Targeting E-selectin to Tackle Cancer Using Uproleselan

 ,
,  , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Role of E-Selectin in Cancer Pathophysiology
2.1. Inhibition of Selectins as a Therapeutic Strategy in Cancer
2.2. The Role of E-selectin in Cancer Progression
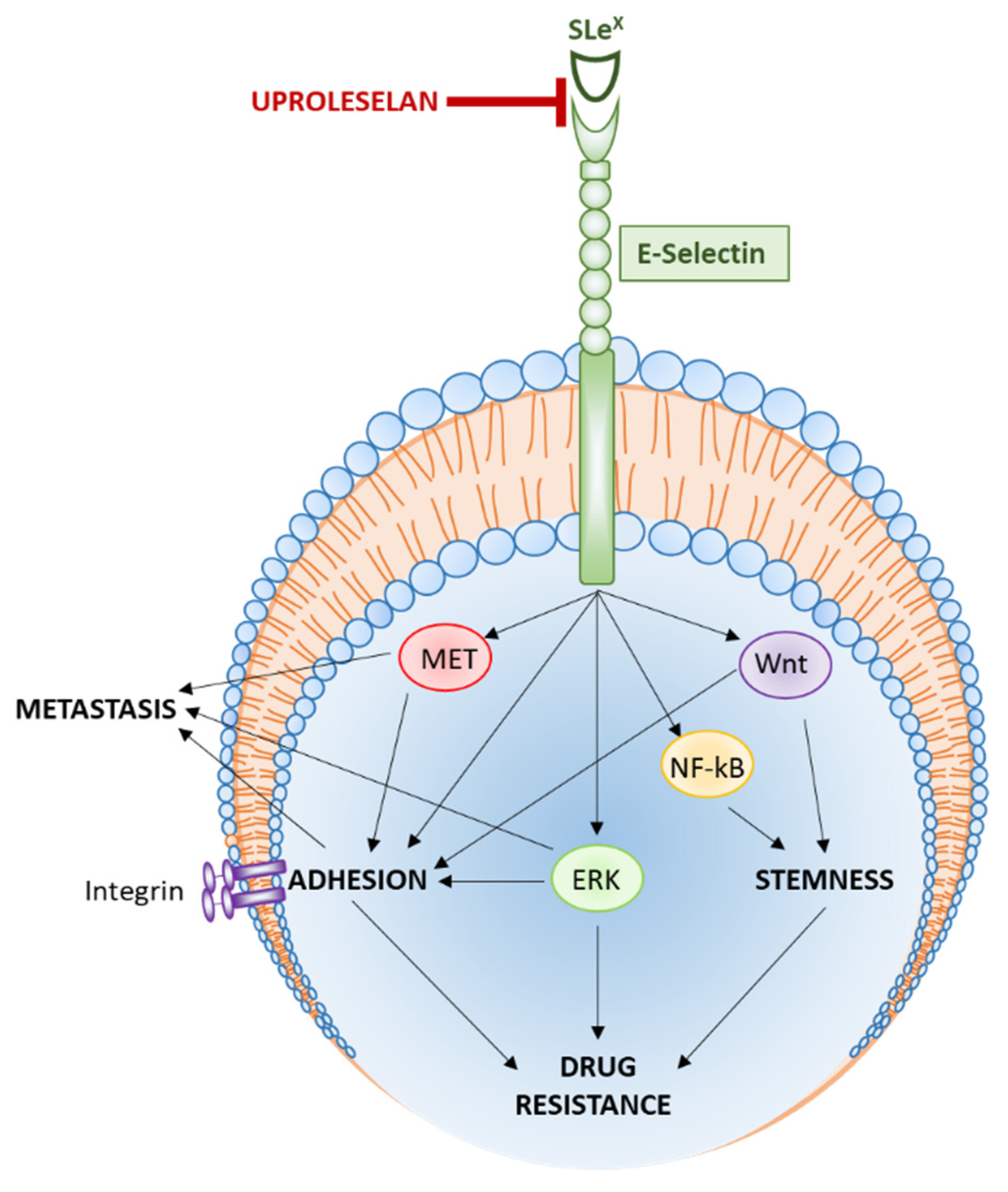
2.3. Signaling Pathways Regulated by E-Selectin
2.3.1. Cell Trafficking and Metastasis
2.3.2. Adhesion and Tumor Growth
2.3.3. Stemness and Self-Renewal
2.3.4. Drug Resistance
3. Uproleselan in Cancer Therapy
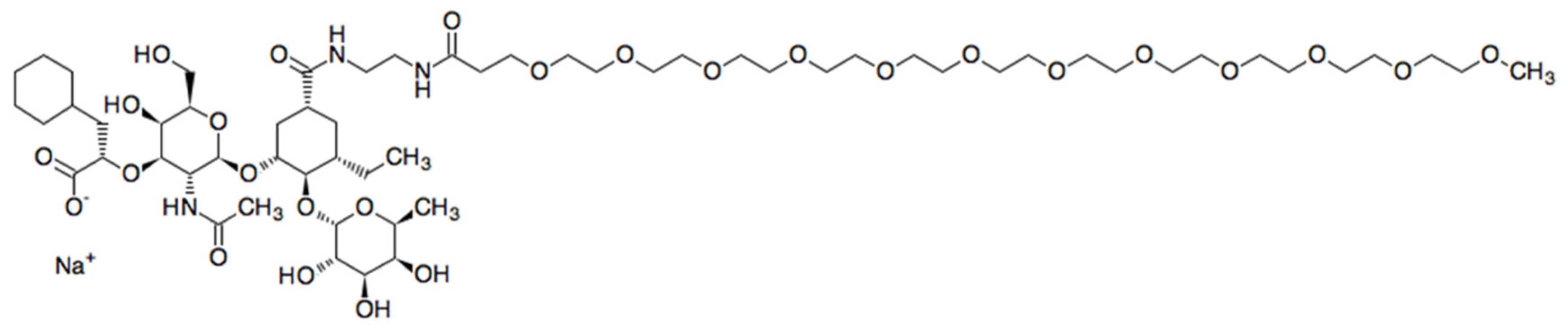
3.1. Uproleselan—Chemical Structure and Properties
3.2. The Role of Uproleselan in Cancer Therapy
3.2.1. Uproleselan Inhibits Metastasis
3.2.2. Uproleselan Decreases Adhesion and Activates Cancer Cell Mobilization
3.2.3. Uproleselan Causes Maturity of Cancer Stem Cells
3.2.4. Uproleselan Resensitizes Cancer Cells to Therapies in Pre-Clinical Models
4. Clinical Trials Using Uproleselan
4.1. Pharmacokinetics of Uproleselan
4.2. Safety of Uproleselan (Phase I and Phase I/II Trials)
4.3. Clinical Efficacy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bevilacqua, M.P.; Nelson, R.M. Selectins. J. Clin. Investig. 1993, 91, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Sai, B.; Xiang, J. Disseminated tumour cells in bone marrow are the source of cancer relapse after therapy. J. Cell. Mol. Med. 2018, 22, 5776–5786. [Google Scholar] [CrossRef]
- Pals, S.T.; Kersten, M.J.; Spaargaren, M. Targeting cell adhesion and homing as strategy to cure Waldenström’s macroglobulinemia. Best Pr. Res. Clin. Haematol. 2016, 29, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. et Biophys. Acta (BBA) Bioenerg. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Martínez, E.; Luengo-Gil, G.; Benito, A.C.; González-Billalabeitia, E.; Conesa, M.A.V.; García, T.G.; García-Garre, E.; Vicente, V.; De La Peña, F.A. Tumor-infiltrating immune cell profiles and their change after neoadjuvant chemotherapy predict response and prognosis of breast cancer. Breast Cancer Res. 2014, 16, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Leslie, M.; Kameyama, H.; Lokesh, G.L.R.; Ichimura, N.; Davis, R.; Hills, N.; Hasan, N.; Zhang, R.; Kondo, Y.; et al. Functional Blockade of E-Selectin in Tumor-Associated Vessels Enhances Anti-Tumor Effect of Doxorubicin in Breast Cancer. Cancers 2020, 12, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsig, L. Selectins Facilitate Carcinoma Metastasis and Heparin Can Prevent Them. Physiology 2004, 19, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucia, M.; Jankowski, K.; Reca, R.; Wysoczynski, M.; Bandura, L.; Allendorf, D.J.; Zhang, J.; Ratajczak, J.; Ratajczak, M.Z. CXCR4–SDF-1 Signalling, Locomotion, Chemotaxis and Adhesion. J. Mol. Histol. 