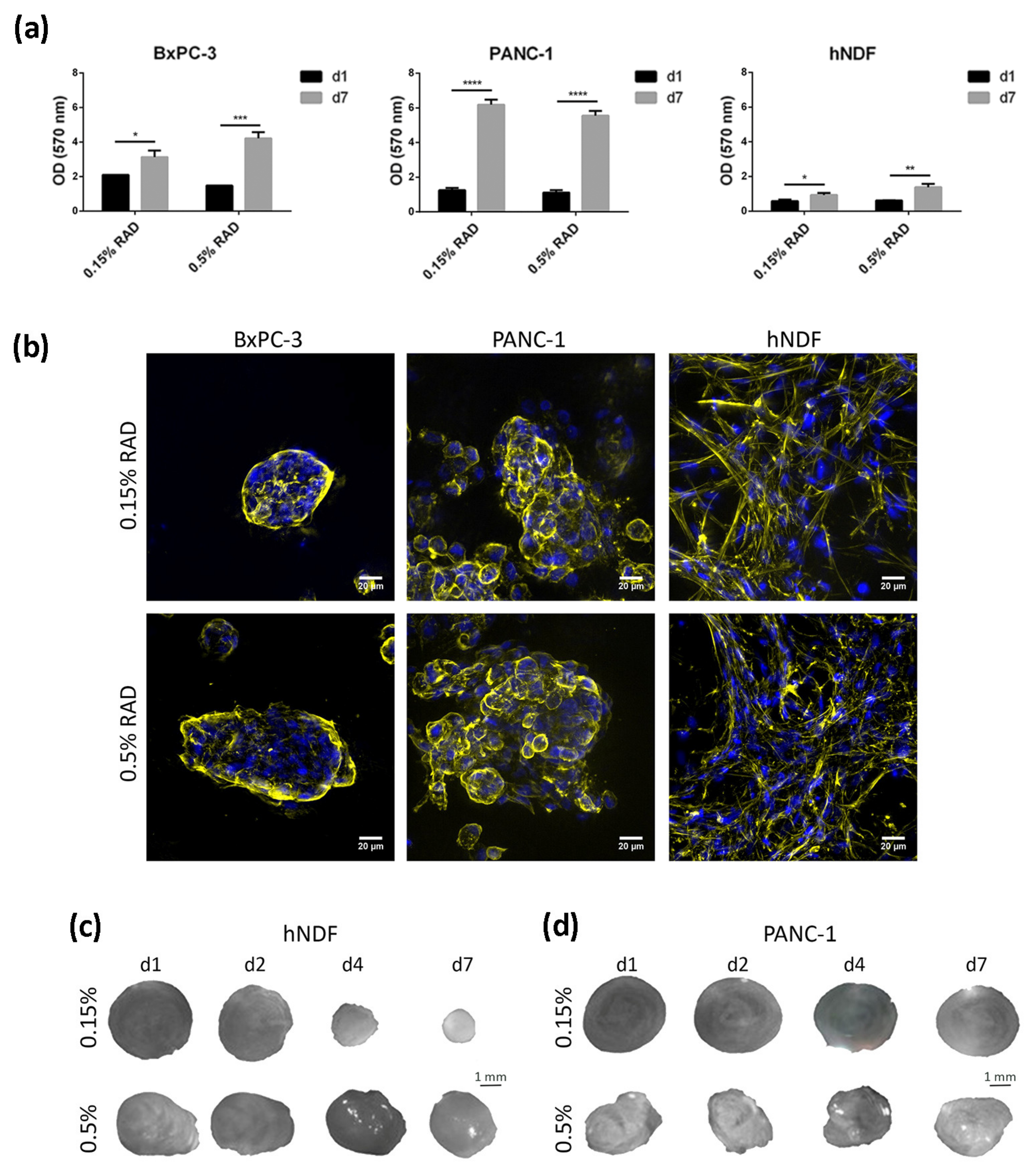
3.1. Cell Culture in RAD16-I Scaffold
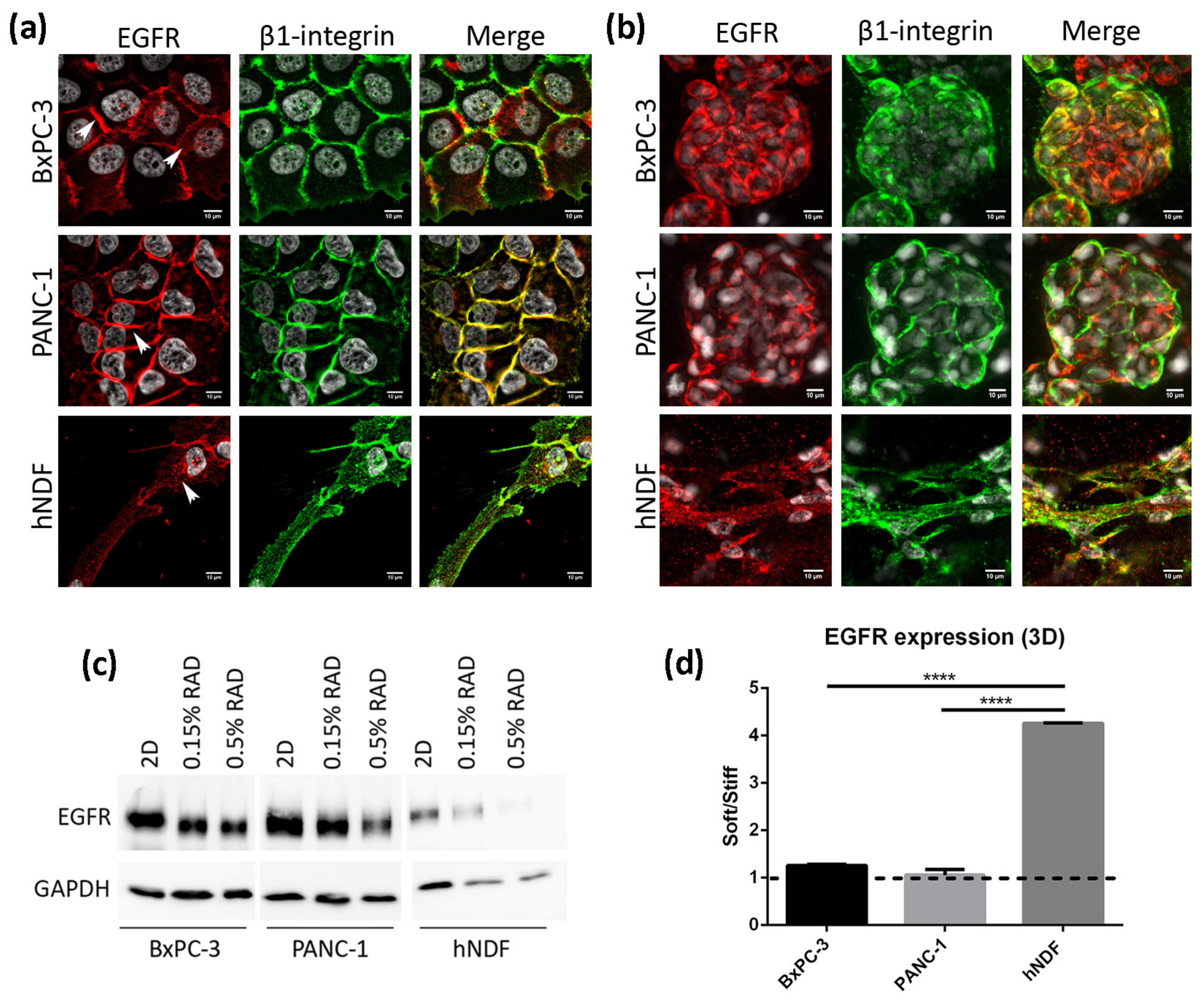
3.2. EGFR Expression in 2D and RAD16-I 3D Cultures
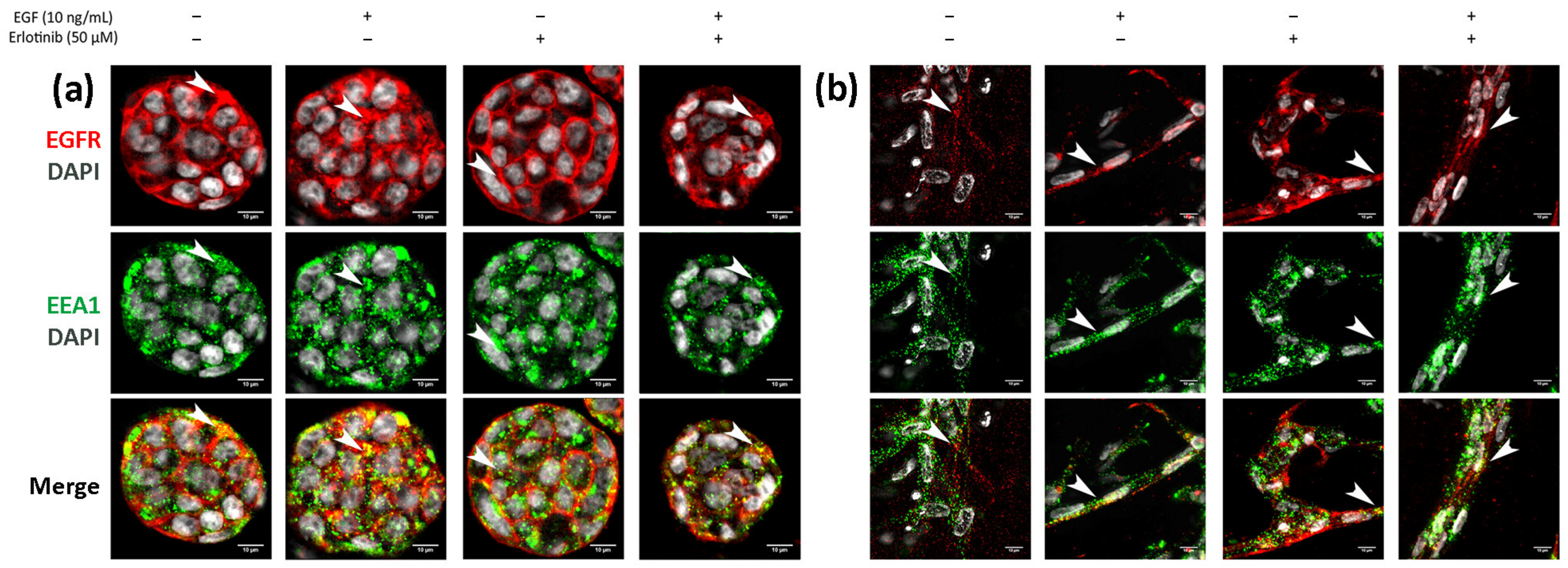
3.3. Effect of EGF and Erlotinib on the Location of the EGFR
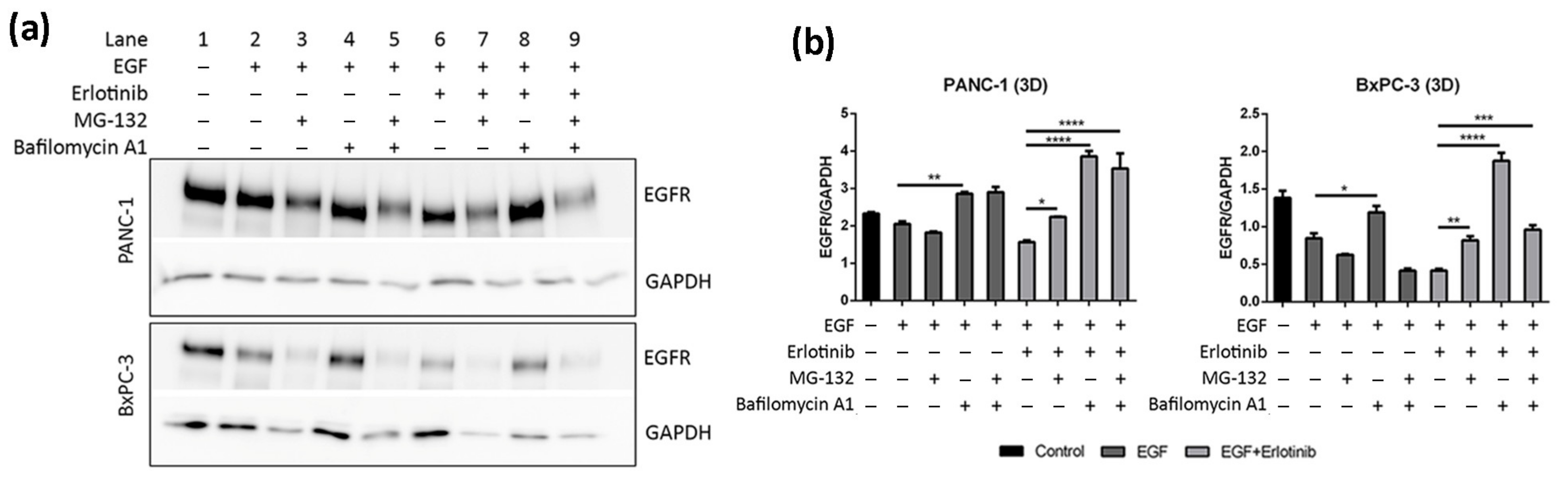
3.4. Effect of EGF and Erlotinib on EGFR Degradation
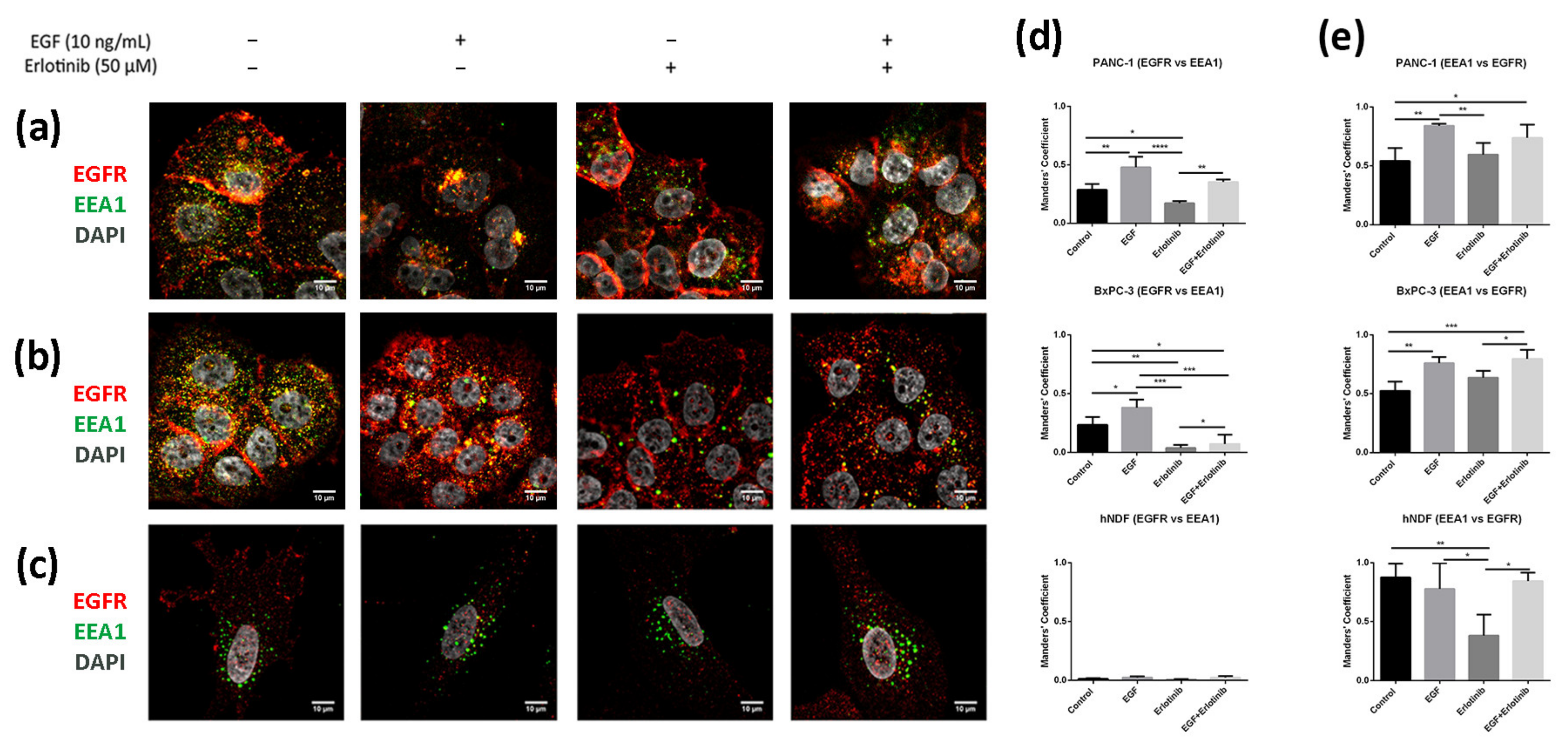
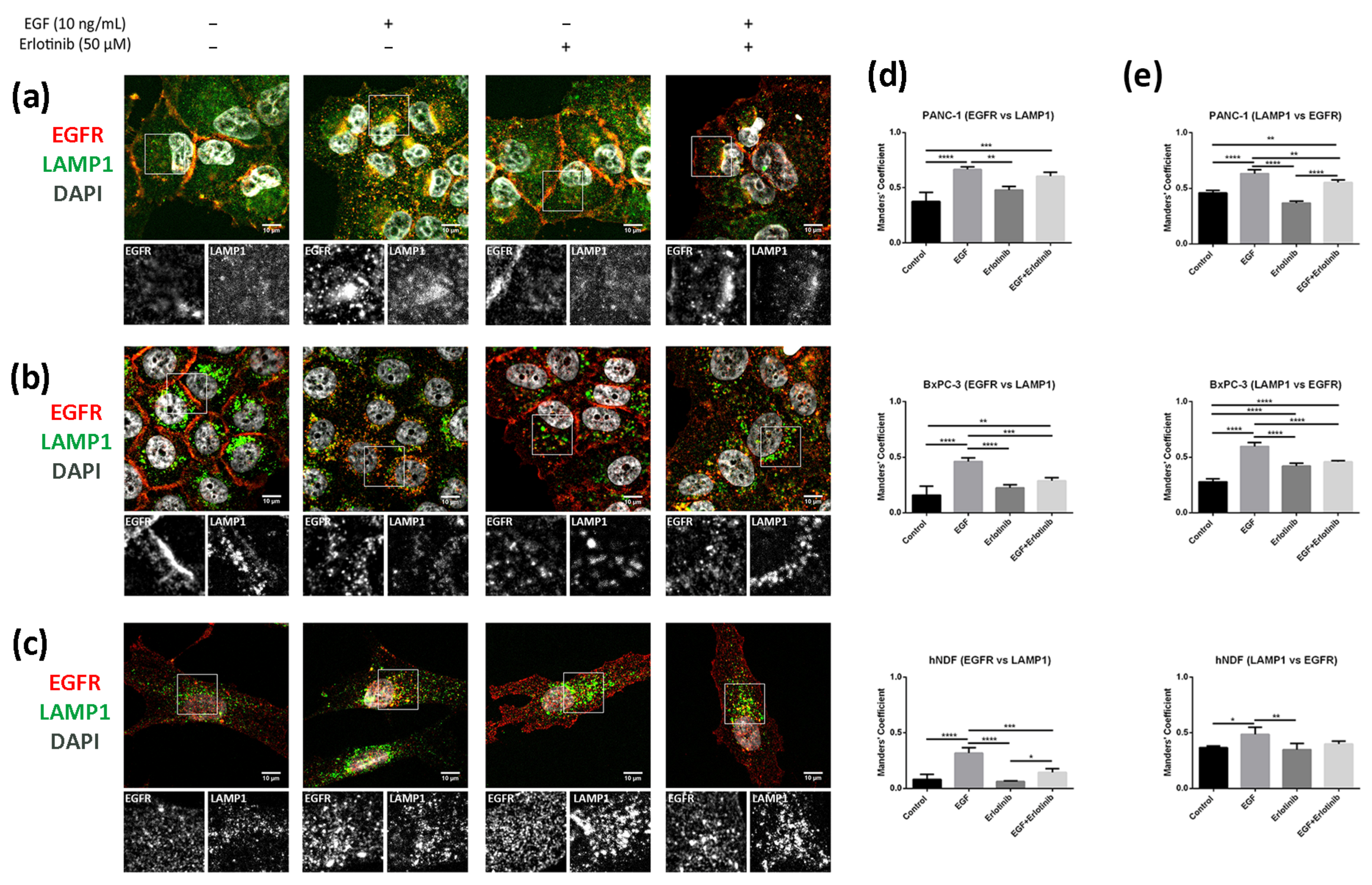
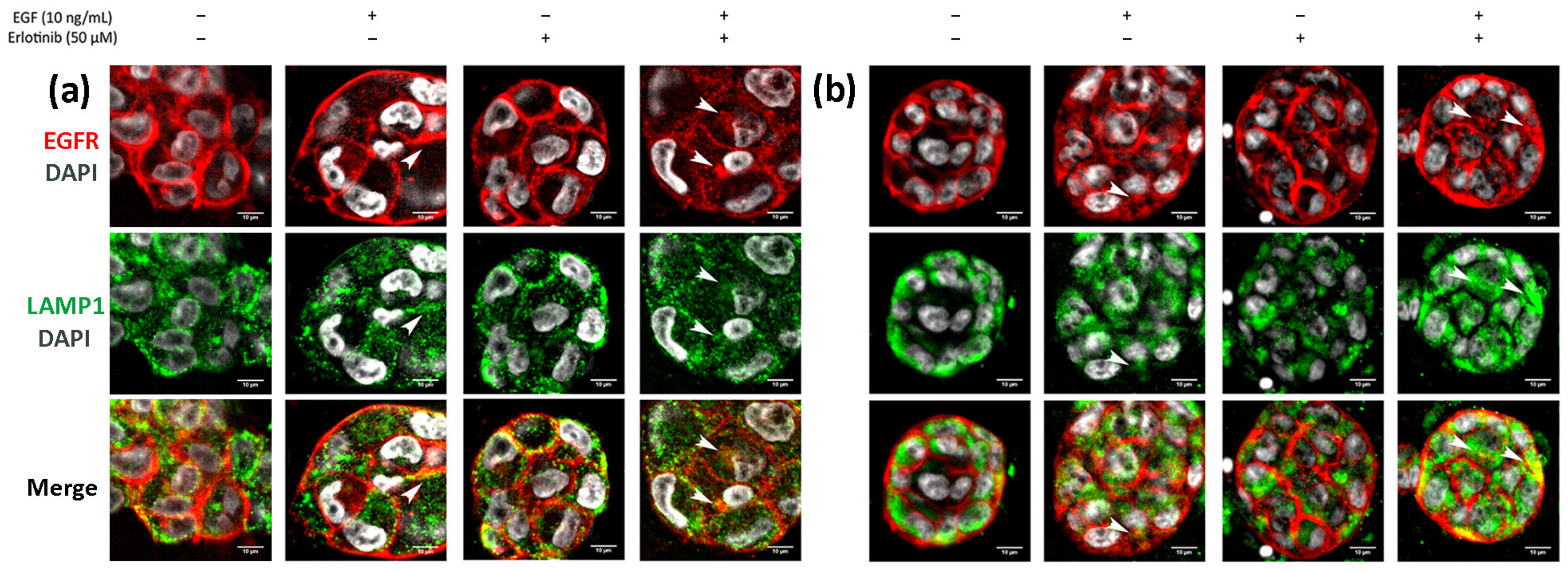
3.5. EGFR Trafficking to Early Endosomes and Lysosomes in 2D and 3D Cultures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendelsohn, J.; Baselga, J. Epidermal Growth Factor Receptor Targeting in Cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Lambert, P.F.; Rapraeger, A.C.; Anderson, R.A. Stress-Induced EGFR Trafficking: Mechanisms, Functions, and Therapeutic Implications. Trends Cell Biol. 2016, 26, 352–366. [Google Scholar] [CrossRef] [Green Version]
- Oksvold, M.P.; Hultfeldt, H.S.; Øtvold, A.C.; Skarpen, E. UV induces tyrosine kinase-independent internalisation and endosome arrest of the EGF receptor. J. Cell Sci. 2002, 115, 793–803. [Google Scholar] [CrossRef]
- Shen, J.; Xia, W.; Khotskaya, Y.; Huo, L.; Nakanishi, K.; Lim, S.O.; Du, Y.; Wang, Y.; Chang, W.-C.; Chen, C.-H.; et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 2013, 497, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tan, J.; Zhang, Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol. Int. 2015, 39, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.J.; Lerdrup, M.; Grøvdal, L.; Willumsen, B.M.; van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, L.; Grandal, M.V.; Knudsen, S.L.J.; van Deurs, B.; Grøvdal, L.M. Internalization Mechanisms of the Epidermal Growth Factor Receptor after Activation with Different Ligands. PLoS ONE 2013, 8, e58148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandin, A.F.; Cruz Da Silva, E.; Mercier, M.C.; Glushonkov, O.; Didier, P.; Dedieu, S.; Schneider, C.; Devy, J.; Etienne-Selloum, N.; Dontenwill, M.; et al. Gefitinib induces EGFR and α5β1 integrin co-endocytosis in glioblastoma cells. Cell Mol. Life Sci. 2020, 78, 2940–2962. [Google Scholar] [CrossRef]
