
Adrenocortical Carcinoma in Childhood: A Systematic Review

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
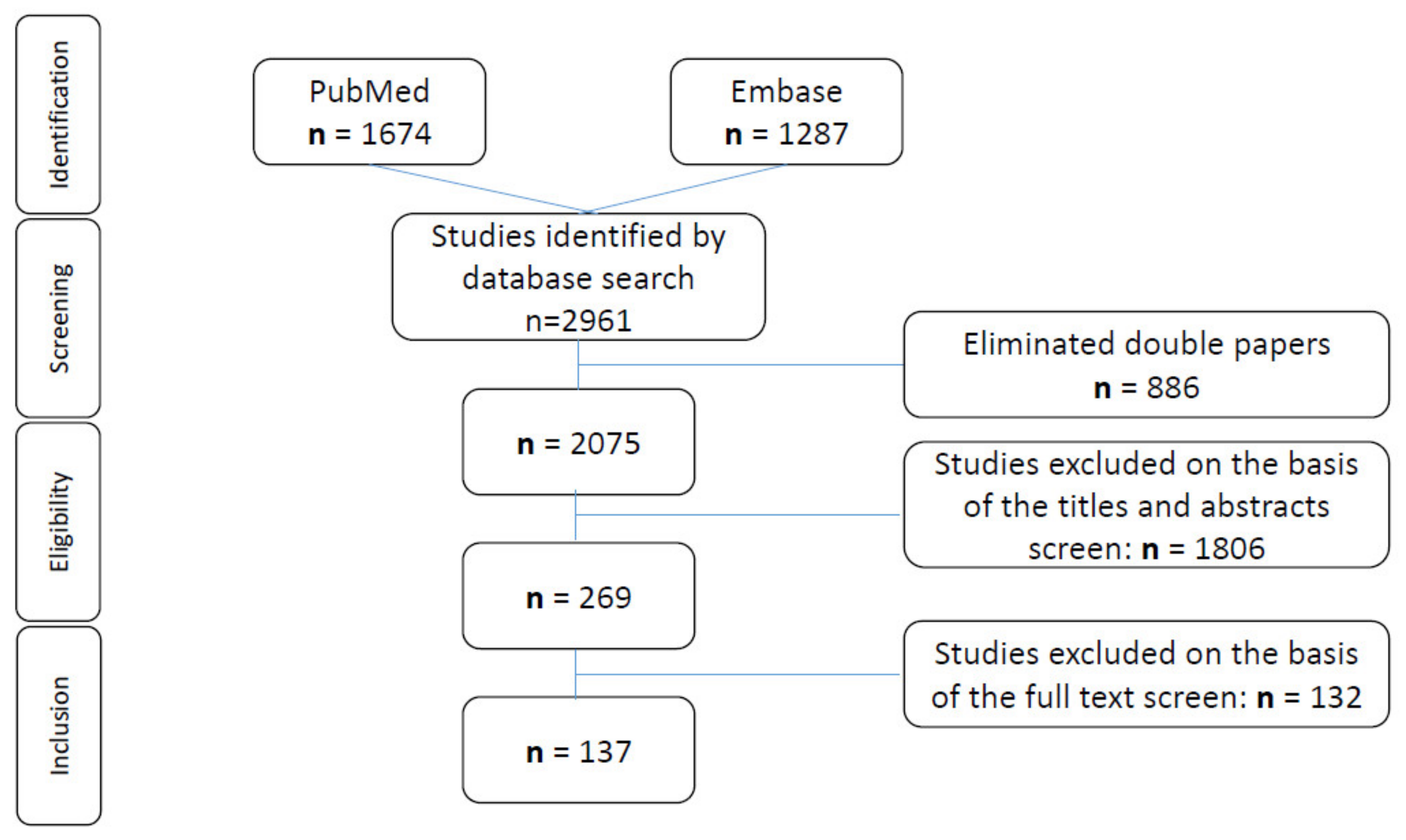
2. Methods
3. Results
3.1. Incidence
3.2. Clinical Characteristics
3.2.1. Brazilian Cohort
3.2.2. Non-Brazilian Cohort
3.2.3. Age-Dependent Clinical Characteristics
3.3. Staging
3.4. Metastasis
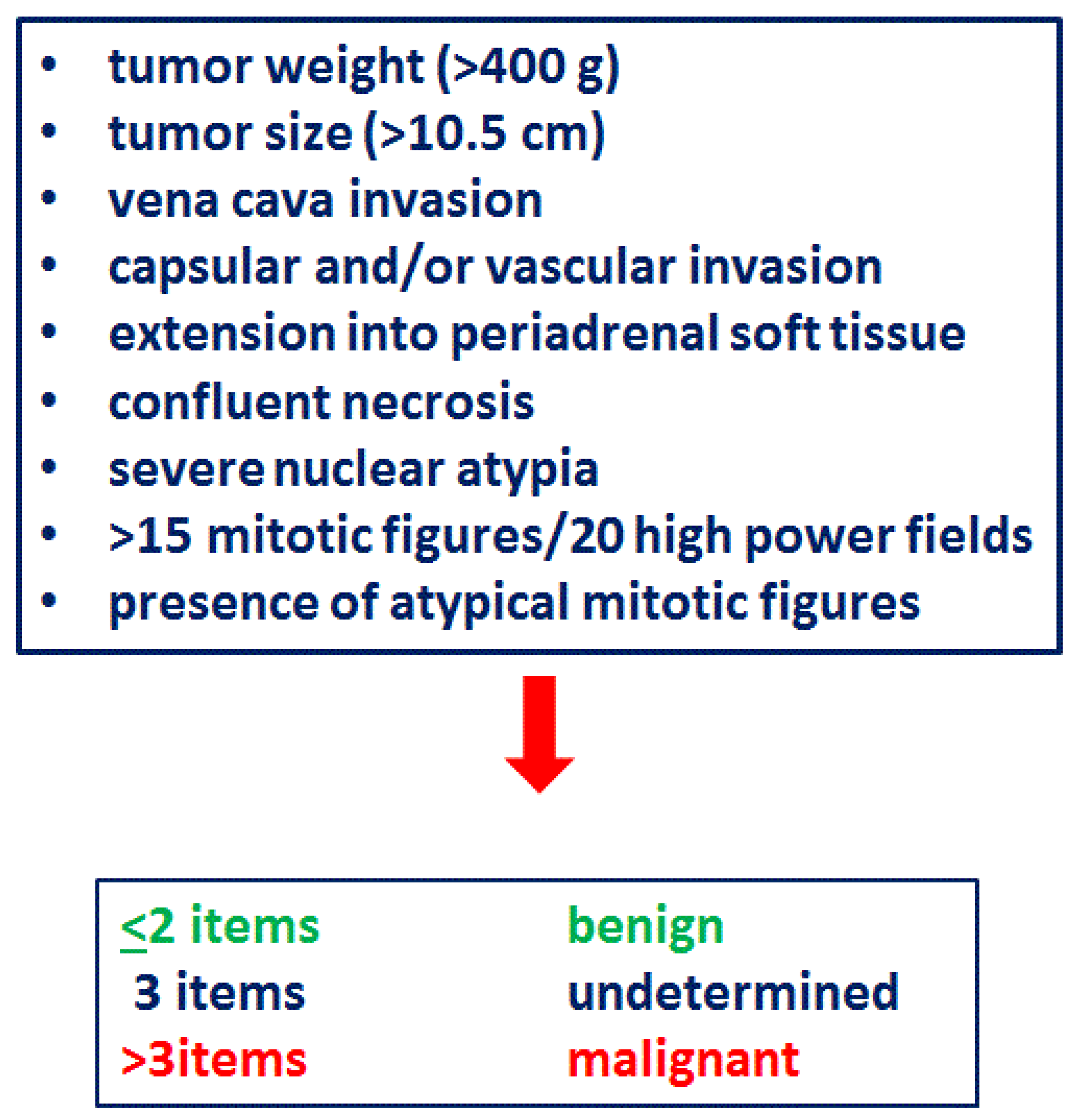
3.5. Pathological Characteristics
- (i)
- (ii)
- (iii)
- increased expression of silver-binding nucleolar organizer regions (agNOR type III, immunohistochemistry) [127];
- (iv)
- placental alkaline phosphatase (PLAP) was detected by immunohistochemical analysis in one third of prepubertal ACC [128];
- (v)
- 1p15 LOH as a widespread finding in pediatric ACT not related to malignancy [129].
3.6. Molecular Changes as Potential Targets
3.7. Prognostic Factors
3.8. Therapy
3.9. Secondary Malignancies and Cancer Predisposition Syndromes
4. Discussion
- What are the best prognostic markers at the time of diagnosis?
- What is the impact of surgery and how can it be optimized?
- Are there alternative tumor markers (to 24 h urine samples) that can be used more easily for follow-up investigations?
- How can we improve the outcomes in pediatric patients at advanced stages?
- How can we reduce the relapse rate?
- Are there specific molecular targets that can be used for tailored therapies?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Hubertus, J.; Boxberger, N.; Redlich, A.; Von Schweinitz, D.; Vorwerk, P. Surgical aspects in the treatment of adrenocortical carcinomas in children: Data of the GPOH-MET 97 trial. Klin. Padiatr. 2012, 224, 143–147. [Google Scholar] [CrossRef]
- Michalkiewicz, E.; Sandrini, R.; Figueiredo, B.; Miranda, E.C.; Caran, E.; Oliveira-Filho, A.G.; Marques, R.; Pianovski, M.A.; Lacerda, L.; Cristofani, L.M.; et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J. Clin. Oncol. 2004, 22, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Kerkhofs, T.M.; Ettaieb, M.H.; Verhoeven, R.H.; Kaspers, G.J.; Tissing, W.J.; Loeffen, J.; Van den Heuvel-Eibrink, M.M.; De Krijger, R.R.; Haak, H.R. Adrenocortical carcinoma in children: First population-based clinicopathological study with long-term follow-up. Oncol. Rep. 2014, 32, 2836–2844. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, G.; Sun, H.; Li, K.; Dong, K.; Ma, Y.; Zheng, S. Clinical characteristics and prognosis of adrenocortical tumors in children. Pediatr. Surg. Int. 2019, 35, 365–371. [Google Scholar] [CrossRef]
- Faria, A.M.; Almeida, M.Q. Differences in the molecular mechanisms of adrenocortical tumorigenesis between children and adults. Mol. Cell. Endocrinol. 2012, 351, 52–57. [Google Scholar] [CrossRef]
- Sedaghati, M.; Lin, D.T.; Cisco, R.M.; Kebebew, E. Pediatric vs Adult Adrenocortical Carcinoma: Different Rate of Surgical Treatment and Patient Outcome. J. Am. Coll. Surg. 2019, 229, S81–S82. [Google Scholar] [CrossRef]
- Zerbini, C.; Kozakewich, H.P.W.; Weinberg, D.S.; Mundt, D.J.; Edwards, J.A.; Lack, E.E. Adrenocortical neoplasms in childhood and adolescence: Analysis of prognostic factors including DNA content. Endocr. Pathol. 1992, 3, 116–128. [Google Scholar] [CrossRef]
- Dehner, L.P.; Hill, D.A. Adrenal cortical neoplasms in children: Why so many carcinomas and yet so many survivors? Pediatr. Dev. Pathol. 2009, 12, 284–291. [Google Scholar] [CrossRef]
- Wieneke, J.A.; Thompson, L.D.R.; Heffess, C.S. Adrenal cortical neoplasms in the pediatric population: A clinicopathologic and immunophenotypic analysis of 83 patients. Am. J. Surg. Pathol. 2003, 27, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.H.; Robinson, K.A.; Saldanha, I.; Anton, B.; Ladenson, P.W. Clinical review: Prevalence and incidence of endocrine and metabolic disorders in the United States: A comprehensive review. J. Clin. Endocrinol. Metab. 2009, 94, 1853–1878. [Google Scholar] [CrossRef] [PubMed]
- Petr, E.J.; Else, T. Adrenocortical carcinoma (ACC): When and why should we consider germline testing? Presse Med. 2018, 47, e119–e125. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, P. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]
- McAteer, J.P.; Huaco, J.A.; Gow, K.W. Predictors of survival in pediatric adrenocortical carcinoma: A Surveillance, Epidemiology, and End Results (SEER) program study. J. Pediatr. Surg. 2013, 48, 1025–1031. [Google Scholar] [CrossRef]