2003, 35, 233–245. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.-T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 Regulates Extra-Medullary Myeloma through Epithelial-Mesenchymal-Transition-like Transcriptional Activation. Cell Rep. 2015, 12, 622–635. [Google Scholar] [CrossRef] [Green Version]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Côté, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nat. Cell Biol. 2005, 435, 969–973. [Google Scholar] [CrossRef]
- Azab, A.K.; Azab, F.; Blotta, S.; Pitsillides, C.M.; Thompson, B.; Runnels, J.M.; Roccaro, A.M.; Ngo, H.T.; Melhem, M.R.; Sacco, A.; et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell–derived factor-1–induced cell adhesion and chemotaxis in multiple myeloma. Blood 2009, 114, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.-S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, E.C.; Picker, L.J. Lymphocyte Homing and Homeostasis. Science 1996, 272, 60–67. [Google Scholar] [CrossRef]
- Angelini, D.E.; Devata, S.; Hawley, A.E.; Blackburn, S.A.; Grewal, S.; Hemmer, M.V.; Flanner, H.; Kramer, W.; E Parker, W.; Li, Y.-L.; et al. E-Selectin Antagonist GMI-1271 Shows a Favorable Safety, PK and Bleeding Profile in Phase I Studies of Healthy Volunteers. Blood 2016, 128, 3826. [Google Scholar] [CrossRef]
- Azab, A.K.; Quang, P.; Azab, F.; Pitsillides, C.; Thompson, B.; Chonghaile, T.N.; Patton, J.T.; Maiso, P.; Monrose, V.; Sacco, A.; et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood 2012, 119, 1468–1478. [Google Scholar] [CrossRef]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Chien, S.S.; Dai, J.; Magnani, J.L.; Sekizaki, T.S.; E Fogler, W.; Thackray, H.M.; Gardner, K.; Estey, E.H.; Becker, P.S. E-Selectin Ligand Expression By Leukemic Blasts Is Associated with Prognosis in Patients with AML. Blood 2018, 132, 1513. [Google Scholar] [CrossRef]