- Cao, X.; Zhu, H.; Ali-Osman, F.; Lo, H.W. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: A potential mechanism of EGFR-driven antagonism of apoptosis. Mol. Cancer 2011, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Bereczky, B.; Ono, M. The EGFR inhibitor gefitinib suppresses ligand-stimulated endocytosis of EGFR via the early/late endocytic pathway in non-small cell lung cancer cell lines. Histochem. Cell Biol. 2007, 127, 541–553. [Google Scholar] [CrossRef]
- Dreier, A.; Barth, S.; Goswami, A.; Weis, J. Cetuximab induces mitochondrial translocalization of EGFRvIII, but not EGFR: Involvement of mitochondria in tumor drug resistance? Tumor. Biol. 2012, 33, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.J.; Carpenter, G. Cetuximab/C225-induced intracellular trafficking of epidermal growth factor receptor. Cancer Res. 2009, 69, 6179–6183. [Google Scholar] [CrossRef] [Green Version]
- Griffith, L.G.; Swartz, M.A. Capturing complex 3D tissue physiology in vitro. Nat. Rev. Mol. Cell Biol. 2006, 7, 211–224. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Thakuri, P.S.; Liu, C.; Luker, G.D.; Tavana, H. Biomaterials-Based Approaches to Tumor Spheroid and Organoid Modeling. Adv. Healthc. Mater. 2018, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in multicellular spheroids formation. J. R. Soc. Interface 2017, 14, 20160877. [Google Scholar] [CrossRef] [PubMed]
- Alemany-Ribes, M.; Semino, C.E. Bioengineering 3D environments for cancer models. Adv. Drug Deliv. Rev. 2014, 79, 40–49. [Google Scholar] [CrossRef]
- Liu, C.; Lewin, M.D.; Chiang, B.; Luker, K.E.; Luker, G.D. Hybrid Collagen Alginate Hydrogel as a Platform for 3D Tumor Spheroid Invasion. Acta Biomater. 2018, 15, 213–225. [Google Scholar] [CrossRef]