- Cronin, K.A.; Ries, L.A.; Edwards, B.K. The Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute. Cancer 2014, 120 (Suppl. 23), 3755–3757. [Google Scholar] [CrossRef]
- Brondani, V.B.; Fragoso, M. Pediatric adrenocortical tumor—Review and management update. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 177–186. [Google Scholar] [CrossRef]
- Ribeiro, R.C.; Pinto, E.M.; Zambetti, G.P.; Rodriguez-Galindo, C. The International Pediatric Adrenocortical Tumor Registry initiative: Contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol. Cell. Endocrinol. 2012, 351, 37–43. [Google Scholar] [CrossRef]
- Rodriguez-Galindo, C.; Pappo, A.S.; Krailo, M.D.; Pashankar, F.; Caran, E.M.M.; Hicks, J.; McCarville, M.B.; Weldon, C.B.; Malkin, D.; Zambetti, G.; et al. Treatment of childhood adrenocortical carcinoma (ACC) with surgery plus retroperitoneal lymph node dissection (RPLND) and multiagent chemotherapy: Results of the children’s oncology group ARAR0332 protocol. J. Clin. Oncol. 2016, 34, 10515. [Google Scholar] [CrossRef]
- Sandrini, F.; Villani, D.P.; Tucci, S.; Moreira, A.C.; de Castro, M.; Elias, L.L. Inheritance of R337H p53 gene mutation in children with sporadic adrenocortical tumor. Horm. Metab. Res. 2005, 37, 231–235. [Google Scholar] [CrossRef]
- Ribeiro, R.C.; Sandrini, F.; Figueiredo, B.; Zambetti, G.P.; Michalkiewicz, E.; Lafferty, A.R.; DeLacerda, L.; Rabin, M.; Cadwell, C.; Sampaio, G.; et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci. USA 2001, 98, 9330–9335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, E.M.; Billerbeck, A.E.; Villares, M.C.; Domenice, S.; Mendonça, B.B.; Latronico, A.C. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq. Bras. Endocrinol. Metabol. 2004, 48, 647–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garritano, S.; Gemignani, F.; Palmero, E.I.; Olivier, M.; Martel-Planche, G.; Le Calvez-Kelm, F.; Brugieres, L.; Vargas, F.R.; Brentani, R.R.; Ashton-Prolla, P.; et al. Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: Evidence for a founder effect. Hum. Mutat. 2010, 31, 143–150. [Google Scholar] [CrossRef]
- Leal, L.F. Wnt/β-Catenin Pathway Deregulation in Childhood Adrenocortical Tumors. J. Clin. Endocrinol. Metab. 2011, 96, 3106–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abduch, R.H.; Bueno, A.C.; Leal, L.F.; Cavalcanti, M.M.; Brandalise, S.R.; Masterallo, M.J.; Yunes, J.A.; Martinelli, C.E.; Scrideli, C.A.; Tone, L.G.; et al. Unraveling the expression of the oncogene YAP1, a wnt/beta-catenin target, in adrenal tumors and its association with poor outcome in pediatric patients. Oncotarget 2016, 7, 84634–84644. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.Q.; Fragoso, M.C.B.V.; Lotfi, C.F.P.; Santos, M.G.; Nishi, M.Y.; Costa, M.H.S.; Lerario, A.M.; Maciel, C.C.; Mattos, G.E.; Jorge, A.A.L.; et al. Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J. Clin. Endocrinol. Metab. 2008, 93, 3524–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, A.S.; Giacaglia, L.R.; Martin, R.M.; Mendonca, B.B.; Lin, C.J. Assessment of the role of transcript for GATA-4 as a marker of unfavorable outcome in human adrenocortical neoplasms. BMC Endocr. Disord. 2004, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, K.S.; Moreno, D.A.; Martinelli, C.E., Jr.; Antonini, S.R.; de Castro, M.; Tucci, S., Jr.; Neder, L.; Ramalho, L.N.; Seidinger, A.L.; Cardinalli, I.; et al. Spindle assembly checkpoint gene expression in childhood adrenocortical tumors (ACT): Overexpression of Aurora kinases A and B is associated with a poor prognosis. Pediatr. Blood Cancer 2013, 60, 1809–1816. [Google Scholar] [CrossRef]
- Brondani, V.B.; Montenegro, L.; Lacombe, A.M.F.; Magalhães, B.M.; Nishi, M.Y.; Funari, M.F.A.; Narcizo, A.M.; Cardoso, L.C.; Siqueira, S.A.C.; Zerbini, M.C.N.; et al. High Prevalence of Alterations in DNA Mismatch Repair Genes of Lynch Syndrome in Pediatric Patients with Adrenocortical Tumors Carrying a Germline Mutation on TP53. Cancers 2020, 12, 621. [Google Scholar] [CrossRef] [Green Version]
- Bergadá, I.; Venara, M.; Maglio, S.; Ciaccio, M.; Diez, B.; Bergadá, C.; Chemes, H. Functional adrenal cortical tumors in pediatric patients: A clinicopathologic and immunohistochemical study of a long term follow-up series. Cancer 1996, 77, 771–777. [Google Scholar] [CrossRef]