- Federico, C.; Alhallak, K.; Sun, J.; Duncan, K.; Azab, F.; Sudlow, G.P.; De La Puente, P.; Muz, B.; Kapoor, V.; Zhang, L.; et al. Tumor microenvironment-targeted nanoparticles loaded with bortezomib and ROCK inhibitor improve efficacy in multiple myeloma. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Jubeli, E.; Moine, L.; Vergnaud, J.; Barratt, G. E-selectin as a target for drug delivery and molecular imaging. J. Control. Release 2012, 158, 194–206. [Google Scholar] [CrossRef]
- Kimura, A.; Kawaishi, K.; Sasaki, A.; Hyodo, H.; Oguma, N. L-Selectin Expression in CD34 Positive Cells in Chronic Myeloid Leukemia. Leuk. Lymphoma 1998, 28, 399–404. [Google Scholar] [CrossRef]
- Laird, C.; Hassanein, W.; O’Neill, N.A.; French, B.M.; Cheng, X.; Fogler, W.; Magnani, J.L.; Parsell, D.; Cimeno, A.; Phelps, C.J.; et al. P- and E-selectin receptor antagonism prevents human leukocyte adhesion to activated porcine endothelial monolayers and attenuates porcine endothelial damage. Xenotransplantation 2018, 25, e12381. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; Azab, F.; De La Puente, P.; Rollins, S.; Alvarez, R.; Kawar, Z.; Azab, A.K. Inhibition of P-Selectin and PSGL-1 Using Humanized Monoclonal Antibodies Increases the Sensitivity of Multiple Myeloma Cells to Bortezomib. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muz, B.; Azab, F.; Fiala, M.; King, J.; Kohnen, D.; Fogler, W.E.; Smith, T.; Magnani, J.L.; Vij, R.; Azab, A.K. Inhibition of E-Selectin (GMI-1271) or E-selectin together with CXCR4 (GMI-1359) re-sensitizes multiple myeloma to therapy. Blood Cancer J. 2019, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Weisberg, E.; Sahin, I.; Liu, F.; Awwad, R.; Azab, F.; Liu, Q.; Griffin, J.D.; Ghobrial, I.M. The influence of hypoxia on CML trafficking through modulation of CXCR4 and E-cadherin expression. Leukemia 2012, 27, 961–964. [Google Scholar] [CrossRef]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; E Forristal, C.; Patton, J.T.; Magnani, J.L.; Lévesque, J.-P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef]
- Shea, D.J.; Li, Y.W.; Stebe, K.J.; Konstantopoulos, K. E-selectin-mediated rolling facilitates pancreatic cancer cell adhesion to hyaluronic acid. FASEB J. 2017, 31, 5078–5086. [Google Scholar] [CrossRef] [Green Version]
- Steele, M.M.; Radhakrishnan, P.; Magnani, J.L.; Hollingsworth, M.A. A Small Molecule Glycomimetic Antagonist of E-selectin (GMI-1271) Prevents Pancreatic Tumor Metastasis and Offers Improved Efficacy of Chemotherapy. Available online: https://cancerres.aacrjournals.org/content/74/19_Supplement/4503 (accessed on 1 January 2020).
