Bromamine T (BAT) Exerts Stronger Anti-Cancer Properties than Taurine (Tau)

 ,
,  ,
,  , ,
, ,
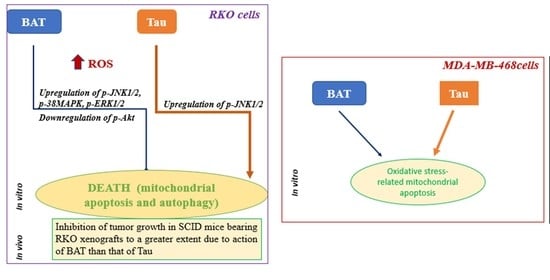
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Anti-Proliferative Effect of BAT and Tau on Cancer Cell Growth
2.2. The Tumor-Inhibitory Effect of BAT and Tau through Induction of Mitochondrial Apoptosis
2.3. The Effect of BAT and Tau via Elevation of ROS in Cancer Cells
2.4. The Effect of BAT and Tau on the MAPK Pathway
2.5. The Tumor-Suppressive Effect of BAT through Induction of Autophagy
2.6. The Tumor-Inhibitory Effect of BAT and Tau on ROS-Related DNA Damage Response (DDR)
2.7. The Tumor-Inhibitory Effect of BAT and Tau In Vivo
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012: Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-Type BRAF Is Required for Response to Panitumumab or Cetuximab in Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-Mediated Reactivation of MAPK Signaling Contributes to Insensitivity of BRAF -Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Schumacker, P.T. Reactive Oxygen Species in Cancer Cells: Live by the Sword, Die by the Sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, W.; Chen, J.; Cai, X.; Yang, J.; Yang, Y.; Yan, H.; Cheng, X.; Ye, J.; Lu, W.; et al. The Natural Diterpenoid Isoforretin A Inhibits Thioredoxin-1 and Triggers Potent ROS-Mediated Antitumor Effects. Cancer Res. 2017, 77, 926–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.-U.; Tan, K.-B.; Lin, H.; Chiu, G.N.C. The Role of Reactive Oxygen Species and Autophagy in Safingol-Induced Cell Death. Cell Death Dis. 2011, 2, e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.R.; Yang, S. Hydrogen Peroxide: A Signaling Messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.S.; Zong, W. Chemotherapeutic Approaches for Targeting Cell Death Pathways. Oncologist 2006, 11, 342–357. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, A.; Le, J.; Sausville, E.; Emadi, A. Autophagy Modulation: A Target for Cancer Treatment Development. Cancer Chemother. Pharmacol. 2015, 75, 439–447. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal Turnover, but Not a Cellular Level, of Endogenous LC3 Is a Marker for Autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Aredia, F.; Guamán Ortiz, L.M.; Giansanti, V.; Scovassi, A.I. Autophagy and Cancer. Cells 2012, 1, 520–534. [Google Scholar] [CrossRef] [Green Version]
- Chesney, R.W. Taurine: Its Biological Role and Clinical Implications. Adv. Pediatr. 1985, 32, 1–42. [Google Scholar] [PubMed]
- Lee, J.Y.; Jung, D.W.; Park, H.A.; Kim, S.J.; Chung, J.H.; Moon, C.K.; Kim, Y.C. Effect of Taurine on Biliary Excretion and Metabolism of Acetaminophen in Male Hamsters. Biol. Pharm. Bull. 2004, 27, 1792–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aerts, L.; Van Assche, F.A. Taurine and Taurine-Deficiency in the Perinatal Period. J. Perinat. Med. 2002, 30, 281–286. [Google Scholar] [CrossRef]
- Blachier, F.; Andriamihaja, M.; Blais, A. Sulfur-Containing Amino Acids and Lipid Metabolism. J. Nutr. 2020, 150, 2524S–2531S. [Google Scholar] [CrossRef]
