
Keeping Myeloma in Check: The Past, Present and Future of Immunotherapy in Multiple Myeloma


Abstract
:Simple Summary
Abstract
1. Introduction
2. IMIDs
2.1. Mechanism of Action
2.2. History
2.3. Thalidomide
2.4. Lenalidomide
2.5. Pomalidomide
2.6. Future Direction
2.6.1. Iberdomide
2.6.2. IMIDs in SMM
3. Antibody Based Therapies
3.1. Monoclonal Antibodies
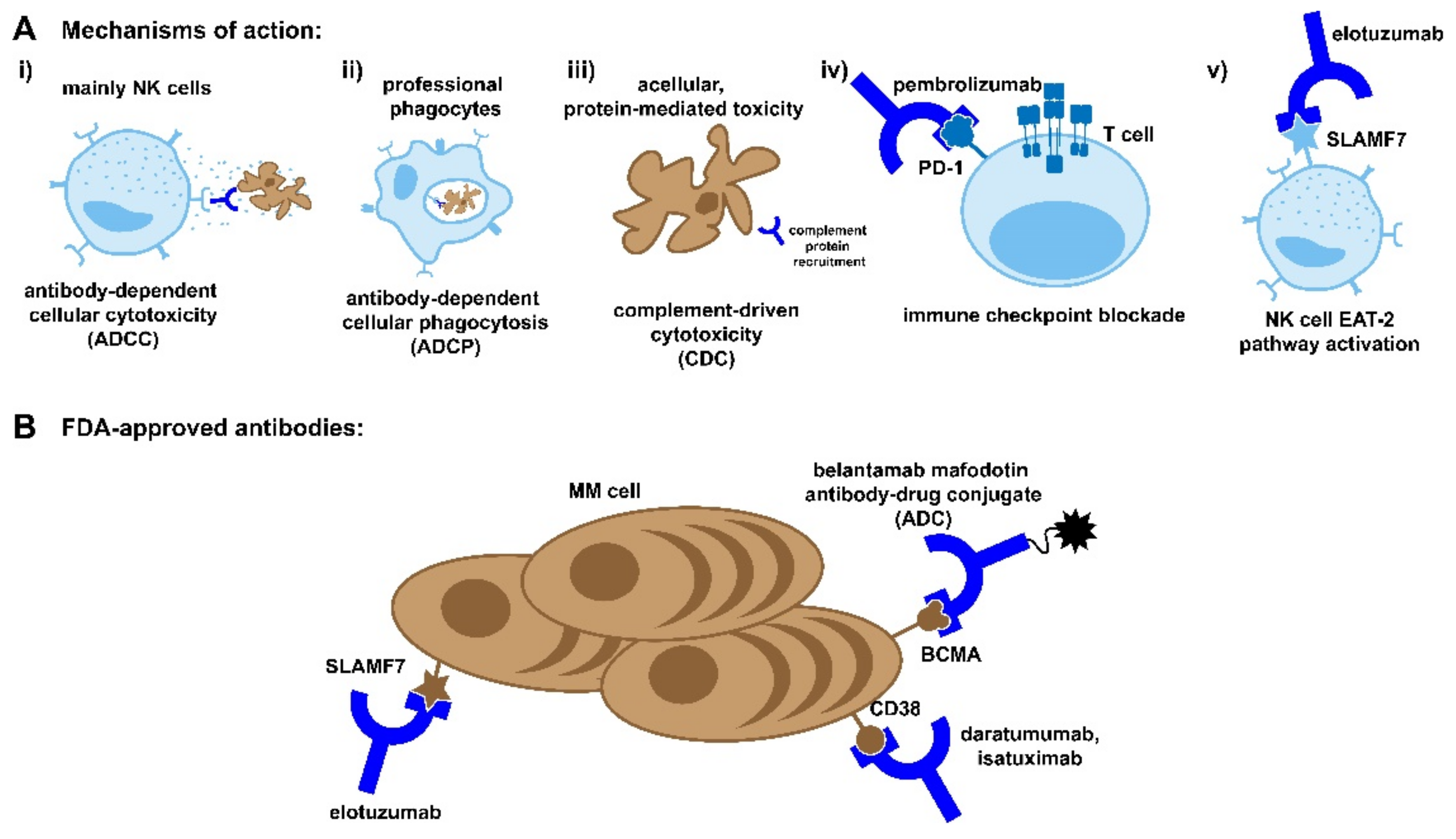
3.1.1. Mechanism of Action
3.1.2. History
3.1.3. Elotuzumab
3.1.4. Daratumumab
3.1.5. Isatuximab
3.2. Immune Checkpoint Inhibitors
3.3. Antibody Drug Conjugates
3.3.1. Mechanism of Action
3.3.2. History