- Bulzico, D.; de Faria, P.A.; de Paula, M.P.; Bordallo, M.A.; Pessoa, C.H.; Corbo, R.; Ferman, S.; Vaisman, M.; Neto, L.V. Recurrence and mortality prognostic factors in childhood adrenocortical tumors: Analysis from the Brazilian National Institute of Cancer experience. Pediatr. Hematol. Oncol. 2016, 33, 248–258. [Google Scholar] [CrossRef]
- Cordeiro, A.M.; Da Graca Bicalho, M.; Marques-Pereira, R.; Franca, P.P.; Moraes, M.M.; Rezende, G.Y.T.; Nesi-Franca, S.; Sandrini, R.; De Lacerda, L. Allelic frequencies of HLA-A, HLA-B and HLA-DRB1 genes in patients with adrenocortical tumor carriers of the germline mutation R337H in the TP53 gene. J. Endocr. Disord. 2014, 1, 1014. [Google Scholar]
- Damiani, D.; Della Manna, T.; Aquino, L.G.; Dichtchekenian, V.; Avancini, V.; Alves, F.; Longatto Filho, A.; Kanamura, C.T.; Setian, N. Proliferating cell nuclear antigen immunoreaction in adrenal tumors. Tumori 1995, 81, 273–277. [Google Scholar] [CrossRef]
- Dias, A.I.; Fachin, C.G.; Avó, L.R.; Frazão, C.V.; Caran, E.M.; Schettini, S.T.; Alves, M.T.; Ribeiro, R.C.; Abib Sde, C. Correlation between selected angiogenic markers and prognosis in pediatric adrenocortical tumors: Angiogenic markers and prognosis in pediatric ACTs. J. Pediatr. Surg. 2015, 50, 1323–1328. [Google Scholar] [CrossRef]
- Lira, R.C.P.; Fedatto, P.F.; Antonio, D.S.M.; Leal, L.F.; Martinelli, C.E.; De Castro, M.; Tucci, S.; Neder, L.; Ramalho, L.; Seidinger, A.L.; et al. IGF2 and IGF1R in pediatric adrenocortical tumors: Roles in metastasis and steroidogenesis. Endocr.-Relat. Cancer 2016, 23, 481–493. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.I.; Suartz, C.V.; Neto, R.P.; Berjeaut, R.H.; Mendonca, B.; Almeida, M.Q.; Fragoso, M.C.V.; Dénes, F.T. Management of functioning pediatric adrenal tumors. J. Pediatr. Surg. 2020, 56, 768–771. [Google Scholar] [CrossRef]
- Latronico, A.C.; Pinto, E.M.; Domenice, S.; Fragoso, M.C.; Martin, R.M.; Zerbini, M.C.; Lucon, A.M.; Mendonca, B.B. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 2001, 86, 4970–4973. [Google Scholar] [CrossRef]
- Junqueira, T.; Matellaro, M.J.; Aguiar, S.S.; Oliveira-Filho, A.G. Primary metastasis and relapses in adrenal corticocarcinoma in children: An overview from a single institution. Pediatr. Blood Cancer 2012, 59, 1070–1071. [Google Scholar] [CrossRef]
- Leite, F.A.; Lira, R.C.; Fedatto, P.F.; Antonini, S.R.; Martinelli, C.E., Jr.; de Castro, M.; Neder, L.; Ramalho, L.N.; Tucci, S., Jr.; Mastelaro, M.J.; et al. Low expression of HLA-DRA, HLA-DPA1, and HLA-DPB1 is associated with poor prognosis in pediatric adrenocortical tumors (ACT). Pediatr. Blood Cancer 2014, 61, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Lorea, C.F.; Moreno, D.A.; Borges, K.S.; Martinelli, C.E., Jr.; Antonini, S.R.; de Castro, M.; Tucci, S., Jr.; Neder, L.; Ramalho, L.N.; Cardinalli, I.; et al. Expression profile of apoptosis-related genes in childhood adrenocortical tumors: Low level of expression of BCL2 and TNF genes suggests a poor prognosis. Eur. J. Endocrinol. 2012, 167, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastellaro, M.J.; Ribeiro, R.C.; Oliveira-Filho, A.G.; Seidinger, A.L.; Cardinalli, I.A.; Miranda, E.C.M.; Aguiar, S.S.; Brandalise, S.R.; Yunes, J.A.; Barros-Filho, A.A. Adrenocortical tumors associated with the TP53 p.R337H germline mutation can be identified during child-care consultations. J. Pediatr. 2018, 94, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Michalkiewicz, E.L.; Sandrini, R.; Bugg, M.F.; Cristofani, L.; Caran, E.; Cardoso, A.M.; de Lacerda, L.; Ribeiro, R.C. Clinical characteristics of small functioning adrenocortical tumors in children. Med. Pediatr. Oncol. 1997, 28, 175–178. [Google Scholar] [CrossRef]
- Mendonca, B.B.; Lucon, A.M.; Menezes, C.A.; Saldanha, L.B.; Latronico, A.C.; Zerbini, C.; Madureira, G.; Domenice, S.; Albergaria, M.A.; Camargo, M.H.; et al. Clinical, hormonal and pathological findings in a comparative study of adrenocortical neoplasms in childhood and adulthood. J. Urol. 1995, 154, 2004–2009. [Google Scholar] [CrossRef]
- Mermejo, L.M.; Leal, L.F.; Colli, L.M.; Martinelli, C.E.; Moreira, A.C.; Tone, L.G.; Scrideli, C.A.; Yunes, J.A.; Mastellaro, M.J.; Seidinger, A.L.; et al. P53 and CTNNB1 mutations reveal different patterns between pediatric and adult adrenocortical tumors. Endocr. Rev. 2011, 32, P1-39. [Google Scholar]
