
Immune Checkpoints in Cancers: From Signaling to the Clinic

 ,
,
Abstract
:Simple Summary
Abstract
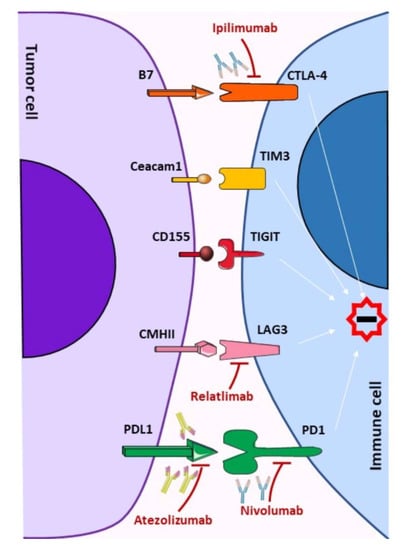
1. Introduction
2. Immune Checkpoints Signaling
2.1. CTLA-4, Cytotoxic T-Lymphocyte-Associated Protein 4
2.2. PD-1, Programmed Death-1
2.3. LAG-3, Lymphocyte Activation Gene 3
2.4. TIM-3, T-Cell Immunoglobulin and Mucin-Domain Containing-3
2.5. TIGIT, T Cell Immunoglobulin and ITIM Domain
2.6. VISTA, V-Domain Ig Suppressor of T Cell Activation
2.7. B7H3, B7 Homolog 3
3. Therapeutic Strategies Targeting Immune Checkpoints
4. Resistance to PD-1 Blockade, ITGBL1 a New Immune Checkpoint?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First global approval. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef]
- Burnet, M. Cancer: A biological approach. III. Viruses associated with neoplastic conditions. IV. Practical applications. Br. Med. J. 1957, 1, 841–847. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bright, R.; Coventry, B.J.; Eardley-Harris, N.; Briggs, N. Clinical Response Rates From Interleukin-2 Therapy for Metastatic Melanoma Over 30 Years’ Experience: A Meta-Analysis of 3312 Patients. J. Immunother. 2017, 40, 21–30. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Lanier, L.L. A renaissance for the tumor immunosurveillance hypothesis. Nat. Med. 2001, 7, 1178–1180. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Cantoni, C.; Rivera, P.; Vitale, M.; Castriconi, R.; Marcenaro, S.; Nanni, M.; Biassoni, R.; Bottino, C.; Moretta, A.; et al. Role of NKG2D in tumor cell lysis mediated by human NK cells: Cooperation with natural cytotoxicity receptors and capability of recognizing tumors of nonepithelial origin. Eur. J. Immunol. 2001, 31, 1076–1086. [Google Scholar] [CrossRef]
- Smyth, M.J.; Hayakawa, Y.; Takeda, K.; Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2002, 2, 850–861. [Google Scholar] [CrossRef]
- Lanier, L.L. Natural killer cell receptor signaling. Curr. Opin. Immunol. 2003, 15, 308–314. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riha, P.; Rudd, C.E. CD28 co-signaling in the adaptive immune response. Self Nonself 2010, 1, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, A.; Barnes, E.; Klenerman, P.; Harlen, M.; Sorensen, P.; Davis, S.J.; Nilsson, P. A theoretical framework for quantitative analysis of the molecular basis of costimulation. J. Immunol. 2005, 175, 1575–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.; Prasad, K.V.; Shoelson, S.E.; Rudd, C.E. CTLA-4 binding to the lipid kinase phosphatidylinositol 3-kinase in T cells. J. Exp. Med. 1995, 181, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Calvo, C.R.; Amsen, D.; Kruisbeek, A.M. Cytotoxic T lymphocyte antigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK) and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphorylation of T cell receptor zeta and ZAP70. J. Exp. Med. 1997, 186, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Smith, X.; Liu, H.; Bismuth, G.; Rudd, C.E. CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur. J. Immunol. 2008, 38, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.; D’Souza, C.A.; Zhang, L. Cell-extrinsic CTLA4-mediated regulation of dendritic cell maturation depends on STAT3. Eur. J. Immunol. 2014, 44, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Bo, L.; Bian, J.; Unsinger, J.; Chang, K.; Hotchkiss, R.S. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock 2011, 36, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inobe, M.; Schwartz, R.H. CTLA-4 engagement acts as a brake on CD4+ T cell proliferation and cytokine production but is not required for tuning T cell reactivity in adaptive tolerance. J. Immunol. 2004, 173, 7239–7248. [Google Scholar] [CrossRef] [Green Version]