3.3.3. Current Therapies
3.3.4. Future
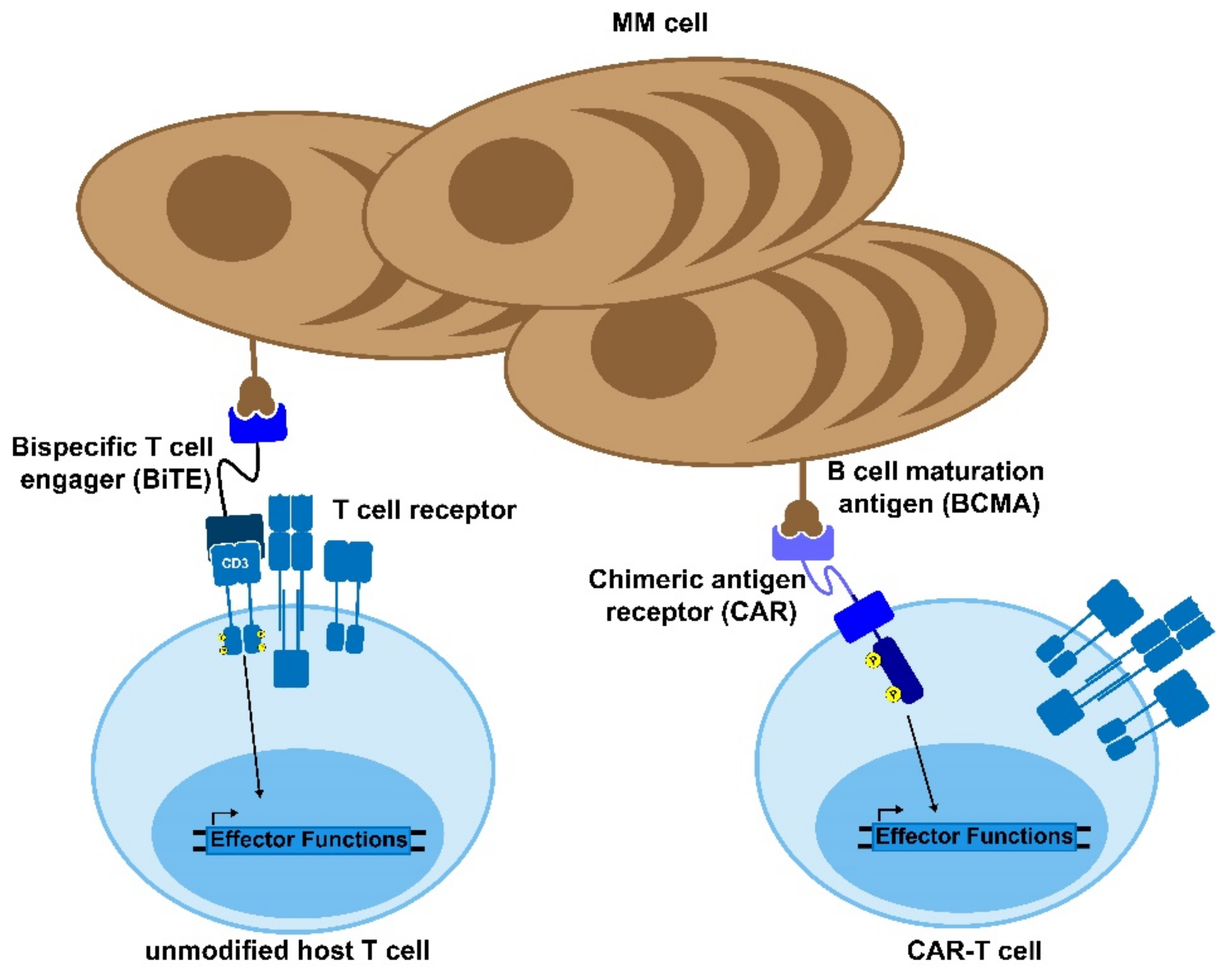
3.4. Bi-Specific Antibodies
3.4.1. Mechanism of Action
3.4.2. History
3.4.3. Future
4. CAR-T Cells
4.1. Mechanism of Action
4.2. History
4.3. CAR-T in Multiple Myeloma
4.4. Current Drawbacks to CAR-T
4.5. Future
5. Vaccines
5.1. Mechanism of Action
5.2. History
5.3. Current Strategies
5.3.1. Dendritic Cell Vaccines
5.3.2. Peptide-Based Vaccines
5.4. Future
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. SEER Cancer Stat Facts: Myeloma. Available online: https://seer.cancer.gov/statfacts/html/mulmy.html (accessed on 22 March 2021).