- Monteiro, N.M.L.; De Sá Rodrigues, K.E.; Vidigal, P.V.T.; De Oliveira, B.M. Adrenal carcinoma in children: Longitudinal study in Minas Gerais, Brazil. Rev. Paul. Pediatr. 2019, 37, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.M.; Pinto, H.F.; Pianovski, M.A.D.; Neto, L.C.; Bressiani, M.; De Carvalho, J.A.R.; Franc¸a, S.N.; Sandrini, R.; De Lacerda, L. Factors influencing prognosis of stage I and II adrenocortical tumor in children and adolescents: Experience of 101 cases. Horm. Res. Paediatr. 2013, 80, 55. [Google Scholar]
- Parise, I.Z.S.; Parise, G.A.; Noronha, L.; Surakhy, M.; Woiski, T.D.; Silva, D.B.; Costa, T.E.B.; Del-Valle, M.; Komechen, H.; Rosati, R.; et al. The Prognostic Role of CD8(+) T Lymphocytes in Childhood Adrenocortical Carcinomas Compared to Ki-67, PD-1, PD-L1, and the Weiss Score. Cancers 2019, 11, 1730. [Google Scholar] [CrossRef] [Green Version]
- Pinto, E.; Rodriguez-Galindo, C.; Choi, J.; Ribeiro, R.; Zambetti, G. MHC-Class II expression in pediatric adrenocortical tumors: Prognostic significance. Horm. Res. Paediatr. 2016, 86, 3–4. [Google Scholar] [CrossRef]
- Pinto, E.M.; Rodriguez-Galindo, C.; Pounds, S.B.; Wang, L.; Clay, M.R.; Neale, G.; Garfinkle, E.A.R.; Lam, C.G.; Levy, C.F.; Pappo, A.S.; et al. Identification of clinical and biologic correlates associated with outcome in children with adrenocortical tumors without germline TP53 mutations: A St Jude adrenocortical tumor registry and children’s oncology group study. J. Clin. Oncol. 2017, 35, 3956–3963. [Google Scholar] [CrossRef]
- Zancanella, P.; Pianovski, M.A.; Oliveira, B.H.; Ferman, S.; Piovezan, G.C.; Lichtvan, L.L.; Voss, S.Z.; Stinghen, S.T.; Callefe, L.G.; Parise, G.A.; et al. Mitotane associated with cisplatin, etoposide, and doxorubicin in advanced childhood adrenocortical carcinoma: Mitotane monitoring and tumor regression. J. Pediatr. Hematol. Oncol. 2006, 28, 513–524. [Google Scholar] [CrossRef]
- Ribeiro, R.C.; Sandrini Neto, R.S.; Schell, M.J.; Lacerda, L.; Sambaio, G.A.; Cat, I. Adrenocortical carcinoma in children: A study of 40 cases. J. Clin. Oncol. 1990, 8, 67–74. [Google Scholar] [CrossRef]
- Sbragia, L.; Oliveira-Filho, A.G.; Vassallo, J.; Pinto, G.A.; Guerra-Junior, G.; Bustorff-Silva, J. Adrenocortical tumors in Brazilian children: Immunohistochemical markers and prognostic factors. Arch. Pathol. Lab. Med. 2005, 129, 1127–1131. [Google Scholar] [CrossRef]
- Tucci, S., Jr.; Martins, A.C.; Suaid, H.J.; Cologna, A.J.; Reis, R.B. The impact of tumor stage on prognosis in children with adrenocortical carcinoma. J. Urol. 2005, 174, 2338–2342. [Google Scholar] [CrossRef] [PubMed]
- Venara, M.; Sanchez Marull, R.; Bergada, I.; Gamboni, M.; Chemes, H. Functional adrenal cortical tumors in childhood: A study of ploidy, p53- protein and nucleolar organizer regions (AgNORs) as prognostic markers. J. Pediatr. Endocrinol. Metab. 1998, 11, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Wajchenberg, B.L.; Albergaria Pereira, M.A.; Medonca, B.B.; Latronico, A.C.; Campos Carneiro, P.; Alves, V.A.; Zerbini, M.C.; Liberman, B.; Carlos Gomes, G.; Kirschner, M.A. Adrenocortical carcinoma: Clinical and laboratory observations. Cancer 2000, 88, 711–736. [Google Scholar] [CrossRef]
- Martins-Filho, S.N.; Almeida, M.Q.; Soares, I.; Wakamatsu, A.; Alves, V.A.F.; Fragoso, M.; Zerbini, M.C.N. Clinical Impact of Pathological Features Including the Ki-67 Labeling Index on Diagnosis and Prognosis of Adult and Pediatric Adrenocortical Tumors. Endocr Pathol. 2021, 32, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Bueno, A.C.; Leal, L.F.; Gomes, D.C.; Montaldi, A.P.; Yunes, J.A.; Cardinalli, I.A.; Masterallo, M.J.; Brandalise, S.R.; Antonini, S.R.; De Castro, M.; et al. Low vitamin d receptor (VDR) impacts pediatric adrenocortical tumor behavior while vdr activation represses WNT/β-catenin signaling and adrenocortical cell proliferation. Horm. Res. Paediatr. 2017, 88, 52. [Google Scholar] [CrossRef]
- Kremer, V.; Goncalves, A.; Gutierrez, F.; Coelho, S.; Carvalho, R.; Azevedo, R. Adrenal cortex tumor in childhood, analysis of 20 years. Pediatr. Blood Cancer 2009, 53, 881. [Google Scholar] [CrossRef]