- Huxtable, R.J. Physiological Actions of Taurine. Physiol. Rev. 1992, 72, 101–163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tu, S.; Wang, Y.; Xu, B.; Wan, F. Mechanism of Taurine-Induced Apoptosis in Human Colon Cancer Cells. Acta Biochim. Biophys. Sin. 2014, 46, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Xia, Y.; Zhang, X.; Liu, L.; Tu, S.; Zhu, W.; Yu, L.; Wan, H.; Yu, B.; Wan, F. Roles of the MST1-JNK Signaling Pathway in Apoptosis of Colorectal Cancer Cells Induced by Taurine. Libyan J. Med. 2018, 13, 1500346. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Zhang, X.; Wan, H.; Xia, Y.; Liu, Z.; Yang, X.; Wan, F. Effect of Taurine on Cell Proliferation and Apoptosis Human Lung Cancer A549 Cells. Oncol. Lett. 2018, 15, 5473–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-H.; Lee, H.; Park, K.K.; Kim, H.W.; Park, T. Taurine-Responsive Genes Related to Signal Transduction as Identified by CDNA Microarray Analyses of HepG2 Cells. J. Med. Food 2006, 9, 33–41. [Google Scholar] [CrossRef]
- Wang, A.S.; Lodi, A.; Rivera, L.B.; Izquierdo-Garcia, J.L.; Firpo, M.A.; Mulvihill, S.J.; Tempero, M.A.; Bergers, G.; Ronen, S.M. HR-MAS MRS of the Pancreas Reveals Reduced Lipid and Elevated Lactate and Taurine Associated with Early Pancreatic Cancer: Pdac Is Associated with Abnormal Lipid, Lactate, and Taurine Levels. NMR Biomed. 2014, 27, 1361–1370. [Google Scholar] [CrossRef] [Green Version]
- Opstad, K.S.; Bell, B.A.; Griffiths, J.R.; Howe, F.A. Taurine: A Potential Marker of Apoptosis in Gliomas. Br. J. Cancer 2009, 100, 789–794. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Kim, A.K. Effect of Taurine on Antioxidant Enzyme System in B16F10 Melanoma Cells. In Taurine 7; Advances in Experimental Medicine and, Biology; Azuma, J., Schaffer, S.W., Ito, T., Eds.; Springer: New York, NY, USA, 2009; Volume 643, pp. 491–499. ISBN 978-0-387-75680-6. [Google Scholar]
- Vanitha, M.K.; Anandakumar, P.; Sakthisekaran, D. Taurine Abrogates Mammary Carcinogenesis through Induction of Apoptosis in Sprague-Dawley Rats. J. Biochem. Mol. Toxicol. 2018, 32, e22204. [Google Scholar] [CrossRef]
- Vanitha, M.K.; Baskaran, K.; Periyasamy, K.; Selvaraj, S.; Ilakkia, A.; Saravanan, D.; Venkateswari, R.; Revathi Mani, B.; Anandakumar, P.; Sakthisekaran, D. Modulatory Effect of Taurine on 7,12-Dimethylbenz(a)Anthracene-Induced Alterations in Detoxification Enzyme System, Membrane Bound Enzymes, Glycoprotein Profile and Proliferative Cell Nuclear Antigen in Rat Breast Tissue: Taurine- and Dmba-Induced Breast Cancer. J. Biochem. Mol. Toxicol. 2016, 30, 414–423. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, H.; Wang, Y.; Liu, C.; Zhu, W.; Zheng, S.; Wan, F. Taurine Induces the Apoptosis of Breast Cancer Cells by Regulating Apoptosis-Related Proteins of Mitochondria. Int. J. Mol. Med. 2015, 35, 218–226. [Google Scholar] [CrossRef]
- Choi, E.-J.; Tang, Y.; Lee, C.B.; Cheong, S.H.; Sung, S.H.; Oh, M.-R.; Jang, S.Y.; Park, P.-J.; Kim, E.-K. Effect of Taurine on In Vitro Migration of MCF-7 and MDA-MB-231 Human Breast Carcinoma Cells. In Taurine 9; Marcinkiewicz, J., Schaffer, S.W., Eds.; Advances in Experimental Medicine and, Biology; Springer: New York, NY, USA, 2015; Volume 803, pp. 191–201. ISBN 978-3-319-15125-0. [Google Scholar]