- Miyazaki, K.; Oyanagi, J.; Hoshino, D.; Kumagai, H.; Miyagi, Y. Cancer cell migration on elongate protrusions of fibroblasts in collagen matrix. Sci. Rep. 2019, 9, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-K.; Jang, S.D.; Kim, H.; Chung, S.; Park, J.K.; Kuh, H.-J. Phenotypic Heterogeneity and Plasticity of Cancer Cell Migration in a Pancreatic Tumor Three-Dimensional Culture Model. Cancers 2020, 12, 1305. [Google Scholar] [CrossRef]
- Ricci, C.; Mota, C.; Moscato, S.; Alessandro, D.D.; Sartoris, S.; Bronte, V.; Boggi, U.; Campani, D.; Funel, N.; Moroni, L.; et al. Interfacing polymeric scaffolds with primary pancreatic ductal adenocarcinoma cells to develop 3D cancer models Interfacing polymeric scaffolds with primary pancreatic ductal adenocarcinoma cells to develop 3D cancer models. Biomatter 2015, 4, e955386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puls, T.J.; Tan, X.; Whittington, C.F.; Voytik-Harbin, S.L. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE 2017, 12, e0188870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Kawano, S.; Kojima, M.; Higuchi, Y.; Sugimoto, M.; Ikeda, K.; Sakuyama, N.; Takahashi, S.; Hayashi, R.; Ochiai, A.; Saito, N. Assessment of elasticity of colorectal cancer tissue, clinical utility, pathological and phenotypical relevance. Cancer Sci. 2015, 106, 1232–1239. [Google Scholar] [CrossRef]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; del Río Hernández, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic Reaction in Pancreatic Cancer: Role of Pancreatic Stellate Cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Takehara, Y.; Kawase, T.; Terashima, K.; Ohkawa, Y.; Hirose, Y.; Koda, A.; Hyodo, N.; Ushio, T.; Hirai, Y.; et al. Feasibility of magnetic resonance elastography for the pancreas at 3T. J. Magn. Reason. Imaging 2016, 43, 384–390. [Google Scholar] [CrossRef]
- Liu, H.Y.; Nguyen, H.D.; Lin, C.C. Dynamic PEG–Peptide Hydrogels via Visible Light and FMN-Induced Tyrosine Dimerization. Adv. Healthc. Mater. 2018, 7, 1800954. [Google Scholar] [CrossRef] [PubMed]
- Marí-Buyé, N.; Luque, T.; Navajas, D.; Semino, C.E. Development of a Three-Dimensional Bone-Like Construct in a Soft Self-Assembling Peptide Matrix. Tissue Eng. Part A 2013, 19, 870–881. [Google Scholar] [CrossRef] [Green Version]