- Cecchetto, G.; Ganarin, A.; Bien, E.; Vorwerk, P.; Bisogno, G.; Godzinski, J.; Dall’Igna, P.; Reguerre, Y.; Schneider, D.; Brugières, L.; et al. Outcome and prognostic factors in high-risk childhood adrenocortical carcinomas: A report from the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). Pediatr. Blood Cancer 2017, 64, e26368. [Google Scholar] [CrossRef]
- Benserai, F.Z.; Kassa, R.; Chilla, D.; Benserai, F.; Asselah, F. Algerian pediatric adrenal cortical neoplasms: An institutional retrospective study. Virchows Arch. 2013, 463, 159–160. [Google Scholar] [CrossRef]
- Brenna, C.T.A.; Michaeli, O.; Wasserman, J.D.; Malkin, D. Clinical Outcomes of Children With Adrenocortical Carcinoma in the Context of Germline TP53 Status. J. Pediatr. Hematol./Oncol. 2020, 43, e635–e641. [Google Scholar] [CrossRef]
- Buyukpamukcu, M.; Yalcin, B.; Ciftci, A.; Kale, G.; Gonc, N.; Ocak, S.; Aydin, B.; Varan, A.; Akyuz, C.; Kutluk, T. Adrenocortical carcinomas in children: Hacettepe experience. Pediatr. Blood Cancer 2011, 57, 802. [Google Scholar] [CrossRef]
- Driver, C.P.; Birch, J.; Gough, D.C.; Bruce, J. Adrenal cortical tumors in childhood. Pediatr. Hematol. Oncol. 1998, 15, 527–532. [Google Scholar] [CrossRef]
- Doghman-Bouguerra, M.; Finetti, P.; Durand, N.; Parise, I.Z.S.; Sbiera, S.; Cantini, G.; Canu, L.; Hescot, S.; Figueiredo, M.M.O.; Komechen, H.; et al. Cancer-testis antigen fate1 expression in adrenocortical tumors is associated with a pervasive autoimmune response and is a marker of malignancy in adult, but not children, acc. Cancers 2020, 12, 689. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.J.; Kim, D.Y.; Kim, S.C.; Kim, T.H.; Kim, I.K. Adrenocortical tumors in children 18 years old and younger. J. Korean Surg. Soc. 2012, 82, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, G.; DasGupta, S.; Mukherjee, G.; Sengupta, M.; Roy, P.; Arun, I.; Datta, C.; Mishra, P.K.; Banerjee, S.; Chatterjee, U. Usefulness of Wieneke criteria in assessing morphologic characteristics of adrenocortical tumors in children. Pediatr. Surg. Int. 2015, 31, 563–571. [Google Scholar] [CrossRef]
- Guntiboina, V.A.; Sengupta, M.; Islam, N.; Barman, S.; Biswas, S.K.; Chatterjee, U.; Mishra, P.K.; Roy, P.; Mallick, M.G.; Datta, C. Diagnostic and prognostic utility of SF1, IGF2 and p57 immunoexpression in pediatric adrenal cortical tumors. J. Pediatr. Surg. 2019, 54, 1906–1912. [Google Scholar] [CrossRef]
- Chen, Y.; Lo, F.S.; Tsai, W.; Sui, H.H. Clinical characteristics of pediatric adrenocortical tumors: A retrospective review of medical centers in North Taiwan. Horm. Res. Paediatr. 2017, 88, 521. [Google Scholar] [CrossRef]
- Chen, Q.L.; Su, Z.; Li, Y.H.; Ma, H.M.; Chen, H.S.; Du, M.L. Clinical characteristics of adrenocortical tumors in children. J. Pediatr. Endocrinol. Metab. 2011, 24, 535–541. [Google Scholar] [CrossRef]
- Ciftci, A.O.; Senocak, M.E.; Tanyel, F.C.; Büyükpamukçu, N. Adrenocortical tumors in children. J. Pediatr. Surg. 2001, 36, 549–554. [Google Scholar] [CrossRef]
- Das, S.; Sengupta, M.; Islam, N.; Roy, P.; Datta, C.; Mishra, P.K.; Banerjee, S.; Chaudhuri, M.K.; Chatterjee, U. Weineke criteria, Ki-67 index and p53 status to study pediatric adrenocortical tumors: Is there a correlation? J. Pediatr. Surg. 2016, 51, 1795–1800. [Google Scholar] [CrossRef]
- Dall’Igna, P.; Virgone, C.; De Salvo, G.L.; Bertorelle, R.; Indolfi, P.; De Paoli, A.; Buffa, P.; Conte, M.; Esposito, G.; Inserra, A.; et al. Adrenocortical tumors in Italian children: Analysis of clinical characteristics and P53 status. Data from the national registries. J. Pediatr. Surg. 2014, 49, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.M.; Pham, T.H.; Askegard-Giesmann, J.R.; Grams, J.M.; Iqbal, C.W.; Stavlo, P.; Moir, C.R. Outcome of adrenocortical tumors in children. J. Pediatr. Surg. 2008, 43, 843–849. [Google Scholar] [CrossRef]
- Federici, S.; Galli, G.; Ceccarelli, P.L.; Ferrari, M.; Cicognani, A.; Cacciari, E.; Domini, R. Adrenocortical tumors in children: A report of 12 cases. Eur. J. Pediatr. Surg. 1994, 4, 21–25. [Google Scholar] [CrossRef]
- Flynt, K.A.; Dillman, J.R.; Davenport, M.S.; Smith, E.A.; Else, T.; Strouse, P.J.; Caoili, E.M. Pediatric adrenocortical neoplasms: Can imaging reliably discriminate adenomas from carcinomas? Pediatr. Radiol. 2015, 45, 1160–1168. [Google Scholar] [CrossRef]
- Gönç, E.N.; Özön, Z.A.; Cakır, M.D.; Alikaşifoğlu, A.; Kandemir, N. Need for comprehensive hormonal workup in the management of adrenocortical tumors in children. J. Clin. Res. Pediatr. Endocrinol. 2014, 6, 68–73. [Google Scholar] [CrossRef]
