Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
- Immunogenic cell death (ICD) of H-1PV-infected tumor cells (indirect immune cell stimulation): PVs are potent triggers of immunogenic stimuli through tumor cell ICD induction. Infected tumor cells release a spectrum of proinflammatory mediators, in particular chemo- and cyto-kines, and pathogen- and danger-associated molecular patterns (PAMPs, DAMPs), which are in turn capable of boosting the maturation and reactivity of distinct immune cell populations. This can be exemplified by H-1PV-infected human melanoma cells, which activate dendritic cell (DC) maturation through the release of heat shock protein 72 [14]. In line with this observation, H-1PV-infected pancreatic and colorectal carcinoma cells were shown to stimulate natural killer (NK) cell tumor-killing capacity through both the overexpression of ligands specific for NK cell activation receptors and the downregulation of MHC I on infected tumor cells [15,16]. Notably, productive infection of tumor cells is not required for immune stimulation. This was demonstrated by co-incubating H-1PV-infected semi-permissive pancreatic carcinoma cells with peripheral blood mononuclear cells (PBMC), under which conditions induction of Th1 signature and release of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) were detected in the PBMC population [17].
- H-1PV infection of immune cells (direct immune cell stimulation): H-1PV infection of human immune cell subpopulations has been documented in various preclinical settings. Virus entry may take place in T, B, NK, DC and monocytic populations; however, infection is aborted at subsequent virus intracellular replication steps [18]. Abortive infection can nevertheless exert multiple immuno-stimulating effects, such as expression of IFN-stimulated genes and proinflammatory cytokine production [17,18]. On the other hand, H-1PV is able to inhibit the immune suppressive activity of regulatory T (Treg) cells [18].
- H-1PV impact on tumor vasculature: It has been demonstrated that endothelial (precursor) cells may constitute direct targets for parvovirus-mediated toxicity. These cells sustain an abortive H-1PV infection in vitro. In animal models, virus treatment inhibits the growth of lymphatic endothelium-derived tumors (Kaposi’s sarcoma). Furthermore, recombinant propagation-deficient parvoviral vectors armed with angiostatic chemokines achieve significant reduction of vascular endothelial growth factor (VEGF) expression in Kaposi’s sarcoma cells [19]. Given the control exerted by the vasculature of tumors over their infiltration with immune cells, these effects are likely to contribute to H-1PV immuno-stimulating activity, as further discussed below. Altogether, these data warrant validation of H-1PV as a tool against highly vascularized cancers, e.g., glioblastoma, one of the most angiogenic human tumors.
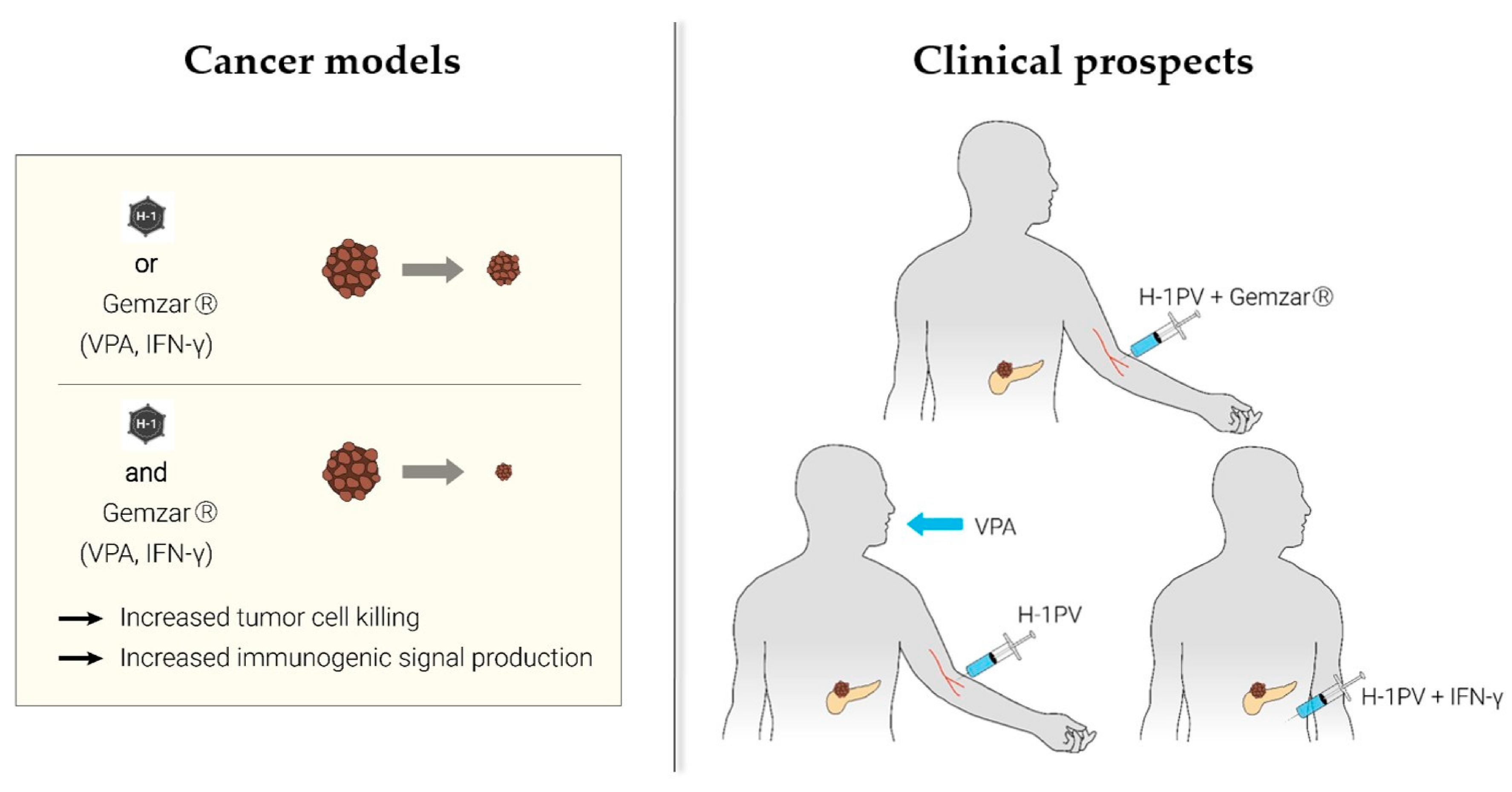
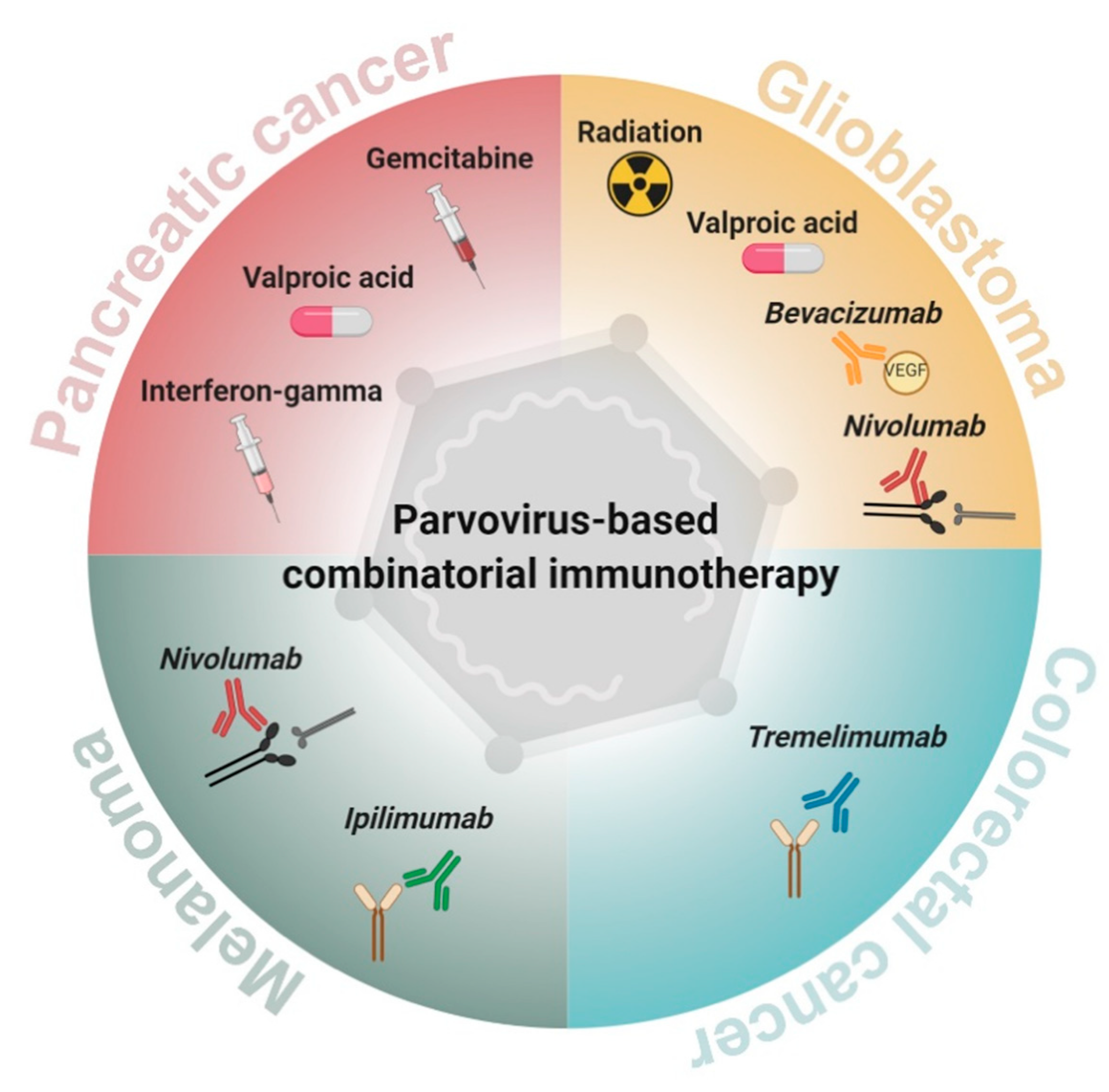