- Recha-Sancho, L.; Semino, C.E. Heparin-based self-assembling peptide scaffold reestablish chondrogenic phenotype of expanded de-differentiated human chondrocytes. J. Biomed. Mater. Res. Part A 2016, 104, 1694–1706. [Google Scholar] [CrossRef] [PubMed]
- Castells-Sala, C.; Recha-Sancho, L.; Llucià-Valldeperas, A.; Soler-Botija, C.; Bayes-Genis, A.; Semino, C.E. Three-Dimensional Cultures of Human Subcutaneous Adipose Tissue-Derived Progenitor Cells Based on RAD16-I Self-Assembling Peptide. Tissue Eng. Part C Methods 2016, 22, 113–124. [Google Scholar] [CrossRef]
- Betriu, N.; Jarrosson-Moral, C.; Semino, C.E. Culture and Differentiation of Human Hair Follicle Dermal Papilla Cells in a Soft 3D Self-Assembling Peptide Scaffold. Biomolecules 2020, 10, 684. [Google Scholar] [CrossRef]
- Song, H.; Cai, G.H.; Liang, J.; Ao, D.S.; Wang, H.; Yang, Z.H. Three-dimensional culture and clinical drug responses of a highly metastatic human ovarian cancer HO-8910PM cells in nanofibrous microenvironments of three hydrogel biomaterials. J. Nanobiotechnol. 2020, 18, 90. [Google Scholar] [CrossRef]
- Miroshnikova, Y.A.; Jorgens, D.M.; Spirio, L.; Auer, M.; Sarang-Sieminski, A.L.; Weaver, V.M. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Phys. Biol. 2011, 8, 026013. [Google Scholar] [CrossRef] [Green Version]
- Betriu, N.; Semino, C.E. Development of a 3D Co-Culture System as a Cancer Model Using a Self-Assembling Peptide Scaffold. Gels 2018, 4, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betriu, N.; Recha-sancho, L.; Semino, C.E. Culturing Mammalian Cells in Three-dimensional Peptide Scaffolds. J. Vis. Exp. 2018, e57259. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Manders, E.M.; Stap, J.; Brakenhoff, G.J.; Van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103, 857–862. [Google Scholar] [CrossRef]