- Gulack, B.C.; Rialon, K.L.; Englum, B.R.; Kim, J.; Talbot, L.J.; Adibe, O.O.; Rice, H.E.; Tracy, E.T. Factors associated with survival in pediatric adrenocortical carcinoma: An analysis of the National Cancer Data Base (NCDB). J. Pediatr. Surg. 2016, 51, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Rivera, M.; Novotny, P.; Rodriguez, V.; Bancos, I.; Lteif, A. Adrenocortical Carcinoma in Children: A Clinicopathological Analysis of 41 Patients at the Mayo Clinic from 1950 to 2017. Horm Res. Paediatr. 2018, 90, 8–18. [Google Scholar] [CrossRef]
- Jehangir, S.; Nanjundaiah, P.; Sigamani, E.; Burad, D.; Manipadam, M.T.; Lea, V.; Ly, T.; Holland, A.J.A. Pathological prognostication of paediatric adrenocortical tumours: Is a gold standard emerging? Pediatr. Blood Cancer 2019, 66, e27989. [Google Scholar] [CrossRef]
- Klein, J.D.; Turner, C.G.; Gray, F.L.; Yu, D.C.; Kozakewich, H.P.; Perez-Atayde, A.R.; Voss, S.D.; Zurakowski, D.; Shamberger, R.C.; Weldon, C.B. Adrenal cortical tumors in children: Factors associated with poor outcome. J. Pediatr. Surg. 2011, 46, 1201–1207. [Google Scholar] [CrossRef]
- Knopfle, G.; Fodisch, H.J.; Holschneider, A. Adrenal cortical carcinoma in childhood and adolescence. Klin. Padiatr. 1986, 198, 250–256. [Google Scholar] [PubMed]
- Letouzé, E.; Rosati, R.; Komechen, H.; Doghman, M.; Marisa, L.; Flück, C.; de Krijger, R.R.; van Noesel, M.M.; Mas, J.C.; Pianovski, M.A.; et al. SNP array profiling of childhood adrenocortical tumors reveals distinct pathways of tumorigenesis and highlights candidate driver genes. J. Clin. Endocrinol. Metab. 2012, 97, E1284–E1293. [Google Scholar] [CrossRef] [Green Version]
- Loncarevic, I.F.; Hering, A.; Posorski, N.; Linden, T.; Hoyer, H.; Bucsky, P. Number of genomic imbalances correlates with the overall survival for adrenocortical cancer in childhood. Pediatr. Blood Cancer 2008, 51, 356–362. [Google Scholar] [CrossRef]
- Magro, G.; Esposito, G.; Cecchetto, G.; Dall’Igna, P.; Marcato, R.; Gambini, C.; Boldrini, R.; Collini, P.; D’Onofrio, V.; Salfi, N.; et al. Pediatric adrenocortical tumors: Morphological diagnostic criteria and immunohistochemical expression of matrix metalloproteinase type 2 and human leucocyte-associated antigen (HLA) class II antigens: Results from the Italian Pediatric Rare Tumor (TREP) Study project. Hum. Pathol. 2012, 43, 31–39. [Google Scholar] [CrossRef]
- Mayer, S.K.; Oligny, L.L.; Deal, C.; Yazbeck, S.; Gagné, I.N.; Blanchard, H. Childhood adrenocortical tumors: Case series and reevaluation of prognosis—A 24-year experience. J. Pediatr. Surg. 1997, 32, 911–915. [Google Scholar] [CrossRef]
- McDonnell, C.M.; Zacharin, M.R. Adrenal cortical tumours: 25 years’ experience at the Royal Children’s Hospital, Melbourne. J. Paediatr. Child. Health 2003, 39, 682–685. [Google Scholar] [CrossRef]
- Miele, E.; Di Giannatale, A.; Crocoli, A.; Cozza, R.; Serra, A.; Castellano, A.; Cacchione, A.; Cefalo, M.G.; Alaggio, R.; De Pasquale, M.D. Clinical, Genetic, and Prognostic Features of Adrenocortical Tumors in Children: A 10-Year Single-Center Experience. Front. Oncol. 2020, 10, 554388. [Google Scholar] [CrossRef]
- Mittal, R.; Ramadan, D.G.; Khalifa, N.M.; Khalifa, S.O.; Mazidi, Z.; Zaki, M. Adrenocortical tumors in children: A Kuwait experience. Gulf J. Oncol. 2012, 2012, 38–46. [Google Scholar]
- Mahendraraj, K.; Sidhu, K.; Chamberlain, R.S. Adrenocortical carcinoma in adults and children: A populationbased outcomes study involving 1,623 patients. Ann. Surg. Oncol. 2014, 21, S93. [Google Scholar] [CrossRef]
- Mattone, M.C.; Gil, S.; Mutti, M.L.G.; Casanovas, A.; Lazzati, J.M.; Zaidman, V.; Belgorosky, A.; Guercio, G. Pediatric adrenocortical tumors. a single tertiary center experience: Clinical, biological and pathologic characteristics analysis. Horm. Res. Paediatr. 2018, 90, 130. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; Agarwal, G.; Misra, A.K.; Agarwal, A.; Mishra, S.K. Functioning adrenal tumours in children and adolescents: An institutional experience. ANZ J. Surg. 2001, 71, 103–107. [Google Scholar] [CrossRef]
- Panamonta, O.; Areemit, S.; Srinakarin, J.; Siritunyaporn, S.; Tuksapun, S. Adrenocortical tumors in children. J. Med Assoc. Thail. 2001, 84, 188–194. [Google Scholar]