- Attal, M.; Harousseau, J.L.; Stoppa, A.M.; Sotto, J.J.; Fuzibet, J.G.; Rossi, J.F.; Casassus, P.; Maisonneuve, H.; Facon, T.; Ifrah, N.; et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N. Engl. J. Med. 1996, 335, 91–97. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, N.S.; Kaufman, J.L.; Dhodapkar, M.V.; Hofmeister, C.C.; Almaula, D.K.; Heffner, L.T.; Gupta, V.A.; Boise, L.H.; Lonial, S.; Nooka, A.K. Long-Term Follow-Up Results of Lenalidomide, Bortezomib, and Dexamethasone Induction Therapy and Risk-Adapted Maintenance Approach in Newly Diagnosed Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franssen, L.E.; Mutis, T.; Lokhorst, H.M.; van de Donk, N. Immunotherapy in myeloma: How far have we come? Adv. Hematol. 2019, 10, 2040620718822660. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lam, K.P. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol. Cell Biol. 2001, 21, 4067–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.T.; Anderson, K.C. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin. Biol. 2019, 19, 1143–1156. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.L.; Mohty, M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2019, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Willenbacher, E.; Balog, A.; Willenbacher, W. Short overview on the current standard of treatment in newly diagnosed multiple myeloma. Memo 2018, 11, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Sonneveld, P.; Broijl, A. Treatment of relapsed and refractory multiple myeloma. Haematologica 2016, 101, 396–406. [Google Scholar] [CrossRef]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Chen, L. Cereblon: A Protein Crucial to the Multiple Functions of Immunomodulatory Drugs as well as Cell Metabolism and Disease Generation. J. Immunol. Res. 2017, 2017, 9130608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, Y.; Yan, F.; Ren, X. The role and mechanism of CRL4 E3 ubiquitin ligase in cancer and its potential therapy implications. Oncotarget 2015, 6, 42590–42602. [Google Scholar] [CrossRef]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.M.; Chunder, N.; Chen, C.; Umetsu, S.E.; Winandy, S.; Wells, A.D. Ikaros enforces the costimulatory requirement for IL2 gene expression and is required for anergy induction in CD4+ T lymphocytes. J. Immunol. 2007, 179, 7305–7315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, S.; Thomas, R.M.; Wertheim, G.B.; Zhang, F.; Shen, H.; Wells, A.D. Ikaros imposes a barrier to CD8+ T cell differentiation by restricting autocrine IL-2 production. J. Immunol. 2014, 192, 5118–5129. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, C.; Prince, H.M.; Neeson, P.J. Understanding the Role of T-Cells in the Antimyeloma Effect of Immunomodulatory Drugs. Front. Immunol. 2021, 12, 632399. [Google Scholar] [CrossRef]
- Henry, J.Y.; Labarthe, M.C.; Meyer, B.; Dasgupta, P.; Dalgleish, A.G.; Galustian, C. Enhanced cross-priming of naive CD8+ T cells by dendritic cells treated by the IMiDs(R) immunomodulatory compounds lenalidomide and pomalidomide. Immunology 2013, 139, 377–385. [Google Scholar] [CrossRef]
- Di Lullo, G.; Marcatti, M.; Heltai, S.; Tresoldi, C.; Paganoni, A.M.; Bordignon, C.; Ciceri, F.; Protti, M.P. Immunomodulatory Drugs in the Context of Autologous Hematopoietic Stem Cell Transplantation Associate With Reduced Pro-tumor T Cell Subsets in Multiple Myeloma. Front. Immunol. 2018, 9, 3171. [Google Scholar] [CrossRef] [Green Version]
- Galustian, C.; Meyer, B.; Labarthe, M.C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Reske, T.; Fulciniti, M.; Munshi, N.C. Mechanism of action of immunomodulatory agents in multiple myeloma. Med. Oncol. 2010, 27, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, M.E.; Macpherson, G.R.; Figg, W.D. Thalidomide. Lancet 2004, 363, 1802–1811. [Google Scholar] [CrossRef] [Green Version]
- Watts, G. Frances Oldham Kelsey. Lancet 2015, 386, 1334. [Google Scholar] [CrossRef]
- Rehman, W.; Arfons, L.M.; Lazarus, H.M. The rise, fall and subsequent triumph of thalidomide: Lessons learned in drug development. Adv. Hematol. 2011, 2, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Delforge, M.; Beksac, M.; Wen, P.; Jongen, J.L.; Sezer, O.; Terpos, E.; Munshi, N.; Palumbo, A.; Rajkumar, S.V.; et al. Management of treatment-emergent peripheral neuropathy in multiple myeloma. Leukemia 2012, 26, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Anagnostopoulos, A.; Weber, D. Treatment of plasma cell dyscrasias with thalidomide and its derivatives. J. Clin. Oncol. 2003, 21, 4444–4454. [Google Scholar] [CrossRef] [PubMed]