- He, F.; Ma, N.; Midorikawa, K.; Hiraku, Y.; Oikawa, S.; Zhang, Z.; Huang, G.; Takeuchi, K.; Murata, M. Taurine Exhibits an Apoptosis-Inducing Effect on Human Nasopharyngeal Carcinoma Cells through PTEN/Akt Pathways in Vitro. Amino Acids 2018, 50, 1749–1758. [Google Scholar] [CrossRef]
- Tang, Y.; Choi, E.-J.; Cheong, S.H.; Hwang, Y.J.; Arokiyaraj, S.; Park, P.-J.; Moon, S.-H.; Kim, E.-K. Effect of Taurine on Prostate-Specific Antigen Level and Migration in Human Prostate Cancer Cells. In Taurine 9; Marcinkiewicz, J., Schaffer, S.W., Eds.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2015; Volume 803, pp. 203–214. ISBN 978-3-319-15125-0. [Google Scholar]
- Chatzakos, V.; Slätis, K.; Djureinovic, T.; Helleday, T.; Hunt, M.C. N-Acyl Taurines Are Anti-Proliferative in Prostate Cancer Cells. Lipids 2012, 47, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, W.-J.; Liu, L.-Q.; Wan, H.-F.; Yang, X.-H.; Zhu, W.-F.; Yu, L.-H.; Zhang, X.-L.; Wan, F.-S. Impact of Taurine on the Proliferation and Apoptosis of Human Cervical Carcinoma Cells and Its Mechanism. Chin. Med. J. Engl. 2019, 132, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Sadzuka, Y.; Matsuura, M.; Sonobe, T. The Effect of Taurine, a Novel Biochemical Modulator, on the Antitumor Activity of Doxorubicin. Biol. Pharm. Bull. 2009, 32, 1584–1587. [Google Scholar] [CrossRef] [Green Version]
- Al-Asmari, A.; Al-Zahrani, A.; Khan, A.; Al-Shahrani, H.; Ali Al Amri, M. Taurine Ameliorates 5-Flourouracil-Induced Intestinal Mucositis, Hepatorenal and Reproductive Organ Damage in Wistar Rats: A Biochemical and Histological Study. Hum. Exp. Toxicol. 2016, 35, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Parvez, S.; Tabassum, H.; Banerjee, B.D.; Raisuddin, S. Taurine Prevents Tamoxifen-Induced Mitochondrial Oxidative Damage in Mice. Basic Clin. Pharmacol. Toxicol. 2008, 102, 382–387. [Google Scholar] [CrossRef]
- Das, J.; Ghosh, J.; Manna, P.; Sil, P.C. Taurine Protects Rat Testes against Doxorubicin-Induced Oxidative Stress as Well as P53, Fas and Caspase 12-Mediated Apoptosis. Amino Acids 2012, 42, 1839–1855. [Google Scholar] [CrossRef]
- Das, J.; Ghosh, J.; Manna, P.; Sil, P.C. Taurine Suppresses Doxorubicin-Triggered Oxidative Stress and Cardiac Apoptosis in Rat via up-Regulation of PI3-K/Akt and Inhibition of P53, P38-JNK. Biochem. Pharmacol. 2011, 81, 891–909. [Google Scholar] [CrossRef]
- Shimizu, M.; Zhao, Z.; Ishimoto, Y.; Satsu, H. Dietary Taurine Attenuates Dextran Sulfate Sodium (DSS)-induced Experimental Colitis in Mice. In Taurine 7; Azuma, J., Schaffer, S.W., Ito, T., Eds.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2009; Volume 643, pp. 265–271. ISBN 978-0-387-75680-6. [Google Scholar]
- Marcinkiewicz, J.; Kontny, E. Taurine and Inflammatory Diseases. Amino Acids 2014, 46, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Logotheti, S.; Khoury, N.; Vlahopoulos, S.A.; Skourti, E.; Papaevangeliou, D.; Liloglou, T.; Gorgoulis, V.; Budunova, I.; Kyriakopoulos, A.M.; Zoumpourlis, V. N-Bromotaurine Surrogates for Loss of Antiproliferative Response and Enhances Cisplatin Efficacy in Cancer Cells with Impaired Glucocorticoid Receptor. Transl. Res. 2016, 173, 58–73. [Google Scholar] [CrossRef] [Green Version]