- Huijts, C.M.; Schneiders, F.L.; Garcia-Vallejo, J.J.; Verheul, H.M.; de Gruijl, T.D.; van der Vliet, H.J. mTOR Inhibition Per Se Induces Nuclear Localization of FOXP3 and Conversion of Invariant NKT (iNKT) Cells into Immunosuppressive Regulatory iNKT Cells. J. Immunol. 2015, 195, 2038–2045. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, A.; Fiegler, N.; Brunner-Weinzierl, M.; Cerwenka, A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK Cell IFN-gamma production in response to mature dendritic cells. J. Immunol. 2014, 192, 4184–4191. [Google Scholar] [CrossRef]
- Sanseviero, E.; O’Brien, E.M.; Karras, J.R.; Shabaneh, T.B.; Aksoy, B.A.; Xu, W.; Zheng, C.; Yin, X.; Xu, X.; Karakousis, G.C.; et al. Anti-CTLA-4 Activates Intratumoral NK Cells and Combined with IL15/IL15Ralpha Complexes Enhances Tumor Control. Cancer Immunol. Res. 2019, 7, 1371–1380. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, T.; Miyatake, S.; Ohno, H.; Nakaseko, C.; Isono, K.; Bonifacino, J.S.; Saito, T. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity 1997, 6, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Bour-Jordan, H.; Esensten, J.H.; Martinez-Llordella, M.; Penaranda, C.; Stumpf, M.; Bluestone, J.A. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol. Rev. 2011, 241, 180–205. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+ CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Youngnak, P.; Kozono, Y.; Kozono, H.; Iwai, H.; Otsuki, N.; Jin, H.; Omura, K.; Yagita, H.; Pardoll, D.M.; Chen, L.; et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem. Biophys. Res. Commun. 2003, 307, 672–677. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Li, L.; Sari, D.; Petkova, V.; Boussiotis, V.A. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol. Cell. Biol. 2013, 33, 3091–3098. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota, G.; Niogret, C.; Dang, A.T.; Barros, C.R.; Fonta, N.P.; Alfei, F.; Morgado, L.; Zehn, D.; Birchmeier, W.; Vivier, E.; et al. Shp-2 Is Dispensable for Establishing T Cell Exhaustion and for PD-1 Signaling In Vivo. Cell Rep. 2018, 23, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wen, R.; Yang, S.; Schuman, J.; Zhang, E.E.; Yi, T.; Feng, G.S.; Wang, D. Identification of Shp-2 as a Stat5A phosphatase. J. Biol. Chem. 2003, 278, 16520–16527. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Amin, H.M.; Franko, B.; Frantz, C.; Shi, X.; Lai, R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood 2006, 108, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.M.; Jack, R.S.; Wunderlich, F.T.; Bruning, J.C.; Muller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.L.; Nairismagi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S.; et al. PD-1 regulates KLRG1+ group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678. [Google Scholar] [CrossRef] [Green Version]