- Sastre, D.; Estadella, I.; Bosch, M.; Felipe, A. Triple-Colocalization Approach to Assess Traffic Patterns and Their Modulation. In Computer Optimized Microscopy, 1st ed.; Rebollo, E., Bosch, M., Eds.; Springer: New York, NY, USA, 2019; pp. 215–233. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Buck, E.; Eyzaguirre, A.; Haley, J.D.; Gibson, N.W.; Cagnoni, P.; Iwata, K.K. Inactivation of Akt by the epidermal growth factor receptor inhibitor erlotinib is mediated by HER-3 in pancreatic and colorectal tumor cell lines and contributes to erlotinib sensitivity. Mol. Cancer Ther. 2006, 5, 2051–2059. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Sieminski, A.L.; Was, A.S.; Kim, G.; Gong, H.; Kamm, R.D. The stiffness of three-dimensional ionic self-assembling peptide gels affects the extent of capillary-like network formation. Cell Biochem. Biophys. 2007, 49, 73–83. [Google Scholar] [CrossRef]
- Hadjipanayi, E.; Mudera, V.; Brown, A. Close dependence of fibroblast proliferation on collagen scaffold matrix stiffness. J. Tissue Eng. Regen. Med. 2008, 3, 77–84. [Google Scholar] [CrossRef]
- Fernández-Muiños, T.; Recha-Sancho, L.; López-Chicón, P.; Castells-Sala, C.; Mata, A.; Semino, C.E. Bimolecular based heparin and self-assembling hydrogel for tissue engineering applications. Acta Biomater. 2015, 16, 35–48. [Google Scholar] [CrossRef]
- Recha-Sancho, L.; Semino, C.E. Chondroitin sulfate- and decorin-based self-Assembling scaffolds for cartilage tissue engineering. PLoS ONE 2016, 11, e0157603. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; King, P.J.; Antonescu, C.N.; Sugiyama, M.G.; Bhamra, A.; Surinova, S.; Angelopoulos, N.; Kragh, M.; Pedersen, M.W.; Hartley, J.A.; et al. Targeting of EGFR by a combination of antibodies mediates unconventional EGFR trafficking and degradation. Sci. Rep. 2020, 10, 663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, M.; Brand, T.M.; Starr, M.M.; Li, C.; Huppert, E.J.; Luthar, N.; Pedersen, M.W.; Horak, I.D.; Kragh, M.; Wheeler, D.L. Sym004, a novel EGFR antibody mixture, can overcome acquired resistance to cetuximab. Neoplasia 2013, 15, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Sobhakumari, A.; Schickling, B.M.; Love-homan, L.; Raeburn, A.; Fletcher, E.V.M.; Case, A.J.; Domann, F.E.; Miller, F.J., Jr.; Simons, A.L. NOX4 mediates cytoprotective autophagy induced by the EGFR inhibitor erlotinib in head and neck cancer cells. Toxicol. Appl. Pharmacol. 