- Nair, C.G.; Lalithakumari, R.; Senan, P.I. Bromamine-T as a New Oxidimetric Titrant. Talanta 1978, 25, 525–527. [Google Scholar] [CrossRef]
- Walczewska, M.; Peruń, A.; Białecka, A.; Śróttek, M.; Jamróz, W.; Dorożyński, P.; Jachowicz, R.; Kulinowski, P.; Nagl, M.; Gottardi, W.; et al. Comparative Analysis of Microbicidal and Anti-inflammatory Properties of Novel Taurine Bromamine Derivatives and Bromamine T. In Taurine 10; Lee, D.-H., Schaffer, S.W., Park, E., Kim, H.W., Eds.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2017; Volume 975, pp. 515–534. ISBN 978-94-024-1077-8. [Google Scholar]
- Kyriakopoulos, A.M.; Nagl, M.; Orth-Höller, D.; Marcinkiewicz, J.; Baliou, S.; Zoumbourlis, V. Successful Treatment of a Unique Chronic Multi-Bacterial Scalp Infection with N-Chlorotaurine, N-Bromotaurine and Bromamine T. Access Microbiol. 2020. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Wojas-Pelc, A.; Walczewska, M.; Lipko-Godlewska, S.; Jachowicz, R.; Maciejewska, A.; Białecka, A.; Kasprowicz, A. Topical Taurine Bromamine, a New Candidate in the Treatment of Moderate Inflammatory Acne Vulgaris: A Pilot Study. Eur. J. Dermatol. 2008, 18, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Losa, J.H.; Cobo, C.P.; Viniegra, J.G.; Lobo, V.J.S.-A.; y Cajal, S.R.; Sánchez-Prieto, R. Role of the P38 MAPK Pathway in Cisplatin-Based Therapy. Oncogene 2003, 22, 3998–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, I.H.; Kang, S.W.; Yoon, S.H.; Um, H.-D. Cellular Components Involved in the Cell Death Induced by Cisplatin in the Absence of P53 Activation. Oncol. Rep. 2006, 15, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wei, Q.; Wang, J.; Du, Q.; Yu, J.; Zhang, L.; Dong, Z. Regulation of PUMA-α by P53 in Cisplatin-Induced Renal Cell Apoptosis. Oncogene 2006, 25, 4056–4066. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.J.; Huang, R.; Austin, C.P.; Xia, M. The Future of Toxicity Testing: A Focus on in Vitro Methods Using a Quantitative High-Throughput Screening Platform. Drug Discov. Today 2010, 15, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, A. P53 Protein or BID Protein Select the Route to Either Apoptosis (Programmed Cell Death) or to Cell Cycle Arrest Opposing Carcinogenesis after DNA Damage by ROS. Med. Hypotheses 2006, 67, 296–299. [Google Scholar] [CrossRef]
- Tu, H.-C.; Ren, D.; Wang, G.X.; Chen, D.Y.; Westergard, T.D.; Kim, H.; Sasagawa, S.; Hsieh, J.J.-D.; Cheng, E.H.-Y. The P53-Cathepsin Axis Cooperates with ROS to Activate Programmed Necrotic Death upon DNA Damage. Proc. Natl. Acad. Sci. USA 2009, 106, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hu, Q.; Shen, H.-M. Pharmacological Inhibitors of Autophagy as Novel Cancer Therapeutic Agents. Pharmacol. Res. 2016, 105, 164–175. [Google Scholar] [CrossRef]
- Xu, Y. Regulation of P53 Responses by Post-Translational Modifications. Cell Death Differ. 2003, 10, 400–403. [Google Scholar] [CrossRef]