2. Parvovirus-Based Combinatorial Immunotherapy against Pancreatic Cancer
2.1. H-1PV + Nucleoside Analogues (Gemcitabine)
- One study conducted in gemcitabine-treated PDAC patients revealed the ability of the drug to enhance T cell-mediated and DC-dependent host immune responses [25].
- In keeping with the aforementioned data, it was documented that gemcitabine therapy may promote naïve T cell activation in PDAC patients and enhance their responsiveness to specific vaccination or to other forms of immunotherapy [26].
- The understanding of gemcitabine immunoregulating effects as a complementary constituent of tumor cell toxicity was extended by the demonstration that this drug alleviates pancreatic cancer immune escape through NK cell cytotoxicity enhancement [27].
- Studies conducted in murine orthotopic PDAC models provided yet another insight into gemcitabine-mediated immuno-stimulation, namely by indicating that low chemotherapeutic doses selectively deplete effector/memory Treg cell populations. The latter has a strong impact on PDAC microenvironment, as Tregs usually form large intra-tumoral infiltrates and trigger local immune suppression [28,29].
- Last but not least, in cancer models other than PDAC, gemcitabine enhances the efficacy of OV (e.g., reovirus) therapy. This complementation is achieved through gemcitabine-mediated inhibition of myeloid-derived suppressor cell (MDSC) recruitment to the TME and acceleration of reovirus-induced antitumor T cell immune responses [30].