- Thibult, M.L.; Mamessier, E.; Gertner-Dardenne, J.; Pastor, S.; Just-Landi, S.; Xerri, L.; Chetaille, B.; Olive, D. PD-1 is a novel regulator of human B-cell activation. Int. Immunol. 2013, 25, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haro, M.A.; Littrell, C.A.; Yin, Z.; Huang, X.; Haas, K.M. PD-1 Suppresses Development of Humoral Responses That Protect against Tn-Bearing Tumors. Cancer Immunol. Res. 2016, 4, 1027–1037. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, G.; Wang, Z.; Liu, B.; Han, N.; Li, J.; Lu, C.; Liu, X.; Zhang, Q.; Yang, Q.; et al. PD-1-expressing B cells suppress CD4+ and CD8+ T cells via PD-1/PD-L1-dependent pathway. Mol. Immunol. 2019, 109, 20–26. [Google Scholar] [CrossRef]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.J.; Dunai, C.; Aguilar, E.G.; Vick, S.C.; Sturgill, I.R.; Khuat, L.T.; Stoffel, K.M.; Van Dyke, J.; Longo, D.L.; Darrow, M.A.; et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J. Clin. Investig. 2020, 130, 3051–3068. [Google Scholar] [CrossRef] [Green Version]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Hodi, F.S.; Kluger, H.; Sznol, M.; Carvajal, R.; Lawrence, D.; Atkins, M.; Powderly, J.; Sharfman, W.; Puzanov, I.; Smith, D.; et al. Abstract CT001: Durable, long-term survival in previously treated patients with advanced melanoma (MEL) who received nivolumab (NIVO) monotherapy in a phase I trial. Cancer Res. 2016, 76. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannier, S.; Tournier, M.; Bismuth, G.; Triebel, F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J. Immunol. 1998, 161, 4058–4065. [Google Scholar]
- Bruniquel, D.; Borie, N.; Hannier, S.; Triebel, F. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics 1998, 48, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.J.; Vignali, D.A. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J. Immunol. 2005, 174, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Okazaki, I.M.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef]
- Weber, S.; Karjalainen, K. Mouse CD4 binds MHC class II with extremely low affinity. Int. Immunol. 1993, 5, 695–698. [Google Scholar] [CrossRef] [Green Version]
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dreano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiter, D.J.; Mattijssen, V.; Broecker, E.B.; Ferrone, S. MHC antigens in human melanomas. Semin. Cancer Biol 1991, 2, 35–45. [Google Scholar]
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R.; et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J. Immunol. 2011, 186, 5173–5183. [Google Scholar] [CrossRef] [Green Version]
- Iouzalen, N.; Andreae, S.; Hannier, S.; Triebel, F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. Eur. J. Immunol. 2001, 31, 2885–2891. [Google Scholar] [CrossRef]
- Workman, C.J.; Rice, D.S.; Dugger, K.J.; Kurschner, C.; Vignali, D.A. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur. J. Immunol. 2002, 32, 2255–2263. [Google Scholar] [CrossRef]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrangeli, R.; Micangeli, E.; Donini, S. Cloning of murine LAG-3 by magnetic bead bound homologous probes and PCR (gene-capture PCR). Anal. Biochem. 1996, 241, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, E.; Carbonari, M.; Del Porto, P.; Cibati, M.; Tedesco, T.; Mazzone, A.M.; Paganelli, R.; Fiorilli, M. Lymphocyte activation gene-3 (LAG-3) expression and IFN-gamma production are variably coregulated in different human T lymphocyte subpopulations. J. Immunol. 1998, 161, 489–493. [Google Scholar]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huard, B.; Tournier, M.; Triebel, F. LAG-3 does not define a specific mode of natural killing in human. Immunol. Lett. 1998, 61, 109–112. [Google Scholar] [CrossRef]