- Takada, A.; Ohmori, K.; Yoneda, T.; Tsuyuoka, K.; Hasegawa, A.; Kiso, M.; Kannagi, R. Contribution of carbohydrate antigens sialyl Lewis A and sialyl Lewis X to adhesion of human cancer cells to vascular endothelium. Cancer Res. 1993, 53, 354–361. [Google Scholar]
- Festuccia, C.; Mancini, A.; Gravina, G.L.; Colapietro, A.; Vetuschi, A.; Pompili, S.; Ventura, L.; Monache, S.D.; Iorio, R.; Del Fattore, A.; et al. Dual CXCR4 and E-Selectin Inhibitor, GMI-1359, Shows Anti-Bone Metastatic Effects and Synergizes with Docetaxel in Prostate Cancer Cell Intraosseous Growth. Cells 2019, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Dimitroff, C.J.; Lechpammer, M.; Long-Woodward, D.; Kutok, J.L. Rolling of Human Bone-Metastatic Prostate Tumor Cells on Human Bone Marrow Endothelium under Shear Flow Is Mediated by E-Selectin. Cancer Res. 2004, 64, 5261–5269. [Google Scholar] [CrossRef] [Green Version]
- Gout, S.; Morin, C.; Houle, F.; Huot, J. Death Receptor-3, a New E-Selectin Counter-Receptor that Confers Migration and Survival Advantages to Colon Carcinoma Cells by Triggering p38 and ERK MAPK Activation. Cancer Res. 2006, 66, 9117–9124. [Google Scholar] [CrossRef] [Green Version]
- Porquet, N.; Poirier, A.; Houle, F.; Pin, A.-L.; Gout, S.; Tremblay, P.-L.; Paquet, E.R.; Klinck, R.; Auger, F.A.; Huot, J. Survival advantages conferred to colon cancer cells by E-selectin-induced activation of the PI3K-NFκB survival axis downstream of Death receptor-3. BMC Cancer 2011, 11, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.T.; Sipkins, D.A. E-Selectin and SDF-1 regulate metastatic trafficking of breast cancer cells within the bone. Mol. Cell. Oncol. 2016, 4, e1214771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Lyerly, H.K.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra73. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-A.; Blache, C.A.; Bajana, S.; Hasan, N.; Kamal, M.; Morita, Y.; Gupta, V.; Tsolmon, B.; Suh, K.S.; Gorenstein, D.G.; et al. The effect of soluble E-selectin on tumor progression and metastasis. BMC Cancer 2016, 16, 331. [Google Scholar] [CrossRef] [Green Version]
- Debreceni, I.B.; Szász, R.; Kónya, Z.; Erdődi, F.; Kiss, F.; Kappelmayer, J. L-Selectin Expression is Influenced by Phosphatase Activity in Chronic Lymphocytic Leukemia. Cytom. Part B Clin. Cytom. 2019, 96, 149–157. [Google Scholar] [CrossRef]
- Steegmaler, M.; Levinovitz, A.; Isenmann, S.; Borges, E.; Lenter, M.; Kocher, H.P.; Kleuser, B.; Vestweber, D. The E-selectin-ligand ESL-1 is a variant of a receptor for fibroblast growth factor. Nat. Cell Biol. 1995, 373, 615–620. [Google Scholar] [CrossRef]
- Picker, L.J.; Warnock, R.; Burns, A.R.; Doerschuk, C.M.; Berg, E.L.; Butchert, E.C. The neutrophil selectin LECAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140. Cell 1991, 66, 921–933. [Google Scholar] [CrossRef]
- Videira, P.A.; Silva, M.; Martin, K.C.; Sackstein, R. Ligation of the CD44 Glycoform HCELL on Culture-Expanded Human Monocyte-Derived Dendritic Cells Programs Transendothelial Migration. J. Immunol. 2018, 201, 1030–1043. [Google Scholar] [CrossRef]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; Von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.L.; Eaton, S.F.; E Lyons, D.; Lichenstein, H.S.; Cummings, R.D.; McEver, R.P. The P-selectin glycoprotein ligand from human neutrophils displays sialylated, fucosylated, O-linked poly-N-acetyllactosamine. J. Biol. Chem. 1994, 269, 23318–23327. [Google Scholar]