- Latif, T.; Chauhan, N.; Khan, R.; Moran, A.; Usmani, S.Z. Thalidomide and its analogues in the treatment of Multiple Myeloma. Exp. Hematol. Oncol. 2012, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Chand, N.K.; Subramanya, H.B.; Rao, G.V. Management of patients who refuse blood transfusion. Indian J. Anaesth. 2014, 58, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Sheng, Z.; Niu, S.; Wang, H.; Yu, J.; Xu, J. Lenalidomide versus thalidomide based regimens as first-line therapy for patients with multiple myeloma. Leuk. Lymphoma 2013, 54, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Schlossman, R.L.; Weller, E.; Hideshima, T.; Mitsiades, C.; Davies, F.; LeBlanc, R.; Catley, L.P.; Doss, D.; Kelly, K.; et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood 2002, 100, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Weller, E.; Lonial, S.; Jakubowiak, A.J.; Jagannath, S.; Raje, N.S.; Avigan, D.E.; Xie, W.; Ghobrial, I.M.; Schlossman, R.L.; et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 2010, 116, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Lacy, M.Q.; Hayman, S.R.; Gertz, M.A.; Dispenzieri, A.; Buadi, F.; Kumar, S.; Greipp, P.R.; Lust, J.A.; Russell, S.J.; Dingli, D.; et al. Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J. Clin. Oncol. 2009, 27, 5008–5014. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.C.; Aneja, E.; Boyer, A.; Jayabalan, D.; Mark, T.M.; Pearse, R.; Pekle, K.; Perry, A.; Coleman, M.; Niesvizky, R. Effect of Renal and Hepatic Function on Pomalidomide Dose in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2014, 124, 4754. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Donk, N.W.C.J.v.d.; Popat, R.; Zonder, J.A.; Minnema, M.C.; Larsen, J.; Nguyen, T.V.; Chen, M.S.; Bensmaine, A.; Cota, M.; et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2019, 37, 8006. [Google Scholar] [CrossRef]
- Mateos, M.V.; Landgren, O. MGUS and Smoldering Multiple Myeloma: Diagnosis and Epidemiology. Cancer Treat. Res. 2016, 169, 3–12. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Updated Diagnostic Criteria and Staging System for Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e418–e423. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.S.; Dhodapkar, M.V.; Lonial, S. The Role of Early Intervention in High-Risk Smoldering Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 1–9. [Google Scholar] [CrossRef]
- Mateos, M.V.; Kumar, S.; Dimopoulos, M.A.; Gonzalez-Calle, V.; Kastritis, E.; Hajek, R.; De Larrea, C.F.; Morgan, G.J.; Merlini, G.; Goldschmidt, H.; et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Corral, L.L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): Long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1127–1136. [Google Scholar] [CrossRef]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Murata, K.; McCash, S.I.; Carroll, B.; Lesokhin, A.M.; Hassoun, H.; Lendvai, N.; Korde, N.S.; Mailankody, S.; Landau, H.J.; Koehne, G.; et al. Treatment of multiple myeloma with monoclonal antibodies and the dilemma of false positive M-spikes in peripheral blood. Clin. Biochem. 2018, 51, 66–71. [Google Scholar] [CrossRef]
- Coulson, A.; Levy, A.; Gossell-Williams, M. Monoclonal Antibodies in Cancer Therapy: Mechanisms, Successes and Limitations. West. Indian Med. J. 2014, 63, 650–654. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.S.; Cohen, A.D.; Pazina, T. Mechanisms of NK Cell Activation and Clinical Activity of the Therapeutic SLAMF7 Antibody, Elotuzumab in Multiple Myeloma. Front. Immunol. 2018, 9, 2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurdi, A.T.; Glavey, S.V.; Bezman, N.A.; Jhatakia, A.; Guerriero, J.L.; Manier, S.; Moschetta, M.; Mishima, Y.; Roccaro, A.; Detappe, A.; et al. Antibody-Dependent Cellular Phagocytosis by Macrophages is a Novel Mechanism of Action of Elotuzumab. Mol. Cancer 2018, 17, 1454–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, L.; Wang, Y.; Siegel, D.S.; Wang, M.L. Daratumumab: A first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol. 2016, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Salles, G.; Barrett, M.; Foa, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [Green Version]
- Zojer, N.; Kirchbacher, K.; Vesely, M.; Hubl, W.; Ludwig, H. Rituximab treatment provides no clinical benefit in patients with pretreated advanced multiple myeloma. Leuk. Lymphoma 2006, 47, 1103–1109. [Google Scholar] [CrossRef]