- Narayanan, S.; Ahl, P.J.; Bijin, V.A.; Kaliaperumal, N.; Lim, S.G.; Wang, C.-I.; Fairhurst, A.-M.; Connolly, J.E. LAG3 is a Central Regulator of NK Cell Cytokine Production. BioRxiv 2020. [Google Scholar] [CrossRef]
- Byun, H.J.; Jung, W.W.; Lee, D.S.; Kim, S.; Kim, S.J.; Park, C.G.; Chung, H.Y.; Chun, T. Proliferation of activated CD1d-restricted NKT cells is down-modulated by lymphocyte activation gene-3 signaling via cell cycle arrest in S phase. Cell Biol. Int. 2007, 31, 257–262. [Google Scholar] [CrossRef]
- Juno, J.A.; Stalker, A.T.; Waruk, J.L.; Oyugi, J.; Kimani, M.; Plummer, F.A.; Kimani, J.; Fowke, K.R. Elevated expression of LAG-3, but not PD-1, is associated with impaired iNKT cytokine production during chronic HIV-1 infection and treatment. Retrovirology 2015, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Goding, S.R.; Wilson, K.A.; Antony, P.A. Combination of adoptive cell transfer, anti-PD-L1 and anti-LAG-3 antibodies for the treatment of recurrent tumors. OncoImmunology 2013, 2, e25050. [Google Scholar] [CrossRef] [Green Version]
- Anonymous. LAG3-PD-1 Inhibitor Combo Impresses in Melanoma. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef] [Green Version]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [Green Version]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Takamura, S.; Tsuji-Kawahara, S.; Yagita, H.; Akiba, H.; Sakamoto, M.; Chikaishi, T.; Kato, M.; Miyazawa, M. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J. Immunol. 2010, 184, 4696–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Zhu, H.G.; Feng, Z.H.; Geng, H.; Zhang, G.M. Expression of Tim-3 in tumor tissue and its role in the induction of tumor immune tolerance. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin. J. Cell. Mol. Immunol. 2005, 21, 403–407. [Google Scholar]
- Sabatos, C.A.; Chakravarti, S.; Cha, E.; Schubart, A.; Sanchez-Fueyo, A.; Zheng, X.X.; Coyle, A.J.; Strom, T.B.; Freeman, G.J.; Kuchroo, V.K. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003, 4, 1102–1110. [Google Scholar] [CrossRef]
- Sanchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa Linhares, A.; Kellner, F.; Jutz, S.; Zlabinger, G.J.; Gabius, H.J.; Huppa, J.B.; Leitner, J.; Steinberger, P. TIM-3 and CEACAM1 do not interact in cis and in trans. Eur. J. Immunol. 2020, 50, 1126–1141. [Google Scholar] [CrossRef] [Green Version]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, D.; Schraven, B.; Veillette, A. PAG-associated FynT regulates calcium signaling and promotes anergy in T lymphocytes. Mol. Cell. Biol. 2007, 27, 1960–1973. [Google Scholar] [CrossRef] [Green Version]
- Tang, R.; Rangachari, M.; Kuchroo, V.K. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin. Immunol. 2019, 42, 101302. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shi, Q.; Dou, S.; Li, G.; Shi, X.; Jiang, X.; Wang, Z.; Yu, D.; Chen, G.; Wang, R.; et al. Negative regulation of Nod-like receptor protein 3 inflammasome activation by T cell Ig mucin-3 protects against peritonitis. Immunology 2018, 153, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef]