- Narasimhan, K.L.; Samujh, R.; Bhansali, A.; Marwaha, R.K.; Chowdhary, S.K.; Radotra, B.D.; Rao, K.L. Adrenocortical tumors in childhood. Pediatr. Surg. Int. 2003, 19, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.K.; Ransley, P.G.; McCullagh, M.; Malone, M.; Spitz, L. Functioning adrenocortical neoplasms in children. BJU Int. 2002, 89, 562–565. [Google Scholar] [CrossRef] [Green Version]
- Picard, C.; Faure-Conter, C.; Leblond, P.; Brugières, L.; Thomas-Teinturier, C.; Hameury, F.; Defachelles, A.S.; Verschuur, A.; Brisse, H.J.; Sarnacki, S.; et al. Exploring heterogeneity of adrenal cortical tumors in children: The French pediatric rare tumor group (Fracture) experience. Pediatr. Blood Cancer 2020, 67, e28086. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Orbach, D.; Carton, M.; Brugieres, L.; Renaudin, K.; Aubert, S.; Berrebi, D.; Galmiche, L.; Dujardin, F.; Leblond, P.; et al. Revisiting the role of the pathological grading in pediatric adrenal cortical tumors: Results from a national cohort study with pathological review. Mod. Pathol. 2019, 32, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Redlich, A.; Boxberger, N.; Strugala, D.; Frühwald, M.C.; Leuschner, I.; Kropf, S.; Bucsky, P.; Vorwerk, P. Systemic treatment of adrenocortical carcinoma in children: Data from the German GPOH-MET 97 trial. Klin. Padiatr. 2012, 224, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Ru, W.; Yang, M.; Xu, S.; Li, M.; Tang, D. Management and prognosis of adrenocortical tumors in children: Can we find out an appropriate points-scoring system to predict prognosis? Pediatr. Surg. Int. 2017, 33, 705–711. [Google Scholar] [CrossRef]
- Sakoda, A.; Mushtaq, I.; Levitt, G.; Sebire, N.J. Clinical and histopathological features of adrenocortical neoplasms in children: Retrospective review from a single specialist center. J. Pediatr. Surg. 2014, 49, 410–415. [Google Scholar] [CrossRef]
- Stewart, J.N.; Flageole, H.; Kavan, P. A Surgical Approach to Adrenocortical Tumors in Children: The Mainstay of Treatment. J. Pediatr. Surg. 2004, 39, 759–763. [Google Scholar] [CrossRef]
- Teinturier, C.; Brugières, L.; Lemerle, J.; Chaussain, J.L.; Bougnères, P.F. Adrenocortical carcinoma in children: Retrospective study of 54 cases. Arch. Pediatrie 1996, 3, 235–240. [Google Scholar] [CrossRef]
- Waldmann, J.; Patsalis, N.; Fendrich, V.; Langer, P.; Saeger, W.; Chaloupka, B.; Ramaswamy, A.; Fassnacht, M.; Bartsch, D.K.; Slater, E.P. Clinical impact of TP53 alterations in adrenocortical carcinomas. Langenbecks Arch. Surg. 2012, 397, 209–216. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Novokmet, A.; Eichler-Jonsson, C.; Ribeiro, R.C.; Rodriguez-Galindo, C.; Zambetti, G.P.; Malkin, D. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: A children’s oncology group study. J. Clin. Oncol. 2015, 33, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolthers, O.D.; Cameron, F.J.; Scheimberg, I.; Honour, J.W.; Hindmarsh, P.C.; Savage, M.O.; Stanhope, R.G.; Brook, C.G. Androgen secreting adrenocortical tumours. Arch. Dis. Child. 1999, 80, 46–50. [Google Scholar] [CrossRef]
- Wu, X.; Xu, J.; Wang, J.; Gu, W.; Zou, C. Childhood adrenocortical tumor: A clinical and immunohistochemical study of 13 cases. Medicine 2019, 98, e17921. [Google Scholar] [CrossRef]
- Sabaretnam, M.; Mishra, A.; Agarwal, G.; Agarwal, A.; Verma, A.K.; Mishra, S.K. Adrenocortical carcinoma in children and adults: Two decades experience in a single institution. Indian. J. Cancer 2016, 53, 317–321. [Google Scholar] [CrossRef]
- Yalçin, Ş.; Çiftçi, A.Ö.; Şenocak, M.E.; Tanyel, F.C.; Büyükpamukçu, N. Adrenocortical tumors of childhood. Cocuk Cerrahisi Derg. 2006, 20, 166–169. [Google Scholar]
- Zekri, W.; Hammad, M.; Rashed, W.M.; Ahmed, G.; Elshafie, M.; Adly, M.H.; Elborai, Y.; Abdalla, B.; Taha, H.; Elkinaae, N.; et al. The outcome of childhood adrenocortical carcinoma in Egypt: A model from developing countries. Pediatr. Hematol. Oncol. 2020, 37, 198–210. [Google Scholar] [CrossRef]
- Fassnacht, M.; Johanssen, S.; Quinkler, M.; Bucsky, P.; Willenberg, H.S.; Beuschlein, F.; Terzolo, M.; Mueller, H.H.; Hahner, S.; Allolio, B.; et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: Proposal for a Revised TNM Classification. Cancer 2009, 115, 243–250. [Google Scholar] [CrossRef]