2.2. H-1PV + Histone Deacetylase Inhibitors (Valproic Acid)
2.3. H-1PV + Proinflammatory Cytokines (Interferon-Gamma)
3. Parvovirus-Based Combinatorial Immunotherapy against Glioblastoma
3.1. H-1PV + Ionizing Radiation
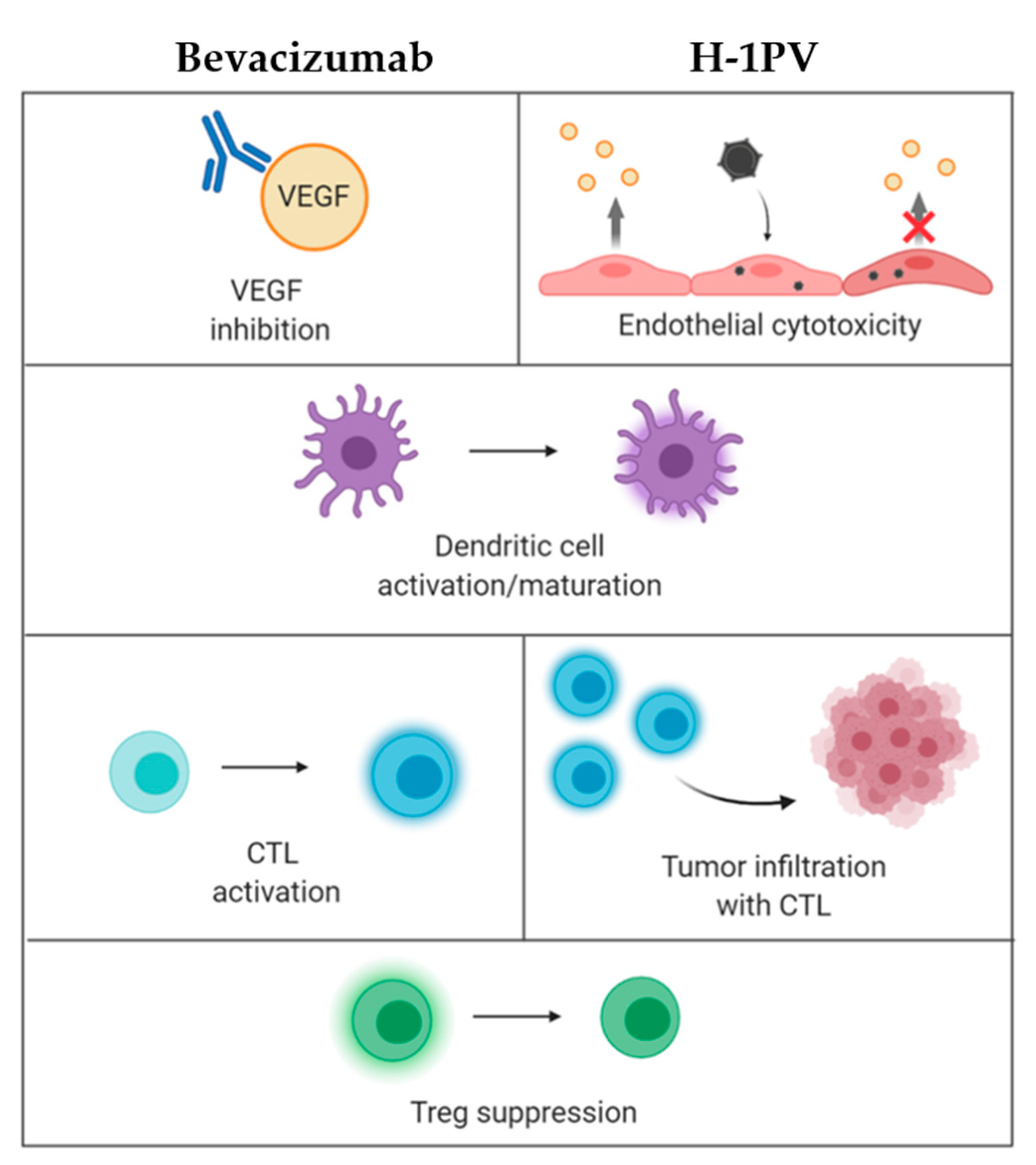
3.2. H-1PV + Tumor Angiogenesis Inhibitors (Bevacizumab)
3.3. H-1PV + PD-1 Immune Checkpoint Inhibitors (Nivolumab)
4. Parvovirus-Based Combinatorial Immunotherapy against Colorectal Cancer
H-1PV + CTLA-4 Immune Checkpoint Blockade (Tremelimumab)
5. Parvovirus-Based Combinatorial Immunotherapy against Melanoma
H-1PV + CTLA-4 (Ipilimumab)/PD-1 (Nivolumab) Immune Checkpoint Blockade
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bretscher, C.; Marchini, A. H-1 parvovirus as a cancer-killing agent: Past, present, and future. Viruses 2019, 11, 562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toolan, H.W.; Dalldore, G.; Barclay, M.; Chandra, S.; Moore, A.E. An unidentified, filtrable agent isolated from transplanted human tumors. Proc. Natl. Acad. Sci. USA 1960, 46, 1256–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besselsen, D.G.; Besch-Williford, C.L.; Pintel, D.J.; Franklin, C.L.; Hook, R.R.; Riley, L.K. Detection of H-1 parvovirus and Kilham rat virus by PCR. J. Clin. Microbiol. 1995, 33, 1699–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rommelaere, J.; Giese, N.; Cziepluch, C.; Cornelis, J.J. Parvoviruses as anticancer agents. In Viral Therapy of Human Cancers; Sinkovics, J.G., Horvath, J.C., Eds.; Marcel Dekker: New York, NY, USA, 2005; pp. 627–675. [Google Scholar]
- Rommelaere, J.; Geletneky, K.; Angelova, A.L.; Daeffler, L.; Dinsart, C.; Kiprianova, I.; Schlehofer, J.R.; Raykov, Z. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 2010, 21, 185–195. [Google Scholar] [CrossRef]
- Angelova, A.L.; Geletneky, K.; Nüesch, J.P.F.; Rommelaere, J. Tumor selectivity of oncolytic parvoviruses: From in vitro and animal models to cancer patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Newman, S.J.; McCallin, P.F.; Sever, J.L. Attempts to isolate H-1 virus from spontaneous human abortions: A negative report. Teratology 1970, 3, 279–281. [Google Scholar] [CrossRef]
- Toolan, H.W.; Saunders, E.L.; Southam, C.M.; Moore, A.E.; Levin, A.G. H-1 virus viremia in the human. Exp. Biol. Med. 1965, 119, 711–715. [Google Scholar] [CrossRef]
- Le Cesne, A.; Dupressoir, T.; Janin, N.; Spielmann, M.; Le Chevalier, T.; Sancho-Garnier, H.; Paoletti, C.; Rommelaere, J.; Stehelin, D.; Tursz, T.; et al. Intralesional administration of a live virus, parvovirus H1 (PVH1) in cancer patients: A feasibility study. Proc. Am. Soc. Clin. Oncol. 1993, 12, 297. [Google Scholar]