- da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurya, N.; Gujar, R.; Gupta, M.; Yadav, V.; Verma, S.; Sen, P. Immunoregulation of dendritic cells by the receptor T cell Ig and mucin protein-3 via Bruton’s tyrosine kinase and c-Src. J. Immunol. 2014, 193, 3417–3425. [Google Scholar] [CrossRef]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Schwartz, J.A.; Clayton, K.L.; Mujib, S.; Zhang, H.; Rahman, A.K.; Liu, J.; Yue, F.Y.; Benko, E.; Kovacs, C.; Ostrowski, M.A. Tim-3 is a Marker of Plasmacytoid Dendritic Cell Dysfunction during HIV Infection and Is Associated with the Recruitment of IRF7 and p85 into Lysosomes and with the Submembrane Displacement of TLR9. J. Immunol. 2017, 198, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Phong, B.L.; Avery, L.; Sumpter, T.L.; Gorman, J.V.; Watkins, S.C.; Colgan, J.D.; Kane, L.P. Tim-3 enhances FcepsilonRI-proximal signaling to modulate mast cell activation. J. Exp. Med. 2015, 212, 2289–2304. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Casado, J.G.; Pawelec, G.; Morgado, S.; Sanchez-Correa, B.; Delgado, E.; Gayoso, I.; Duran, E.; Solana, R.; Tarazona, R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol. Immunother. 2009, 58, 1517–1526. [Google Scholar] [CrossRef]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.F.; Joller, N.; Tan, D.J.; Teng, M.W.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef] [Green Version]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef]
- Zhou, X.M.; Li, W.Q.; Wu, Y.H.; Han, L.; Cao, X.G.; Yang, X.M.; Wang, H.F.; Zhao, W.S.; Zhai, W.J.; Qi, Y.M.; et al. Intrinsic Expression of Immune Checkpoint Molecule TIGIT Could Help Tumor Growth in vivo by Suppressing the Function of NK and CD8+ T Cells. Front. Immunol. 2018, 9, 2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjunpaa, H.; Blake, S.J.; Ahern, E.; Allen, S.; Liu, J.; Yan, J.; Lutzky, V.; Takeda, K.; Aguilera, A.R.; Guillerey, C.; et al. Deficiency of host CD96 and PD-1 or TIGIT enhances tumor immunity without significantly compromising immune homeostasis. Oncoimmunology 2018, 7, e1445949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Flies, D.B.; Sandler, B.J.; Sznol, M.; Chen, L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J. Biol. Med. 2011, 84, 409–421. [Google Scholar] [PubMed]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Flies, D.B.; Han, X.; Higuchi, T.; Zheng, L.; Sun, J.; Ye, J.J.; Chen, L. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J. Clin. Investig. 2014, 124, 1966–1975. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wu, G.; Manick, B.; Hernandez, V.; Renelt, M.; Erickson, C.; Guan, J.; Singh, R.; Rollins, S.; Solorz, A.; et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 2019, 156, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [Green Version]
- Prasad, D.V.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 is a negative regulator of T cells. J. Immunol. 2004, 173, 2500–2506. [Google Scholar] [CrossRef] [Green Version]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Chen, C.; Shen, Y.; Qu, Q.X.; Chen, X.Q.; Zhang, X.G.; Huang, J.A. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp. Cell Res. 2013, 319, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Tekle, C.; Nygren, M.K.; Chen, Y.W.; Dybsjord, I.; Nesland, J.M.; Maelandsmo, G.M.; Fodstad, O. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int. J. Cancer 2012, 130, 2282–2290. [Google Scholar] [CrossRef]