- Redon, C.E.; Dickey, J.S.; Bonner, W.M.; Sedelnikova, O.A. γ-H2AX as a Biomarker of DNA Damage Induced by Ionizing Radiation in Human Peripheral Blood Lymphocytes and Artificial Skin. Adv. Space Res. 2009, 43, 1171–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossi, V. P38α MAPK Pathway: A Key Factor in Colorectal Cancer Therapy and Chemoresistance. World J. Gastroenterol. 2014, 20, 9744. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.; Krett, N.L.; Guzman, G.; Khalid, A.; Ozden, O.; Staudacher, J.J.; Bauer, J.; Baik, S.H.; Carroll, T.; Yazici, C.; et al. NFkB Is Essential for Activin-Induced Colorectal Cancer Migration via Upregulation of PI3K-MDM2 Pathway. Oncotarget 2017, 8, 37377–37393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.-M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H.K. Bufalin Induces Autophagy-Mediated Cell Death in Human Colon Cancer Cells through Reactive Oxygen Species Generation and JNK Activation. Free Radic. Biol. Med. 2011, 51, 1365–1375. [Google Scholar] [CrossRef]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A Role for the P38 Mitogen-Acitvated Protein Kinase Pathway in the Transcriptional Activation of P53 on Genotoxic Stress by Chemotherapeutic Agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar]
- Liu, Y.; Bodmer, W.F. Analysis of P53 Mutations and Their Expression in 56 Colorectal Cancer Cell Lines. Proc. Natl. Acad. Sci. USA 2006, 103, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Storr, S.J.; Woolston, C.M.; Zhang, Y.; Martin, S.G. Redox Environment, Free Radical, and Oxidative DNA Damage. Antioxid. Redox Signal. 2013, 18, 2399–2408. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA Damage Induces Phosphorylation of the Amino Terminus of P53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef] [Green Version]
- Jong, C.J.; Azuma, J.; Schaffer, S. Mechanism Underlying the Antioxidant Activity of Taurine: Prevention of Mitochondrial Oxidant Production. Amino Acids 2012, 42, 2223–2232. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Trivieri, M.G.; Khaper, N.; Husain, T.; Wilson, G.J.; Liu, P.; Sole, M.J.; Backx, P.H. Taurine Supplementation Reduces Oxidative Stress and Improves Cardiovascular Function in an Iron-Overload Murine Model. Circulation 2004, 109, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, Y.; Liu, X.; Zuo, J.; Wang, K.; Liu, W.; Ge, J. Exogenous Taurine Attenuates Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Rat Cardiomyocytes. Acta Biochim. Biophys. Sin. 2013, 45, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Rajakumar, D.; Jeraud, M.; Felix, A.J.W.; Balasubramanian, T. Effects of Taurine on Glutathione Peroxidase, Glutathione Reductase and Reduced Glutathione Levels in Rats. Pak. J. Biol. Sci. 2011, 14, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, A. Role of JNK Activation in Apoptosis: A Double-Edged Sword. Cell Res. 2005, 15, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 Regulates ER Ca2+ Homeostasis and Apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sorensen, T.; Rafn, B.; Bottzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jaattela, M. Sensitization to the Lysosomal Cell Death Pathway by Oncogene-Induced Down-Regulation of Lysosome-Associated Membrane Proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK Activation in Cisplatin-Induced Apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, S.; Schnellmann, R.G. A Death-Promoting Role for Extracellular Signal-Regulated Kinase. J. Pharmacol. Exp. Ther. 2006, 319, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V.; Schulte, T.; Nguyen, P.; Trepel, J.; Neckers, L.M. Taxol-Induced Apoptosis and Phosphorylation of Bcl-2 Protein Involves c-Raf-1 and Represents a Novel c-Raf-1 Signal Transduction Pathway. Cancer Res. 1996, 56, 1851–1854. [Google Scholar]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-Induced Apoptosis Depends on MAP Kinase Pathways (ERK and P38) and Is Independent of P53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Cagnol, S.; Chambard, J.-C. ERK and Cell Death: Mechanisms of ERK-Induced Cell Death—Apoptosis, Autophagy and Senescence: ERK and Cell Death. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- She, Q.-B.; Chen, N.; Dong, Z. ERKs and P38 Kinase Phosphorylate P53 Protein at Serine 15 in Response to UV Radiation. J. Biol. Chem. 2000, 275, 20444–20449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Klionsky, D.J. Physiological Functions of Atg6/Beclin 1: A Unique Autophagy-Related Protein. Cell Res. 2007, 17, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.-Y.; Yoon, C.-H.; An, S.; Park, I.-C.; Kang, C.-M.; Kim, M.-J.; Lee, S.-J. The Rac1/MKK7/JNK Pathway Signals Upregulation of Atg5 and Subsequent Autophagic Cell Death in Response to Oncogenic Ras. Carcinogenesis 2009, 30, 1880–1888. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Agnihotri, N. Piperlongumine, a Piper Alkaloid Targets Ras/PI3K/Akt/MTOR Signaling Axis to Inhibit Tumor Cell Growth and Proliferation in DMH/DSS Induced Experimental Colon Cancer. Biomed. Pharmacother. 2019, 109, 1462–1477. [Google Scholar] [CrossRef]
- Chen, J.; Dai, J.; Kang, Z.; Yang, T.; Zhao, Q.; Zheng, J.; Zhang, X.; Zhang, J.; Xu, J.; Sun, G.; et al. A Combinatorial Strategy for Overcoming Primary and Acquired Resistance of MEK Inhibition in Colorectal Cancer. Exp. Cell Res. 2020, 393, 112060. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baliou, S.; Goulielmaki, M.; Ioannou, P.; Cheimonidi, C.; Trougakos, I.P.; Nagl, M.; Kyriakopoulos, A.M.; Zoumpourlis, V. Bromamine T (BAT) Exerts Stronger Anti-Cancer Properties than Taurine (Tau). Cancers 2021, 13, 182. https://doi.org/10.3390/cancers13020182
Baliou S, Goulielmaki M, Ioannou P, Cheimonidi C, Trougakos IP, Nagl M, Kyriakopoulos AM, Zoumpourlis V. Bromamine T (BAT) Exerts Stronger Anti-Cancer Properties than Taurine (Tau). Cancers. 2021; 13(2):182. https://doi.org/10.3390/cancers13020182
Chicago/Turabian StyleBaliou, Stella, Maria Goulielmaki, Petros Ioannou, Christina Cheimonidi, Ioannis P. Trougakos, Markus Nagl, Anthony M. Kyriakopoulos, and Vassilis Zoumpourlis. 2021. "Bromamine T (BAT) Exerts Stronger Anti-Cancer Properties than Taurine (Tau)" Cancers 13, no. 2: 182. https://doi.org/10.3390/cancers13020182
APA StyleBaliou, S., Goulielmaki, M., Ioannou, P., Cheimonidi, C., Trougakos, I. P., Nagl, M., Kyriakopoulos, A. M., & Zoumpourlis, V. (2021). Bromamine T (BAT) Exerts Stronger Anti-Cancer Properties than Taurine (Tau). Cancers, 13(2), 182. https://doi.org/10.3390/cancers13020182