- Fancher, K.M.; Bunk, E.J. Elotuzumab: The First Monoclonal Antibody for the Treatment of Multiple Myeloma. J. Adv. Pract. Oncol. 2016, 7, 542–547. [Google Scholar] [CrossRef]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Weisel, K.; San-Miguel, J.; Shpilberg, O.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: Final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Moreau, P.; Touzeau, C. Update on elotuzumab for the treatment of relapsed/refractory multiple myeloma: Patients’ selection and perspective. Onco Targets 2019, 12, 5813–5822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; Leblanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus pomalidomide and dexamethasone for relapsed/refractory multiple myeloma: Efficacy results after additional follow-up of the phase 2, randomized eloquent-3 study. In Proceedings of the European Hematology Association (EHA) Annual Meeting, Amsterdam, The Netherlands, 13–16 June 2019. [Google Scholar]
- Costa, F.; Dalla Palma, B.; Giuliani, N. CD38 Expression by Myeloma Cells and Its Role in the Context of Bone Marrow Microenvironment: Modulation by Therapeutic Agents. Cells 2019, 8, 1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopleva, M.; Estrov, Z.; Zhao, S.; Andreeff, M.; Mehta, K. Ligation of cell surface CD38 protein with agonistic monoclonal antibody induces a cell growth signal in myeloid leukemia cells. J. Immunol. 1998, 161, 4702–4708. [Google Scholar]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Weiss, B.M.; Plesner, T.; Bahlis, N.J.; Belch, A.; Lonial, S.; Lokhorst, H.M.; Voorhees, P.M.; Richardson, P.G.; Chari, A.; et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016, 128, 37–44. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D., Jr.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Laubach, J.P.; Sborov, D.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.W.; Costa, L.J.; Anderson, L.D.; Nathwani, N.; et al. Daratumumab (DARA) Plus Lenalidomide, Bortezomib, and Dexamethasone (RVd) in Patients with Transplant-Eligible Newly Diagnosed Multiple Myeloma (NDMM): Updated Analysis of Griffin after 12 Months of Maintenance Therapy. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Deckert, J.; Wetzel, M.C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallee, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Dimopoulos, M.A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Spicka, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Lancman, G.; Arinsburg, S.; Jhang, J.; Cho, H.J.; Jagannath, S.; Madduri, D.; Parekh, S.; Richter, J.; Chari, A. Blood Transfusion Management for Patients Treated With Anti-CD38 Monoclonal Antibodies. Front. Immunol. 2018, 9, 2616. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Sunakawa-Kii, M.; Inokuchi, K. PD-L1-PD-1 Pathway in the Pathophysiology of Multiple Myeloma. Cancers 2020, 12, 924. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Bal, S.; Badros, A.Z. Lessons Learned from Checkpoint Blockade Targeting PD-1 in Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 1224–1229. [Google Scholar] [CrossRef]
- Moser-Katz, T.; Joseph, N.S.; Dhodapkar, M.V.; Lee, K.P.; Boise, L.H. Game of Bones: How Myeloma Manipulates Its Microenvironment. Front. Oncol. 2020, 10, 625199. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- Bruins, W.S.C.; Zweegman, S.; Mutis, T.; van de Donk, N. Targeted Therapy With Immunoconjugates for Multiple Myeloma. Front. Immunol. 2020, 11, 1155. [Google Scholar] [CrossRef] [PubMed]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Chari, R.V. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J. The “Utility” of Highly Toxic Marine-Sourced Compounds. Mar. Drugs 2019, 17, 342. [Google Scholar] [CrossRef] [Green Version]
- Oroudjev, E.; Lopus, M.; Wilson, L.; Audette, C.; Provenzano, C.; Erickson, H.; Kovtun, Y.; Chari, R.; Jordan, M.A. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer 2010, 9, 2700–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Jagannath, S.; Heffner, L.T., Jr.; Ailawadhi, S.; Munshi, N.C.; Zimmerman, T.M.; Rosenblatt, J.; Lonial, S.; Chanan-Khan, A.; Ruehle, M.; Rharbaoui, F.; et al. Indatuximab Ravtansine (BT062) Monotherapy in Patients With Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, K.; Herbener, P.; Zuber, C.; Hader, T.; Bernoster, K.; Uherek, C.; Schuttrumpf, J. Activity of Indatuximab Ravtansine against Triple-Negative Breast Cancer in Preclinical Tumor Models. Pharm. Res. 2018, 35, 118. [Google Scholar] [CrossRef] [Green Version]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Anderson, L.D., Jr.; Sutherland, H.J.; Yong, K.; et al. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): A dose escalation and expansion phase 1 trial. Lancet Oncol. 2018, 19, 1641–1653. [Google Scholar] [CrossRef]