- Hartley, A.; Kavishwar, G.; Salvato, I.; Marchini, A. A roadmap for the success of oncolytic parvovirus-based anticancer therapies. Annu. Rev. Virol. 2020, 7, 537–557. [Google Scholar] [CrossRef]
- Geletneky, K.; Nüesch, J.P.; Angelova, A.L.; Kiprianova, I.; Rommelaere, J. Double-faceted mechanism of parvoviral oncosuppression. Curr. Opin. Virol. 2015, 13, 17–24. [Google Scholar] [CrossRef]
- Angelova, A.L.; Rommelaere, J. Immune System Stimulation by Oncolytic Rodent Protoparvoviruses. Viruses 2019, 11, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune conversion of tumor microenvironment by oncolytic viruses: The protoparvovirus H-1PV case study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Zeidler, M.; Schede, J.; Rommelaere, J.; Galle, P.R.; Cornelis, J.J.; Heike, M. Oncolytic parvovirus H1 induces release of heat-shock protein HSP72 in susceptible human tumor cells but may not affect primary immune cells. Cancer Gene Ther. 2003, 10, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Dempe, S.; Dinsart, C.; Rommelaere, J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int. J. Cancer 2010, 128, 908–919. [Google Scholar] [CrossRef]
- Bhat, R.; Rommelaere, J. NK-cell-dependent killing of colon carcinoma cells is mediated by natural cytotoxicity receptors (NCRs) and stimulated by parvovirus infection of target cells. BMC Cancer 2013, 13, 367. [Google Scholar] [CrossRef] [Green Version]
- Grekova, S.P.; Aprahamian, M.; Giese, N.; Schmitt, S.; Giese, T.; Falk, C.S.; Daeffler, L.; Cziepluch, C.; Rommelaere, J.; Raykov, Z. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol. Ther. 2010, 10, 1280–1289. [Google Scholar] [CrossRef] [Green Version]
- Moralès, O.; Richard, A.; Martin, N.; Mrizak, D.; Sénéchal, M.; Miroux, C.; Pancré, V.; Rommelaere, J.; Caillet-Fauquet, P.; De Launoit, Y.; et al. Activation of a Helper and Not Regulatory Human CD4+ T Cell Response by Oncolytic H-1 Parvovirus. PLoS ONE 2012, 7, e32197. [Google Scholar] [CrossRef] [Green Version]
- Lavie, M.; Struyf, S.; Stroh-Dege, A.; Rommelaere, J.; Van Damme, J.; Dinsart, C. Capacity of wild-type and chemokine-armed parvovirus H-1PV for inhibiting neo-angiogenesis. Virology 2013, 447, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, M.; Hruban, R.H.; Wood, L.D. New developments in the molecular mechanisms of pancreatic tumorigenesis. Adv. Anat. Pathol. 2018, 25, 131–142. [Google Scholar] [CrossRef]