- Chen, J.T.; Chen, C.H.; Ku, K.L.; Hsiao, M.; Chiang, C.P.; Hsu, T.L.; Chen, M.H.; Wong, C.H. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chong, K.K.; Nakamura, Y.; Nguyen, L.; Huang, S.K.; Kuo, C.; Zhang, W.; Yu, H.; Morton, D.L.; Hoon, D.S. B7-H3 associated with tumor progression and epigenetic regulatory activity in cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 2050–2058. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Tekle, C.; Fodstad, O. The immunoregulatory protein human B7H3 is a tumor-associated antigen that regulates tumor cell migration and invasion. Curr. Cancer Drug Targets 2008, 8, 404–413. [Google Scholar] [CrossRef]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S. Overcoming immunological tolerance to melanoma: Targeting CTLA-4. Asia-Pac. J. Clin. Oncol. 2010, 6 (Suppl. S1), S23. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, M.L.; Makohon-Moore, A.; Lipson, E.J.; Taube, J.M.; McMiller, T.L.; Berger, A.E.; Fan, J.; Kaunitz, G.J.; Cottrell, T.R.; Kohutek, Z.A.; et al. Transcriptional Mechanisms of Resistance to Anti-PD-1 Therapy. Clin. Cancer Res. 2017, 23, 3168–3180. [Google Scholar] [CrossRef] [Green Version]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dehghani, B.; Li, Y.; Kaler, L.J.; Vandenbark, A.A.; Offner, H. Oestrogen modulates experimental autoimmune encephalomyelitis and interleukin-17 production via programmed death 1. Immunology 2009, 126, 329–335. [Google Scholar] [CrossRef]
- Lin, P.Y.; Sun, L.; Thibodeaux, S.R.; Ludwig, S.M.; Vadlamudi, R.K.; Hurez, V.J.; Bahar, R.; Kious, M.J.; Livi, C.B.; Wall, S.R.; et al. B7-H1-dependent sex-related differences in tumor immunity and immunotherapy responses. J. Immunol. 2010, 185, 2747–2753. [Google Scholar] [CrossRef]
- Conforti, F.; Pala, L.; Bagnardi, V.; De Pas, T.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746. [Google Scholar] [CrossRef]
- Wu, Y.; Ju, Q.; Jia, K.; Yu, J.; Shi, H.; Wu, H.; Jiang, M. Correlation between sex and efficacy of immune checkpoint inhibitors (PD-1 and CTLA-4 inhibitors). Int. J. Cancer 2018, 143, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 2021, 39, 96–108. [Google Scholar] [CrossRef]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell 2021, 39, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.; Cammarata, I.; Martire, C.; Focaccetti, C.; Piconese, S.; Buccilli, M.; Mancone, C.; Buzzacchino, F.; Berrios, J.R.G.; D’Alessandris, N.; et al. Combination of chemotherapy and PD-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun. Biol. 2020, 3, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, S.; Mortier, L.; Dutriaux, C.; Maubec, E.; Boileau, M.; Dereure, O.; Leccia, M.T.; Arnault, J.P.; Brunet-Possenti, F.; Aubin, F.; et al. Combined Therapy with Anti-PD1 and BRAF and/or MEK Inhibitor for Advanced Melanoma: A Multicenter Cohort Study. Cancers 2020, 12, 1666. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Ballotti, R.; Cheli, Y.; Bertolotto, C. The complex relationship between MITF and the immune system: A Melanoma ImmunoTherapy (response) Factor? Mol. Cancer 2020, 19, 170. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Ohta, A. Oxygen-dependent regulation of immune checkpoint mechanisms. Int. Immunol. 2018, 30, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wu, S.; Chen, X.; Ye, Y.; Weng, Y.; Pan, Y.; Chen, Z.; Chen, L.; Qiu, X.; Qiu, S. Characterization of Hypoxia Signature to Evaluate the Tumor Immune Microenvironment and Predict Prognosis in Glioma Groups. Front. Oncol. 2020, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Cheli, Y.; Giuliano, S.; Botton, T.; Rocchi, S.; Hofman, V.; Hofman, P.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 2011, 30, 2307–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Ohanna, M.; Cerezo, M.; Nottet, N.; Bille, K.; Didier, R.; Beranger, G.; Mograbi, B.; Rocchi, S.; Yvan-Charvet, L.; Ballotti, R.; et al. Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes Dev. 2018, 32, 448–461. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF-the first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [Green Version]