- Popat, R.; Nooka, A.; Stockerl-Goldstein, K.; Abonour, R.; Ramaekers, R.; Khot, A.; Forbes, A.; Lee, C.; Augustson, B.; Spencer, A.; et al. DREAMM-6: Safety, Tolerability and Clinical Activity of Belantamab Mafodotin (Belamaf) in Combination with Bortezomib/Dexamethasone (BorDex) in Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 19–20. [Google Scholar] [CrossRef]
- Wahab, A.; Rafae, A.; Mushtaq, K.; Masood, A.; Ehsan, H.; Khakwani, M.; Khan, A. Ocular Toxicity of Belantamab Mafodotin, an Oncological Perspective of Management in Relapsed and Refractory Multiple Myeloma. Front. Oncol. 2021, 11, 678634. [Google Scholar] [CrossRef]
- Eaton, J.S.; Miller, P.E.; Mannis, M.J.; Murphy, C.J. Ocular Adverse Events Associated with Antibody-Drug Conjugates in Human Clinical Trials. J. Ocul. Pharm. 2015, 31, 589–604. [Google Scholar] [CrossRef]
- Kumar, S.K.; Migkou, M.; Bhutani, M.; Spencer, A.; Ailawadhi, S.; Kalff, A.; Walcott, F.; Pore, N.; Gibson, D.; Wang, F.; et al. Phase 1, First-in-Human Study of MEDI2228, a BCMA-Targeted ADC in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Dahlen, E.; Veitonmaki, N.; Norlen, P. Bispecific antibodies in cancer immunotherapy. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schurch, C.M. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front. Immunol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Lancman, G.; Sastow, D.L.; Cho, H.J.; Jagannath, S.; Madduri, D.; Parekh, S.S.; Richard, S.; Richter, J.; Sanchez, L.; Chari, A. Bispecific Antibodies in Multiple Myeloma: Present and Future. Blood Cancer Discov. 2021, 2, 423–433. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2021, 1–12. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Nisonoff, A.; Rivers, M.M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef]
- Newman, M.J.; Benani, D.J. A review of blinatumomab, a novel immunotherapy. J. Oncol. Pharm. Pract. 2016, 22, 639–645. [Google Scholar] [CrossRef]
- Zhou, X.; Einsele, H.; Danhof, S. Bispecific Antibodies: A New Era of Treatment for Multiple Myeloma. J. Clin. Med. 2020, 9, 2166. [Google Scholar] [CrossRef]
- Lancman, G.; Richter, J.; Chari, A. Bispecifics, trispecifics, and other novel immune treatments in myeloma. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Maples, K.T.; Johnson, C.; Lonial, S. Antibody treatment in multiple myeloma. Clin. Adv. Hematol. Oncol. 2021, 19, 166–174. [Google Scholar] [PubMed]
- Cho, S.F.; Lin, L.; Xing, L.J.; Wen, K.; Yu, T.T.; Hsieh, P.A.; Li, Y.Y.; Munshi, N.C.; Wahl, J.; Matthes, K.; et al. AMG 701 Potently Induces Anti-Multiple Myeloma (MM) Functions of T Cells and IMiDs Further Enhance Its Efficacy to Prevent MM Relapse In Vivo. Blood 2019, 134, 135. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermudez, A.; de la Rubia, J.; Mateos, M.V.; Ocio, E.M.; Rodriguez-Otero, P.; San-Miguel, J.; Li, S.Y.; Sarmiento, R.; et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Levy, M.Y.; Dalovisio, A.P.; Bahlis, N.J.; Solh, M.; Sebag, M.; Jakubowiak, A.; Jethava, Y.S.; Costello, C.L.; Chu, M.P.; et al. Preliminary Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics of Subcutaneously (SC) Administered PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 8–9. [Google Scholar] [CrossRef]
- Alfred, L.G.; Saad, Z.U.; María-Victoria, M.; Hareth, N.; Van De Donk, N.W.C.J.; Jesus, F.S.M.; Albert, O.R.; Laura, R.; Ajai, C.; Manisha, B.; et al. Updated Phase 1 Results of Teclistamab, a B-Cell Maturation Antigen (BCMA) x CD3 Bispecific Antibody, in Relapsed and/or Refractory Multiple Myeloma (RRMM). In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5 December 2020. [Google Scholar]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Benmebarek, M.-R.; Karches, C.; Cadilha, B.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.K.; Chen, Y.; Smith, C.C.; Montgomery, S.A.; Vincent, B.G.; Dotti, G.; Savoldo, B. CD30-Redirected Chimeric Antigen Receptor T Cells Target CD30+ and CD30− Embryonal Carcinoma via Antigen-Dependent and Fas/FasL Interactions. Cancer Immunol. Res. 2018, 6, 1274–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell Maturation Antigen Is a Promising Target for Adoptive T-cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfall, A.L.; Maus, M.V.; Hwang, W.-T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Chng, W.J. CAR T-cell therapy in multiple myeloma: More room for improvement. Blood Cancer J. 2021, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 2018, 132, 1011. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone Marrow Microenvironment in Multiple Myeloma Progression. J. Biomed. Biotechnol. 2012, 2012, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, e99048. [Google Scholar] [CrossRef] [Green Version]