2013, 272, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Ling, Y.; Sironi, J.; Schwartz, E.L. The Autophagy Inhibitor Chloroquine Overcomes the Innate Resistance of Wild-Type EGFR Non-Small-Cell Lung Cancer Cells to Erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of Autophagy Is an Early Response to Gefitinib and a Potential Therapeutic Target in Breast Cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, Y.; Matsukuma, S.; Nakamura, Y.; Yoshihara, M.; Koizume, S.; Sekiguchi, H.; Saito, H.; Nakayama, H.; Kameda, Y.; Yokose, T.; et al. Enhanced autophagy is required for survival in EGFR-independent EGFR-mutant lung adenocarcinoma cells. Lab. Investig. 2013, 93, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar] [CrossRef]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melikova, M.S.; Kondratov, K.A.; Kornilova, E.S. Two different stages of epidermal growth factor (EGF) receptor endocytosis are sensitive to free ubiquitin depletion produced by proteasome inhibitor MG132. Cell Biol. Int. 2006, 30, 31–43. [Google Scholar] [CrossRef]
- Albornoz, N.; Bustamante, H.; Soza, A.; Burgos, P. Cellular responses to proteasome inhibition: Molecular mechanisms and beyond. Int. J. Mol. Sci. 2019, 20, 3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Zhang, X.; Long, C.; Xu, H.; Cheng, X.; Chang, J.; Zhang, C.; Zhang, C.; Wang, X. Collagen-based three-dimensional culture microenvironment promotes epithelial to mesenchymal transition and drug resistance of human ovarian cancer in vitro. RSC Adv. 2018, 8, 8910–8919. [Google Scholar] [CrossRef] [Green Version]
- Firuzi, O.; Che, P.P.; El Hassouni, B.; Buijs, M.; Coppola, S.; Löhr, M.; Funel, N.; Heuchel, R.; Carnevale, I.; Schmidt, T.; et al. Role of c-MET inhibitors in overcoming drug resistance in spheroid models of primary human pancreatic cancer and stellate cells. Cancers 2019, 11, 638. [Google Scholar] [CrossRef] [Green Version]