- Cheli, Y.; Tulic, M.K.; El Hachem, N.; Nottet, N.; Jacquel, A.; Gesson, M.; Strub, T.; Bille, K.; Picard-Gauci, A.; Montaudie, H.; et al. ITGBL1 is a new immunomodulator that favors development of melanoma tumors by inhibiting natural killer cells cytotoxicity. Mol. Cancer 2021, 20, 12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Immune Checkpoint | Compound Name | Combination | Phase | Stage | ClinicalTrials.gov ID |
|---|---|---|---|---|---|
| LAG-3 | IMP-321 LAG-3Ig fusion protein and APC activator | Pembrolizumab | I | Unresectable or Metastatic Melanoma | NCT02676869 |
| Tumor Antigenic Peptides and Montanide | I/II | Stage II-IV Melanoma | NCT01308294 | ||
| Immunological peptides and immunological adjuvants + HLA-A2 peptides + Montanide ISA51 | I/II | Disease-Free Melanoma | NCT00365937 | ||
| Cyclophosphamide, fludarabine, Melan-A VLP vaccine | I | Metastatic Melanoma | NCT00324623 | ||
| +/− Avelumab | I | Solid Tumors | NCT03252938 | ||
| Relatlimab (BMS-986213) Fully human IgG4 mAb | Nivolumab | I | Advanced Solid Tumors | NCT03335540 | |
| II | MSI-H Solid Tumors | NCT03607890 | |||
| Stage IIIB/IV Melanomas | NCT02519322 | ||||
| Locally Advanced, Unresectable, or Metastatic Melanoma | NCT03724968 | ||||
| II/III | Advanced Melanoma | NCT03470922 | |||
| +/− Nivolumab | I | Advanced Solid Tumors | NCT02966548 | ||
| I/II | Solid Tumors Including Melanoma | NCT01968109 | |||
| II | Metastatic Melanoma | NCT03743766 | |||
| Nivolumab + rHuPH20 | I | Cancer including Melanoma | NCT04112498 | ||
| Nivolumab + BMS-986205 + Ipilimumab | I/II | Solid Cancers | NCT03459222 | ||
| Ipilimumab | I | Advanced Melanoma | NCT03978611 | ||
| Ieramilimab (LAG525) Fully human IgG4 mAb | PDR001 | I/II | Advanced Solid Tumor Including Melanoma | NCT02460224 | |
| PDR001, capmatinib, canakinumab, ribociclib | II | Previously Treated Unresectable or Metastatic Melanoma | NCT03484923 | ||
| MK-4280 Fully human IgG4 mAb | Pembrolizumab + Oxaliplatin+ Irinotecan + Leucovorin (Calcium Folinate) + Fluorouracil [5-FU] + MK-4280A | I | Advanced Solid Tumors | NCT02720068 | |
| REGN3767 Fully human mAb | +/− cemiplimab | I | Advanced Cancers | NCT03005782 | |
| TSR-033 Fully human IgG4 mAb | Anti-PD-1 | I | Advanced Solid Tumors | NCT03250832 | |
| BI 754111 Humanized IgG4 mAb | BI 754091(anti-pd1) | Early Phase I | Neoplasms | NCT03433898 | |
| II | Advanced Solid Tumors | NCT03697304 | |||
| +/− BI 754091(anti-pd1) | I | Advanced Cancers | NCT03156114 | ||
| BI 754091(anti-pd1) + BI907828 | I | Advanced Solid Tumors | NCT03964233 | ||
| Sym022 Fully human Fc-inert mAb | - | I | Advanced Solid Tumor | NCT03489369 | |
| Sym021 (PD1) | I | Advanced Solid Tumor | NCT03311412 | ||
| FS118 Bispecific antibody binding both LAG-3 and PD-L1 | - | I | Advanced and Metastatic Cancer | NCT03440437 | |
| MGD013 Bispecific DART protein binding both LAG-3 and PD-1 | +/− margetuximab (anti-HER2) | I | Advanced Solid Tumors | NCT03219268 | |