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Dwarshuis, N.J.; Song, H.W.; Patel, A.; Kotanchek, T.; Roy, K. Functionalized microcarriers improve T cell manufacturing by facilitating migratory memory T cell production and increasing CD4/CD8 ratio. bioRxiv 2019, 646760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.K.Y.; Cheung, A.S.; Mooney, D.J. Activation and expansion of human T cells using artificial antigen-presenting cell scaffolds. Nat. Protoc. 2020, 15, 773–798. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morenweiser, R. Downstream processing of viral vectors and vaccines. Gene Ther. 2005, 12, S103–S110. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cho, S.-F.; Xing, L.; Wen, K.; Li, Y.; Yu, T.; Hsieh, P.A.; Chen, H.; Kurtoglu, M.; Zhang, Y.; et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia 2021, 35, 752–763. [Google Scholar] [CrossRef]
- Lyman, G.H.; Nguyen, A.; Snyder, S.; Gitlin, M.; Chung, K.C. Economic Evaluation of Chimeric Antigen Receptor T-Cell Therapy by Site of Care Among Patients With Relapsed or Refractory Large B-Cell Lymphoma. JAMA Netw. Open 2020, 3, e202072. [Google Scholar] [CrossRef]
- Lee, L.; Draper, B.; Chaplin, N.; Philip, B.; Chin, M.; Galas-Filipowicz, D.; Onuoha, S.; Thomas, S.; Baldan, V.; Bughda, R.; et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood 2018, 131, 746–758. [Google Scholar] [CrossRef]
- Fernández De Larrea, C.; Staehr, M.; Lopez, A.V.; Ng, K.Y.; Chen, Y.; Godfrey, W.D.; Purdon, T.J.; Ponomarev, V.; Wendel, H.-G.; Brentjens, R.J.; et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 146–154. [Google Scholar] [CrossRef]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasim, W.; Amrolia, P.J.; Samarasinghe, S.; Ghorashian, S.; Zhan, H.; Stafford, S.; Butler, K.; Ahsan, G.; Gilmour, K.; Adams, S.; et al. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL. Blood 2015, 126, 2046. [Google Scholar] [CrossRef]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [CrossRef] [Green Version]
- Soekojo, C.Y.; Ooi, M.; de Mel, S.; Chng, W.J. Immunotherapy in Multiple Myeloma. Cells 2020, 9, 601. [Google Scholar] [CrossRef] [Green Version]
- Hoyos, V.; Borrello, I. The immunotherapy era of myeloma: Monoclonal antibodies, vaccines, and adoptive T-cell therapies. Blood 2016, 128, 1679–1687. [Google Scholar] [CrossRef]
- Klausen, U.; Holmberg, S.; Holmstrom, M.O.; Jorgensen, N.G.D.; Grauslund, J.H.; Svane, I.M.; Andersen, M.H. Novel Strategies for Peptide-Based Vaccines in Hematological Malignancies. Front. Immunol. 2018, 9, 2264. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Raje, N.; Fowler, J.A.; Mezzi, K.; Scott, E.C.; Dhodapkar, M.V. How to Train Your T Cells: Overcoming Immune Dysfunction in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 1541–1554. [Google Scholar] [CrossRef] [Green Version]
- Hanna, M.G., Jr.; Peters, L.C. Specific immunotherapy of established visceral micrometastases by BCG-tumor cell vaccine alone or as an adjunct to surgery. Cancer 1978, 42, 2613–2625. [Google Scholar] [CrossRef]
- Rhee, F. Idiotype vaccination strategies in myeloma: How to overcome a dysfunctional immune system. Clin. Cancer Res. 2007, 13, 1353–1355. [Google Scholar] [CrossRef] [Green Version]
- Lacy, M.Q.; Mandrekar, S.; Dispenzieri, A.; Hayman, S.; Kumar, S.; Buadi, F.; Dingli, D.; Litzow, M.; Wettstein, P.; Padley, D.; et al. Idiotype-pulsed antigen-presenting cells following autologous transplantation for multiple myeloma may be associated with prolonged survival. Am. J. Hematol. 2009, 84, 799–802. [Google Scholar] [CrossRef] [Green Version]
- Garfall, A.L.; Stadtmauer, E.A. Cellular and vaccine immunotherapy for multiple myeloma. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Hoang, M.D.; Jung, S.H.; Lee, H.J.; Lee, Y.K.; Nguyen-Pham, T.N.; Choi, N.R.; Vo, M.C.; Lee, S.S.; Ahn, J.S.; Yang, D.H.; et al. Dendritic Cell-Based Cancer Immunotherapy against Multiple Myeloma: From Bench to Clinic. Chonnam Med. J. 2015, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. 2013, 19, 3640–3648. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Avivi, I.; Binyamini, N.; Uhl, L.; Somaiya, P.; Stroopinsky, D.; Palmer, K.A.; Coll, M.D.; Katz, T.; Bisharat, L.; et al. Blockade of PD-1 in Combination with Dendritic Cell/Myeloma Fusion Cell Vaccination Following Autologous Stem Cell Transplantation Is Well Tolerated, Induces Anti-Tumor Immunity and May Lead to Eradication of Measureable Disease. Blood 2015, 126, 4218. [Google Scholar] [CrossRef]