- Winkler, I.G.; Erbani, J.M.; Barbier, V.; Davies, J.M.; Tay, J.; Fiveash, C.E.; Lowe, J.; Tallack, M.; Magnani, J.L.; Levesque, J.-P. Vascular E-Selectin Mediates Chemo-Resistance in Acute Myeloid Leukemia Initiating Cells Via Canonical Receptors PSGL-1 (CD162) and Hcell (CD44) and AKT Signaling. Available online: http://glycomimetics.com/wp-content/uploads/2018/11/ASH2017_Winkler-talk-12dec2017_for-GMI_with-text2-1.pdf (accessed on 1 January 2020).
- Nonomura, C.; Kikuchi, J.; Kiyokawa, N.; Ozaki, H.; Mitsunaga, K.; Ando, H.; Kanamori, A.; Kannagi, R.; Fujimoto, J.; Muroi, K.; et al. CD43, but not P-Selectin Glycoprotein Ligand-1, Functions as an E-Selectin Counter-Receptor in Human Pre-B–Cell Leukemia NALL-1. Cancer Res. 2008, 68, 790–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Atarashi, K.; Umemoto, E.; Furukawa, Y.; Shigeta, A.; Miyasaka, M.; Hirata, T. CD43 Functions as a Ligand for E-Selectin on Activated T Cells. J. Immunol. 2005, 175, 8042–8050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natoni, A.; Smith, T.A.G.; Keane, N.; McEllistrim, C.; Connolly, C.; Jha, A.; Andrulis, M.; Ellert, E.; Raab, M.S.; Glavey, S.V.; et al. E-selectin ligands recognised by HECA452 induce drug resistance in myeloma, which is overcome by the E-selectin antagonist, GMI-1271. Leukemia 2017, 31, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- Natoni, A.; Smith, T.A.; Keane, N.; Locatelli-Hoops, S.C.; Oliva, I.; Fogler, W.E.; Magnani, J.L.; O’Dwyer, M. E-Selectin Ligand Expression Increases with Progression of Myeloma and Induces Drug Resistance in a Murine Transplant Model, Which Is Overcome By the Glycomimetic E-Selectin Antagonist, GMI-1271. Blood 2015, 126, 1805. [Google Scholar] [CrossRef]
- Yasmin-Karim, S.; King, M.R.; Messing, E.M.; Lee, Y.-F. E-selectin ligand-1 controls circulating prostate cancer cell rolling/adhesion and metastasis. Oncotarget 2014, 5, 12097–12110. [Google Scholar] [CrossRef] [Green Version]
- Xiaobo, M.; Zhong, Y.-P.; Zheng, Y.; Jiang, J.; Wang, Y.-P. Coexpression of CD 5 and CD 43 predicts worse prognosis in diffuse large B-cell lymphoma. Cancer Med. 2018, 7, 4284–4295. [Google Scholar] [CrossRef]
- Dimitroff, C.J.; Descheny, L.; Trujillo, N.; Kim, R.; Nguyen, V.; Huang, W.; Pienta, K.J.; Kutok, J.L.; Rubin, M.A. Identification of Leukocyte E-Selectin Ligands, P-Selectin Glycoprotein Ligand-1 and E-Selectin Ligand-1, on Human Metastatic Prostate Tumor Cells. Cancer Res. 2005, 65, 5750–5760. [Google Scholar] [CrossRef] [Green Version]
- Sorigue, M.; Juncà, J.; Sarrate, E.; Grau, J. Expression of CD43 in chronic lymphoproliferative leukemias. Cytom. Part B Clin. Cytom. 2017, 94, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Cash, S.E.; Andersen, J.J.; Kennedy, C.R.; Oldenburg, D.G.; Zander, V.B.; Foley, G.R.; Shelley, C.S. CD43 in the nucleus and cytoplasm of lung cancer is a potential therapeutic target. Int. J. Cancer 2012, 132, 1761–1770. [Google Scholar] [CrossRef]