| INCAGN02385 Fc engineered IgG1k antibody | - | I | Advanced Malignancies | NCT03538028 | |
| TIM-3 | TSR-022 Humanized mAb | TSR-042, TSR-033 | I | Advanced Solid Tumors | NCT02817633 |
| Niraparib, TSR-042, Bevacizumab, Platinum-Based chemotherapy | I | Advanced Solid Tumors | NCT03307785 | ||
| MBG453 Humanized IgG4 monoclonal antibody | PDR001 | I/II | Advanced Malignancies. | NCT02608268 | |
| Sym023 Fully human mAb | - | I | Advanced Solid Tumor | NCT03489343 | |
| Sym021, Sym022 | I | Advanced Solid Tumor or Lymphomas | NCT03311412 | ||
| INCAGN2390 mAb | - | I | Advanced Malignancies | NCT03652077 | |
| LY3321367 mAb | LY3300054 | I | Advanced Solid Tumor | NCT03099109 | |
| LY3300054, Ramucirumab, Abemaciclib, Merestinib | I | Advanced Solid Tumor | NCT02791334 | ||
| BMS-986258 Fully human mAb | Nivolumab, rHuPH20 | I/II | Advanced Solid Tumor | NCT03446040 | |
| SHR-1702 | Camrelizumab | I | Advanced Solid Tumor | NCT03871855 | |
| RO7121661 Anti-PD-1/TIM3 bispecific Ab | - | I | Advanced Solid Tumor | NCT03708328 | |
| BGB-A425 Humanized, IgG1-variant monoclonal antibody | Tislelizumab | I/II | Locally Advanced or Metastatic Solid Tumors | NCT03744468 | |
| LY3415244 Anti PD-L1/TIM-3 Bispecific Antibody | - | I | Advanced Solid Tumor | NCT03752177 | |
| TIGIT | MK-7684 IgG1 mAb | Pembrolizumab | I | Advanced Solid Tumor | NCT02964013 |
| Etigilimab/OMP-313 M32 mAb | Nivolumab | I | Advanced Solid Tumor | NCT03119428 | |
| Tiragolumab/MTIG7192A/RG-6058 Fully human IgG1 mAb | Atezolizumab | I | Advanced Solid Tumor | NCT02794571 | |
| BMS-986207 IgG1 mAb (FcyR-null) | Nivolumab | I/II | Advanced Solid Tumor | NCT02913313 | |
| AB-154 Fully humanized IgG1 mAb | AB122 | I | Advanced Malignancies | NCT03628677 | |
| ASP-8374 IgG1 mAb (FcyR-null) | Pembrolizumab | I | Advanced Solid Tumors | NCT03260322 | |
| - | I | Advanced Solid Tumor | NCT03945253 | ||
| VISTA | JNJ-61610588 Complete human mAb | - | I | Advanced Solid Tumor | NCT02671955 |
| CA-170d Small orally available molecule that directly targets PD-L1/PD-L2, and VISTA | - | I | Advanced Solid Tumors | NCT02812875 | |
| B7-H3 | Enoblituzumab/MGA271 Humanized IgG1κ mAb | - | I | Advanced Solid Tumors | NCT01391143 |
| Ipilimumab | I | Advanced Solid Tumors | NCT02381314 | ||
| Pembrolizumab | I | Advanced Solid Tumors | NCT02475213 | ||
| - | I | Children with B7-H3-Expressing Solid Tumors | NCT02982941 | ||
| MGD009 Humanized, bispecific DART molecule that recognizes both B7-H3 and CD3 | MGA012 | I | Advanced Solid Tumors | NCT03406949 | |
| - | I | B7-H3-Expressing Tumors | NCT02628535 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisibon, C.; Ouertani, A.; Bertolotto, C.; Ballotti, R.; Cheli, Y. Immune Checkpoints in Cancers: From Signaling to the Clinic. Cancers 2021, 13, 4573. https://doi.org/10.3390/cancers13184573
Pisibon C, Ouertani A, Bertolotto C, Ballotti R, Cheli Y. Immune Checkpoints in Cancers: From Signaling to the Clinic. Cancers. 2021; 13(18):4573. https://doi.org/10.3390/cancers13184573
Chicago/Turabian StylePisibon, Céline, Amira Ouertani, Corine Bertolotto, Robert Ballotti, and Yann Cheli. 2021. "Immune Checkpoints in Cancers: From Signaling to the Clinic" Cancers 13, no. 18: 4573. https://doi.org/10.3390/cancers13184573
APA StylePisibon, C., Ouertani, A., Bertolotto, C., Ballotti, R., & Cheli, Y. (2021). Immune Checkpoints in Cancers: From Signaling to the Clinic. Cancers, 13(18), 4573. https://doi.org/10.3390/cancers13184573