- Perche, F.; Torchilin, V.P. Cancer cell spheroids as a model to evaluate chemotherapy protocols. Cancer Biol. Ther. 2012, 13, 1205–1213. [Google Scholar] [CrossRef]
- Chitcholtan, K.; Asselin, E.; Parent, S.; Sykes, P.H.; Evans, J.J. Differences in growth properties of endometrial cancer in three dimensional (3D) culture and 2D cell monolayer. Exp. Cell Res. 2013, 319, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [Green Version]
- Dangi-Garimella, S.; Krantz, S.B.; Barron, M.R.; Shields, M.A.; Heiferman, M.J.; Grippo, P.J.; Bentrem, D.J.; Munshi, H.G. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011, 71, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinilla-Macua, I.; Grassart, A.; Duvvuri, U.; Watkins, S.C.; Sorkin, A. EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. eLife 2017, 6, e31993. [Google Scholar] [CrossRef]
- Heukers, R.; Vermeulen, J.F.; Fereidouni, F.; Bader, A.N.; Voortman, J.; Roovers, R.C.; Gerritsen, H.C.; van Bergen en Henegouwen, P.M.P. Endocytosis of EGFR requires its kinase activity and N-terminal transmembrane dimerization motif. J. Cell Sci. 2013, 126, 4900–4912. [Google Scholar] [CrossRef] [Green Version]
- Ménard, L.; Floc’h, N.; Martin, M.J.; Cross, D.A.E. Reactivation of mutant-EGFR degradation through clathrin inhibition overcomes resistance to EGFR tyrosine kinase inhibitors. Cancer Res. 2018, 78, 3267–3279. [Google Scholar] [CrossRef] [Green Version]
- De Wit, M.; Gao, Y.; Mercieca, D.; de Heer, I.; Valkenburg, B.; van Royen, M.E.; Aerts, J.; Smitt, P.S.; French, P. Mutation and drug-specific intracellular accumulation of EGFR predict clinical responses to tyrosine kinase inhibitors. EBio Med. 2020, 56, 102796. [Google Scholar] [CrossRef]
- Beardmore, J.M.; Richards, R.C. Concentrations of epidermal growth factor in mouse milk throughout lactation. J. Endocrinol. 1983, 96, 287–292. [Google Scholar] [CrossRef]
- Grau, M.; Rodríguez, C.; Soley, M.; Ramírez, I. Relationship between epidermal growth factor in mouse submandibular glands, plasma, and bile: Effects of catecholamines and fasting. Endocrinology 1994, 135, 1854–1862. [Google Scholar] [CrossRef]