- Krause, D.S.; Lazarides, K.; Von Andrian, U.H.; A Van Etten, R. Requirement for CD44 in homing and engraftment of BCR-ABL–expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar] [CrossRef]
- Godavarthy, P.S.; Kumar, R.; Herkt, S.C.; Pereira, R.S.; Hayduk, N.; Weissenberger, E.S.; Aggoune, D.; Manavski, Y.; Lucas, T.; Pan, K.-T.; et al. The vascular bone marrow niche influences outcome in chronic myeloid leukemia via the E-selectin - SCL/TAL1 - CD44 axis. Haematologica 2019, 105, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.-C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.-H.; et al. Bone vascular niche E-selectin induces mesenchymal–epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Läubli, H.; Borsig, L. Selectins as Mediators of Lung Metastasis. Cancer Microenviron. 2010, 3, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.; Haq, S.U.; Pawlus, P.M.; Moon, P.R.T.; Estey, M.E.H.; Appelbaum, F.R.; Othus, M.; Magnani, J.L.; Becker, P.S. Adhesion Of Acute Myeloid Leukemia Blasts To E-Selectin In The Vascular Niche Enhances Their Survival By Mechanisms Such As Wnt Activation. Blood 2013, 122, 61. [Google Scholar] [CrossRef]
- Muz, B.; De La Puente, P.; Azab, F.; Luderer, M.; Azab, A.K. Hypoxia promotes stem cell-like phenotype in multiple myeloma cells. Blood Cancer J. 2014, 4, e262. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Moreno, M.; Leiva, M.; Aguilera-Montilla, N.; Sevilla-Movilla, S.; De Val, S.I.; Arellano-Sánchez, N.; Gutiérrez, N.C.; Maldonado, R.; Martínez-López, J.; Buño, I.; et al. In vivo adhesion of malignant B cells to bone marrow microvasculature is regulated by α4β1 cytoplasmic-binding proteins. Leukemia 2016, 30, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Winkler, I.G.; Barbier, V.; Perkins, A.C.; Magnani, J.L.; Levesque, J.-P. Mobilisation of Reconstituting HSC Is Boosted By Synergy Between G-CSF and E-Selectin Antagonist GMI-1271. Blood 2014, 124, 317. [Google Scholar] [CrossRef]
- Chien, S.; Zhao, X.; Brown, M.; Saxena, A.; Patton, J.T.; Magnani, J.L.; Becker, P.S. A Novel Small Molecule E-Selectin Inhibitor GMI-1271 Blocks Adhesion of AML Blasts to E-Selectin and Mobilizes Blood Cells in Nodscid IL2Rgc-/- Mice Engrafted with Human AML. Blood 2012, 120. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Q.; Mu, H.; Battula, V.L.; Patel, N.; Schober, W.; Han, X.; Fogler, W.E.; Magnani, J.L.; Andreeff, M. Dual E-Selectin/CXCR4 Antagonist GMI-1359 Exerts Efficient Anti-Leukemia Effects in a FLT3 ITD Mutated Acute Myeloid Leukemia Patient-Derived Xenograft Murine Model. Blood 2016, 128, 3519. [Google Scholar] [CrossRef]
- Muz, B.; De La Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, I.G.; Barbier, V.; Nutt, H.; Hasnain, S.Z.; Levesque, J.P.; Magnani, J.L.; McGuckin, M.A. Administration of E-selectin Antagonist GMI-1271 Improves Survival to High-Dose Chemotherapy by Alleviating Mucositis and Accelerating Neutrophil Recovery. Available online: http://glycomimetics.com/wp-content/uploads/2018/11/Ingrid-mucositis-poster-layout-29nov13-5pm-Compatibility-Mode.pdf (accessed on 1 January 2020).