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2017. CA A Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Aprahamian, M.; Grekova, S.P.; Hajri, A.; Leuchs, B.; Giese, N.A.; Dinsart, C.; Herrmann, A.; Balboni, G.; Rommelaere, J.; et al. Improvement of Gemcitabine-Based Therapy of Pancreatic Carcinoma by Means of Oncolytic Parvovirus H-1PV. Clin. Cancer Res. 2009, 15, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, A.L.; Grekova, S.P.; Heller, A.; Kuhlmann, O.; Soyka, E.; Giese, T.; Aprahamian, M.; Bour, G.; Rüffer, S.; Cziepluch, C.; et al. Complementary Induction of Immunogenic Cell Death by Oncolytic Parvovirus H-1PV and Gemcitabine in Pancreatic Cancer. J. Virol. 2014, 88, 5263–5276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plate, J.M.D.; Harris, J.E. Effects of gemcitabine treatment on immune cells and functions in pancreatic cancer patients. Cancer Res. 2004, 64 (Suppl. 7), 500. [Google Scholar]
- Plate, J.M.D.; Plate, A.E.; Shott, S.; Bograd, S.; Harris, J.E. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol. Immunother. 2005, 54, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Huang, M.; Xie, F.; Zhou, H.; Yang, J.; Huang, Q. Gemcitabine inhibits immune escape of pancreatic cancer by down regulating the soluble ULBP2 protein. Oncotarget 2016, 7, 70092–70099. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, I.; Karakhanova, S.; Soltek, S.; Link, J.; Bayry, J.; Werner, J.; Umansky, V.; Bazhin, A.V. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int. J. Cancer 2013, 133, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Taniguchi, K.; Nakazawa, M.; Matsuyama, R.; Mori, R.; Takeda, K.; Ichikawa, Y.; Tanaka, K.; Endo, I. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin. Transl. Oncol. 2014, 16, 330–335. [Google Scholar] [CrossRef]
- Gujar, S.A.; Clements, D.A.; Dielschneider, R.; Helson, E.; Marcato, P.S.; Lee, P.W. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br. J. Cancer 2014, 110, 83–93. [Google Scholar] [CrossRef]
- Hajda, J.; Lehmann, M.; Krebs, O.; Kieser, M.; Geletneky, K.; Jäger, D.; Dahm, M.; Huber, B.; Schöning, T.; Sedlaczek, O.; et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer 2017, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- ParvOryx02: A Phase II Trial of Intravenous and Intratumoral Administration of H-1 Parvovirus in Patients with Metastatic Pancreatic Cancer. Available online: http://oryx-medicine.com/fileadmin/user_upload/uploads/News/Publications/201910_IOVC_Ungerechts_ParvOryx02.pdf (accessed on 16 November 2020).