- Mullin, J.M. Epithelial Barriers, Compartmentation and Cancer. Sci. Signal. 2004, 2004, pe2. [Google Scholar] [CrossRef] [PubMed]
- Thøgersen, V.B.; Sørensen, B.S.; Poulsen, S.S.; Ørntoft, T.F.; Wolf, H.; Nexo, E. A Subclass of HER1 Ligands Are Prognostic Markers for Survival in Bladder Cancer Patients. Cancer Res. 2001, 61, 6227–6233. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; De Luca, A.; Caponigro, F.; Salomon, D.S. The ErbB Receptors and their Ligands in Cancer: An Overview. Curr. Drug Targets 2005, 6, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Révillion, F.; Lhotellier, V.; Hornez, L.; Bonneterre, J.; Peyrat, J.-P. ErbB/HER ligands in human breast cancer, and relationships with their receptors, the bio-pathological features and prognosis. Ann. Oncol. 2008, 19, 73–80. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | BxPC-3 | PANC-1 | hNDF | |
|---|---|---|---|---|
| Doubling time (h) | 2D | 39 | 30 | 24.3 |
| 0.15% RAD | ND | 63.6 | No proliferation | |
| 0.5% RAD | 95 | 62.3 | 65.3 | |
| Erlotinib IC50 (µM) | 2D | 10 | 45 | 10 |
| 3D | 10 | 100 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betriu, N.; Andreeva, A.; Semino, C.E. Erlotinib Promotes Ligand-Induced EGFR Degradation in 3D but Not 2D Cultures of Pancreatic Ductal Adenocarcinoma Cells. Cancers 2021, 13, 4504. https://doi.org/10.3390/cancers13184504
Betriu N, Andreeva A, Semino CE. Erlotinib Promotes Ligand-Induced EGFR Degradation in 3D but Not 2D Cultures of Pancreatic Ductal Adenocarcinoma Cells. Cancers. 2021; 13(18):4504. https://doi.org/10.3390/cancers13184504
Chicago/Turabian StyleBetriu, Nausika, Anna Andreeva, and Carlos E. Semino. 2021. "Erlotinib Promotes Ligand-Induced EGFR Degradation in 3D but Not 2D Cultures of Pancreatic Ductal Adenocarcinoma Cells" Cancers 13, no. 18: 4504. https://doi.org/10.3390/cancers13184504
APA StyleBetriu, N., Andreeva, A., & Semino, C. E. (2021). Erlotinib Promotes Ligand-Induced EGFR Degradation in 3D but Not 2D Cultures of Pancreatic Ductal Adenocarcinoma Cells. Cancers, 13(18), 4504. https://doi.org/10.3390/cancers13184504