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone Marrow Microenvironment in Multiple Myeloma Progression. J. Biomed. Biotechnol. 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Muz, B.; De La Puente, P.; Azab, F.; Luderer, M.; Azab, A.K. The Role of Hypoxia and Exploitation of the Hypoxic Environment in Hematologic Malignancies. Mol. Cancer Res. 2014, 12, 1347–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogler, W.E.; Smith, T.A.; King, R.K.; Magnani, J.L. Abstract 1757: Mobilization of tumor-primed, marrow-infiltrating lymphocytes into peripheral blood with inhibitors of E-selectin or E-selectin and CXCR4. Immunology 2018, 78, 1757. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Jonas, B.A.; Becker, P.S.; O’Dwyer, M.; Advani, A.S.; Marlton, P.; Magnani, J.; Thackray, H.M.; Liesveld, J. GMI-1271, a novel E-selectin antagonist, combined with induction chemotherapy in elderly patients with untreated AML. J. Clin. Oncol. 2017, 35, 2560. [Google Scholar] [CrossRef]
- Winkler, I.G.; Barbier, V.; Pattabiraman, D.R.; Gonda, T.J.; Magnani, J.L.; Levesque, J. Vascular Niche E-Selectin Protects Acute Myeloid Leukemia Stem Cells from Chemotherapy. Blood 2014, 124. [Google Scholar] [CrossRef]
- Fogler, W.E.; Flanner, H.; Wolfgang, C.; Smith, J.A.; Thackray, H.M.; Magnani, J.L. Administration of the Dual E-Selectin/CXCR4 Antagonist, GMI-1359, Results in a Unique Profile of Tumor Mobilization from the Bone Marrow and Facilitation of Chemotherapy in a Murine Model of FLT3 ITD AML. Blood 2016, 128, 2826. [Google Scholar] [CrossRef]
- Zhang, W.; Fogler, W.E.; Magnani, J.L.; Andreeff, M. The Dual E-Selectin/CXCR4 Inhibitor, GMI-1359, Enhances Efficacy of Anti-Leukemia Chemotherapy in FLT3-ITD Mutated Acute Myeloid Leukemia. Blood 2015, 126, 3790. [Google Scholar] [CrossRef]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef] [Green Version]
- Fei, M.; Hang, Q.; Hou, S.; He, S.; Ruan, C. Adhesion to fibronectin induces p27Kip1 nuclear accumulation through down-regulation of Jab1 and contributes to cell adhesion-mediated drug resistance (CAM-DR) in RPMI 8226 cells. Mol. Cell. Biochem. 2013, 386, 177–187. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Jonas, B.A.; Liesveld, J.L.; Bixby, D.L.; Advani, A.S.; Marlton, P.; O’Dwyer, M.E.; Magnani, J.L.; Thackray, H.M.; Becker, P.S. GMI-1271 improves efficacy and safety of chemotherapy in R/R and newly diagnosed older patients with AML: Results of a Phase 1/2 study. Blood 2017, 130. [Google Scholar] [CrossRef]
- Devata, S.; Sood, S.L.; Hemmer, M.M.V.; Flanner, M.H.; Kramer, W.; Nietubicz, C.; Hawley, A.; E Angelini, D.; Myers, D.D.D.; Blackburn, S.; et al. First in Human Phase 1 Single Dose Escalation Studies of the E-Selectin Antagonist GMI-1271 Show a Favorable Safety, Pharmacokinetic, and Biomarker Profile. Blood 2015, 126, 1004. [Google Scholar] [CrossRef]
- Devata, S.; Angelini, D.E.; Rn, M.S.B.; Hawley, A.; Myers, D.D.; Schaefer, J.K.; Ma, M.H.; Magnani, J.L.; Thackray, H.M.; Wakefield, T.W.; et al. Use of GMI-1271, an E-selectin antagonist, in healthy subjects and in 2 patients with calf vein thrombosis. Res. Pr. Thromb. Haemost. 2020, 4, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeAngelo, D.J.; Erba, H.; Jonas, B.A.; O’Dwyer, M.E.; Marlton, P.; Huls, G.; Liesveld, J.L.; Cooper, B.; Bhatnagar, B.; Armstrong, M.; et al. Trials in Progress: A phase 3 trial to evaluate the efficacy of Uproleselan (GMI-1271) with chemotherapy in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Myers, J.D.; Lester, P.; Adili, R.; Hawley, A.; Durham, L.; Dunivant, V.; Reynolds, G.; Crego, K.; Zimmerman, Z.; Sood, S.; et al. A new way to treat proximal deep venous thrombosis using E-selectin inhibition. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 268–278. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| E-Selectin Ligand | Full Name | Expression in Cancer | References |