- Li, J.; Bonifati, S.; Hristov, G.; Marttila, T.; Valmary-Degano, S.; Stanzel, S.; Schnölzer, M.; Mougin, C.; Aprahamian, M.; Grekova, S.P.; et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1 PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol. Med. 2013, 5, 1537–1555. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Breckenridge, C.A.; Yu, J.; Price, R.L.; Wei, M.; Wang, Y.; Nowicki, M.O.; Ha, Y.P.; Bergin, S.M.; Hwang, C.; Fernandez, S.A.; et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J. Virol. 2012, 86, 4566–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuki, A.; Patel, A.; Kasai, K.; Suzuki, M.; Kurozumi, K.; Chiocca, E.A.; Saeki, Y. Histone Deacetylase Inhibitors Augment Antitumor Efficacy of Herpes-based Oncolytic Viruses. Mol. Ther. 2008, 16, 1546–1555. [Google Scholar] [CrossRef]
- VanOosten, R.L.; Earel, J.K., Jr.; Griffith, T.S. Histone deacetylase inhibitors enhance Ad5-TRAIL killing of TRAIL-resistant prostate tumor cells through increased caspase-2 activity. Apoptosis 2006, 12, 561–571. [Google Scholar] [CrossRef]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Ago, Y.; Okada, N.; Tachibana, M. Valproic acid attenuates immunosuppressive function of myeloid-derived suppressor cells. J. Pharmacol. Sci. 2018, 137, 359–365. [Google Scholar] [CrossRef]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural Killer Cell–Mediated Lysis of Hepatoma Cells via Specific Induction of NKG2D Ligands by the Histone Deacetylase Inhibitor Sodium Valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef] [Green Version]
- Lugrin, J.; Ding, X.C.; Le Roy, D.; Chanson, A.-L.; Sweep, F.C.; Calandra, T.; Roger, T. Histone deacetylase inhibitors repress macrophage migration inhibitory factor (MIF) expression by targeting MIF gene transcription through a local chromatin deacetylation. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Grekova, S.P.; Aprahamian, M.; Daeffler, L.; Leuchs, B.; Angelova, A.; Giese, T.; Galabov, A.; Heller, A.; Giese, N.A.; Rommelaere, J.; et al. Interferon γ improves the vaccination potential of oncolytic parvovirus H-1PV for the treatment of peritoneal carcinomatosis in pancreatic cancer. Cancer Biol. Ther. 2011, 12, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.-Y.; Gao, P.; Sun, Y.; Duan, Y. Development of targeted therapies in treatment of glioblastoma. Cancer Biol. Med. 2015, 12, 223–237. [Google Scholar] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Calle, M.H.Y.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic Activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and trail-resistant glioma cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef] [Green Version]
- Geletneky, K.; Kiprianova, I.; Ayache, A.; Koch, R.; Herrero y Calle, M.; Deleu, L.; Sommer, C.; Thomas, N.; Rommelaere, J.; Schlehofer, J.R. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol. 2010, 12, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Kiprianova, I.; Thomas, N.; Ayache, A.; Fischer, M.; Leuchs, B.; Klein, M.; Rommelaere, J.; Schlehofer, J.R. Regression of glioma in rat models by intranasal application of parvovirus H-1. Clin. Cancer Res. 2011, 17, 5333–5342. [Google Scholar] [CrossRef] [Green Version]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [Green Version]
- Angelova, A.L.; Barf, M.; Geletneky, K.; Unterberg, A.; Rommelaere, J. Immunotherapeutic potential of oncolytic H-1 parvovirus: Hints of glioblastoma microenvironment conversion towards immunogenicity. Viruses 2017, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Chonan, M.; Saito, R.; Shoji, T.; Shibahara, I.; Kanamori, M.; Sonoda, Y.; Watanabe, M.; Kikuchi, T.; Ishii, N.; Tominaga, T. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol. 2015, 17, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Geletneky, K.; Hartkopf, A.D.; Krempien, R.; Rommelaere, J.; Schlehofer, J.R. Improved killing of human high-grade glioma cells by combining ionizing radiation with oncolytic parvovirus H-1 infection. J. Biomed. Biotechnol. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slone, H.B.; Peters, L.J.; Milas, L. Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J. Natl. Cancer Inst. 1979, 63, 1229–1235. [Google Scholar] [CrossRef]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: Using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Limbergen, E.J.; De Ruysscher, D.K.; Pimentel, V.O.; Marcus, D.; Berbee, M.; Hoeben, A.; Rekers, N.H.; Theys, J.; Yaromina, A.; Dubois, L.J.; et al. Combining radiotherapy with immunotherapy: The past, the present and the future. Br. J. Radiol. 2017, 90, 20170157. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Shao, C. Radiotherapy-Mediated Immunomodulation and Anti-Tumor Abscopal Effect Combining Immune Checkpoint Blockade. Cancers 2020, 12, 2762. [Google Scholar] [CrossRef]
- Touchefeu, Y.; Vassaux, G.; Harrington, K.J. Oncolytic viruses in radiation oncology. Radiother. Oncol. 2011, 99, 262–270. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Geletneky, K.; Angelova, A.; Leuchs, B.; Bartsch, A.; Capper, D.; Hajda, J.; Rommelaere, J. ATNT-07. Favorable response of patients with glioblastoma at second or third recurrence to repeated injection of oncolytic parvovirus H-1 in combination with bevacicumab. Neuro Oncol. 2015, 17, v11. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; De Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro Oncol. 2017, 19, iii21. [Google Scholar] [CrossRef] [Green Version]
- Desai, K.; Hubben, A.; Ahluwalia, M. The role of checkpoint inhibitors in glioblastoma. Target. Oncol. 2019, 14, 375–394. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Weiss, C.; Bernhard, H.; Capper, D.; Leuchs, B.; Marchini, A.; Rommelaere, J. ATIM-29. First clinical observation of improved anti-tumor effects of viro-immunotherapy with oncolytic parvovirus H-1 in combination with PD-1 checkpoint blockade and bevacicumab in patients with recurrent glioblastoma. Neuro Oncol. 2016, 18, vi24. [Google Scholar] [CrossRef] [Green Version]
- Geletneky, K.; Bartsch, A.; Weiss, C.; Bernhard, H.; Marchini, A.; Rommelaere, J. ATIM-40. High rate of objective anti-tumor response in 9 patients with glioblastoma after viro-immunotherapy with oncolytic parvovirus H-1 in combination with bevacicumab and PD-1 checkpoint blockade. Neuro Oncol. 2018, 20, vi10. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; DeSantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA A Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Mazzolini, G. Immunotherapy and immunoescape in colorectal cancer. World J. Gastroenterol. 2007, 13, 5822–5831. [Google Scholar] [CrossRef]
- Heinrich, B.; Goepfert, K.; Delic, M.; Galle, P.R.; Moehler, M. Influence of the oncolytic parvovirus H-1, CTLA-4 antibody tremelimumab and cytostatic drugs on the human immune system in a human in vitro model of colorectal cancer cells. OncoTargets Ther. 2013, 6, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef]
- Goepfert, K.; Dinsart, C.; Rommelaere, J.; Foerster, F.; Moehler, M. Rational combination of parvovirus h1 with ctla-4 and pd-1 checkpoint inhibitors dampens the tumor induced immune silencing. Front. Oncol. 2019, 9, 425. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelova, A.; Ferreira, T.; Bretscher, C.; Rommelaere, J.; Marchini, A. Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers. Cancers 2021, 13, 342. https://doi.org/10.3390/cancers13020342
Angelova A, Ferreira T, Bretscher C, Rommelaere J, Marchini A. Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers. Cancers. 2021; 13(2):342. https://doi.org/10.3390/cancers13020342
Chicago/Turabian StyleAngelova, Assia, Tiago Ferreira, Clemens Bretscher, Jean Rommelaere, and Antonio Marchini. 2021. "Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers" Cancers 13, no. 2: 342. https://doi.org/10.3390/cancers13020342
APA StyleAngelova, A., Ferreira, T., Bretscher, C., Rommelaere, J., & Marchini, A. (2021). Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers. Cancers, 13(2), 342. https://doi.org/10.3390/cancers13020342