- Bae, J.; Smith, R.; Daley, J.; Mimura, N.; Tai, Y.T.; Anderson, K.C.; Munshi, N.C. Myeloma-specific multiple peptides able to generate cytotoxic T lymphocytes: A potential therapeutic application in multiple myeloma and other plasma cell disorders. Clin. Cancer Res. 2012, 18, 4850–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nooka, A.K.; Wang, M.L.; Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma: A Nonrandomized Clinical Trial. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Raje, N.; Yee, A.J. How We Approach Smoldering Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1119–1125. [Google Scholar] [CrossRef]
- Luca Biavati, M.; Carol, A.H.; Anna, F.; Lakshmi, R.; Cristina, Z.; Rachel, G.; Sarah, J.; Catherine, S.; Abbas, A.A.; Kimberly, N.; et al. Treating MRD Positivity in Multiple Myeloma: An Allogeneic GM-CSF-Based Vaccine in Combination with Lenalidomide Induces Long-Term Remissions in Patients with Low Disease Burden. ASH Publ. 2020, 136, 8. [Google Scholar] [CrossRef]
- Borrello, I.; Noonan, K.; Ferguson, A.K.; Sidorski, A.; Rudrarajuraraju, L.; Casildo, A.; Huff, C.A. Allogeneic myeloma GVAX with lenalidomide in near complete remission to enhance progression free survival. J. Clin. Oncol. 2015, 33, 3082. [Google Scholar] [CrossRef]
- Jorgensen, N.G.; Klausen, U.; Grauslund, J.H.; Helleberg, C.; Aagaard, T.G.; Do, T.H.; Ahmad, S.M.; Olsen, L.R.; Klausen, T.W.; Breinholt, M.F.; et al. Peptide Vaccination Against PD-L1 With IO103 a Novel Immune Modulatory Vaccine in Multiple Myeloma: A Phase I First-in-Human Trial. Front. Immunol. 2020, 11, 595035. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, N.G.; Ahmad, S.M.; Abildgaard, N.; Straten, P.T.; Svane, I.M.; Andersen, M.H.; Knudsen, L.M. Peptide vaccination against multiple myeloma using peptides derived from anti-apoptotic proteins: A phase I trial. Stem Cell Investig. 2016, 3, 95. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.S.; O’Halloran, J.A.; Kalaidina, E.; Kim, W.; Schmitz, A.J.; Zhou, J.Q.; Lei, T.; Thapa, M.; Chen, R.E.; Case, J.B.; et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 2021, 596, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Burgess, J.L.; Naleway, A.L.; Tyner, H.; Yoon, S.K.; Meece, J.; Olsho, L.E.W.; Caban-Martinez, A.J.; Fowlkes, A.L.; Lutrick, K.; et al. Prevention and Attenuation of Covid-19 with the BNT162b2 and mRNA-1273 Vaccines. N. Engl. J. Med. 2021, 385, 320–329. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2016, 4, 146–156. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Trial Number | Drug | Tumor Target | T Cell Target | Status |
|---|---|---|---|---|
| NCT03933735 | TNB-383B | BCMA | CD3 | Recruiting |
| NCT03269136 | PF-06863135 | BCMA | CD3 | Active, not recruiting |
| NCT04649359 | PF-06863135 | BCMA | CD3 | Recruiting |
| NCT04798586 | PF-06863135 | BCMA | CD3 | Recruiting |
| NCT03309111 | ISB 1342 | CD38 | CD3 | Recruiting |
| NCT04773522 | Talquetamab | GPRC5D | CD3 | Recruiting |
| NCT03761108 | REGN5458 | BCMA | CD3 | Recruiting |
| NCT04083534 | REGN5459 | BCMA | CD3 | Recruiting |
| NCT04634552 | Talquetamab | GPRC5D | CD3 | Recruiting |
| NCT04722146 | Teclistamab | BCMA | CD3 | Recruiting |
| NCT04557098 | Teclistamab | BCMA | CD3 | Recruiting |
| NCT03399799 | Talquetamab | GPRC5D | CD3 | Recruiting |
| NCT04696809 | Teclistamab | BCMA | CD3 | Recruiting |
| NCT03145181 | Talquetamab | GPRC5D | CD3 | Recruiting |
| NCT04735575 | EMB-06 | BCMA | CD3 | Not yet recruiting |
| NCT04586426 | Talquetamab/ Teclistamab | BCMA/GPRC5D | CD3 | Recruiting |
| NCT04108195 | Talquetamab/ Teclistamab | BCMA/GPRC5D | CD3 | Recruiting |
| NCT04910568 | RG6160 | FcRH5 | CD3 | Not yet recruiting |
| NCT03275103 | RG6160 | FcRH5 | CD3 | Recruiting |
| NCT03486067 | CC-93269 | BCMA | CD3 | Recruiting |
| NCT03287908 | AMG 701 | BCMA | CD3 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ackley, J.; Ochoa, M.A.; Ghoshal, D.; Roy, K.; Lonial, S.; Boise, L.H. Keeping Myeloma in Check: The Past, Present and Future of Immunotherapy in Multiple Myeloma. Cancers 2021, 13, 4787. https://doi.org/10.3390/cancers13194787
Ackley J, Ochoa MA, Ghoshal D, Roy K, Lonial S, Boise LH. Keeping Myeloma in Check: The Past, Present and Future of Immunotherapy in Multiple Myeloma. Cancers. 2021; 13(19):4787. https://doi.org/10.3390/cancers13194787
Chicago/Turabian StyleAckley, James, Miguel Armenta Ochoa, Delta Ghoshal, Krishnendu Roy, Sagar Lonial, and Lawrence H. Boise. 2021. "Keeping Myeloma in Check: The Past, Present and Future of Immunotherapy in Multiple Myeloma" Cancers 13, no. 19: 4787. https://doi.org/10.3390/cancers13194787
APA StyleAckley, J., Ochoa, M. A., Ghoshal, D., Roy, K., Lonial, S., & Boise, L. H. (2021). Keeping Myeloma in Check: The Past, Present and Future of Immunotherapy in Multiple Myeloma. Cancers, 13(19), 4787. https://doi.org/10.3390/cancers13194787