|---|---|---|---|
| ESL-1 | E-selectin ligand | Prostate | [47,49] |
| PSGL-1 | P-selectin glycoprotein ligand-1; CD162 | MM AML | [15,18,22] [40,42] |
| L-selectin | CD62L | CLL | [36] |
| CD43 | Leukosialin, sialophorin, galactoglycoprotein | ALL DLBCL CLL Lung | [43] [48] [50] [51] |
| CD44 | Homing cell adhesion molecule 1 (HCAM1) | AML Breast | [42,52] [35] |
| DR-3 | Death receptor 3 | Colon | [31,32] |
| CLA | Cutaneous lymphocyte-associated antigen | AML MM | [17] [47,49,53] |
| E-Selectin-Mediated Function | Signaling Pathway | Role | Reference |
|---|---|---|---|
| Cell Trafficking and metastasis | p38 ERK/MAPK | Pro-migratory | [31] [32] |
| Adhesion and tumor growth | NF-kB and PI3K ERK/AKT Wnt | Pro-survival Antiapoptotic | [32] [42,55,56] [57] |
| Stemness and self-renewal | Wnt Hedgehog | Maintaining stemness | [54,57] |
| Drug resistance | ERK/AKT NF-kB | Chemoresistance Pro-survival | [32,42,55,56] |
| Role of Uproleselan in Cancer | Results | Reference |
|---|---|---|
| Metastasis | Prevented MM dissemination Inhibited pancreatic ductal adenocarcinoma to the lymph nodes, as well as to the liver, lung and diaphragm in combination with gemcitabine Inhibited breast cancer metastasis to the bone marrow | [23] [27] [54] |
| Adhesion | Decreased the adhesion of cancer cells to stromal and endothelial cells in vitro Reduced adhesion of CML leukemic stem cells to E-selectin in the vascular niche | [23,45] [53] |
| Mobilization | Enhanced mobilization of cancer cells out of the bone marrow into the circulation Mobilized myeloma and leukemic cells from the marrow into the peripheral blood after a single injection Activated the tumor-reactive and tumor-specific marrow infiltrating lymphocytes | [62] [23,45] [68] |
| Cancer stem-cell like | Inhibited cancer (stem) cell quiescence and induced cell maturation Resensitized leukemic stem cell to chemotherapy in AML-bearing mice | [57,64,65,69] [16,70] |
| Chemotherapy sensitization | Improved CML killing in combination with imatinib Sensitized AML in combination with daunorubicin (DNR) and cytarabine (AraC) in different mouse models (syngeneic, xenogeneic and patient blasts) Overcame MM drug resistance and improved the efficacy to proteasome inhibitors (bortezomib and carfilzomib) and IMiDs (lenalidomide) | [53] [16,62] [23,47,49,53] |
| Reducing adverse events | Reduced bone marrow toxicity including neutropenia, protected and increased percentile of HSCs, enhanced neutrophilic recovery, reduced small intestine mucositis by decreasing the number of infiltrating inflammatory macrophages | [16,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muz, B.; Abdelghafer, A.; Markovic, M.; Yavner, J.; Melam, A.; Salama, N.N.; Azab, A.K. Targeting E-selectin to Tackle Cancer Using Uproleselan. Cancers 2021, 13, 335. https://doi.org/10.3390/cancers13020335
Muz B, Abdelghafer A, Markovic M, Yavner J, Melam A, Salama NN, Azab AK. Targeting E-selectin to Tackle Cancer Using Uproleselan. Cancers. 2021; 13(2):335. https://doi.org/10.3390/cancers13020335
Chicago/Turabian StyleMuz, Barbara, Anas Abdelghafer, Matea Markovic, Jessica Yavner, Anupama Melam, Noha Nabil Salama, and Abdel Kareem Azab. 2021. "Targeting E-selectin to Tackle Cancer Using Uproleselan" Cancers 13, no. 2: 335. https://doi.org/10.3390/cancers13020335
APA StyleMuz, B., Abdelghafer, A., Markovic, M., Yavner, J., Melam, A., Salama, N. N., & Azab, A. K. (2021). Targeting E-selectin to Tackle Cancer Using Uproleselan. Cancers, 13(2), 335. https://doi.org/10.3390/cancers13020335