- Lalli, E.; Figueiredo, B.C. Pediatric adrenocortical tumors: What they can tell us on adrenal development and comparison with adult adrenal tumors. Front. Endocrinol. 2015, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Assie, G.; Letouze, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; Rene-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 30, 363. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.M.; Chen, X.; Easton, J.; Finkelstein, D.; Liu, Z.; Pounds, S.; Galindo, C.R.; Figueiredo, B.C.; Zhang, J.; Downing, J.R.; et al. Genomic landscape of pediatric adrenocortical tumors. Endocr. Rev. 2015, 36, 6302. [Google Scholar]
- Ishimoto, H.; Jaffe, R.B. Development and function of the human fetal adrenal cortex: A key component in the feto-placental unit. Endocr Rev. 2011, 32, 317–355. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.M.; Medeiros, L.J.; Vickery, A.L., Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am. J. Surg. Pathol. 1989, 13, 202–206. [Google Scholar] [CrossRef]
- Lau, S.K.; Weiss, L.M. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum. Pathol. 2009, 40, 757–768. [Google Scholar] [CrossRef]
- Filho, S.N.M.; Almeida, M.Q.; Wakamatsu, A.; Alves, V.; Fragoso, M.C.; Zerbini, M. A simple yet powerful algorithm including Ki67 Labeling Index (LI) helps predict outcome in pediatric adrenocortical tumors (ACT). Lab. Investig. 2018, 98, 234. [Google Scholar] [CrossRef] [Green Version]
- Galluzzo Mutti, M.L.; Mattone, M.C.; Casanovas, A.; Nespoli, E.; Belgorosky, A.; Guercio, G.; Lubieniecki, F. Pediatric adrenocortical tumors. Which pathological criteria system is the best? In Laboratory Investigation; Nature Publishing Group: New York, NY, USA, 2019; Volume 99. [Google Scholar]
- Lucon, A.M.; Pereira, M.A.; Mendonça, B.B.; Zerbini, M.C.; Saldanha, L.B.; Arap, S. Adrenocortical tumors: Results of treatment and study of Weiss’s score as a prognostic factor. Rev. Do Hosp. Das Clin. 2002, 57, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Papotti, M.; Libe, R.; Duregon, E.; Volante, M.; Bertherat, J.; Tissier, F. The Weiss score and beyond--histopathology for adrenocortical carcinoma. Horm. Cancer 2011, 2, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Cavalcanti, M.; Leal, L.; Lacchini, F.; Martineli, C.; Scrideli, C.; Tucci, S.; Molina, C.; Yunes, J.; Mastellaro, M.; Brandalise, S.; et al. Analysis of DAX1 and SF1 genes and their interaction with genes involved in stem cell maintenance in adrenocortical tumors. Horm. Res. Paediatr. 2015, 84, 15–16. [Google Scholar] [CrossRef]
- de Sousa, G.R.; Soares, I.C.; Faria, A.M.; Domingues, V.B.; Wakamatsu, A.; Lerario, A.M.; Alves, V.A.; Zerbini, M.C.; Mendonca, B.B.; Fragoso, M.C.; et al. DAX1 Overexpression in Pediatric Adrenocortical Tumors: A Synergic Role with SF1 in Tumorigenesis. Horm. Metab. Res. 2015, 47, 656–661. [Google Scholar] [CrossRef]
- Lira, R.C.P.; Fedatto, P.F.; Martinelli, C.E., Jr.; Antonini, S.R.R.; Castro, M.; Tucci, S., Jr.; Neder, L.; Ramalho, L.Z.; Seidinger, A.L.; Cardinalli, I.; et al. Expression of IGF2 and IGF1R in pediatric adrenocortical tumors (ACT): Overexpression of IGF1R is associated with unfavorable event and metastasis in adrenocortical carcinomas. Growth Horm. IGF Res. 2012, 22, S41. [Google Scholar] [CrossRef]
- Lira, R.C.P.; Corrêa, C.A.P.; Yunes, J.A.; Mastellaro, M.J.; Brandalise, S.R.; Antonini, S.R.R.; Tone, L.G.; Scrideli, C. MIR-149-3PIS indicative of poor prognosis and increased cell viability of adrenocortical tumors. Pediatr. Blood Cancer 2018, 65, S400. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.J.; Lima-Valassi, H.P.; Lerario, A.M.; Mendonca, B.B. A potential role of mevalonate pathway in adrenocortical tumors. FASEB J. 2010, 24, 421.1. [Google Scholar]
- Pinto, E.M.; Billerbeck, A.E.; Fragoso, M.C.; Mendonca, B.B.; Latronico, A.C. Deletion mapping of chromosome 17 in benign and malignant adrenocortical tumors associated with the Arg337His mutation of the p53 tumor suppressor protein. J. Clin. Endocrinol. Metab. 2005, 90, 2976–2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orhan, D.; Kale, G.; Cağlar, M.; Göğüş, S.; Karaağaoğlu, E. Histone mRNA in situ hybridization and Ki 67 immunohistochemistry in pediatric adrenocortical tumors. Virchows Arch.: Int. J. Pathol. 2006, 448, 591–596. [Google Scholar] [CrossRef]
- Sredni, S.T.; Zerbini, M.C.; Latorre, M.R.; Alves, V.A. p53 as a prognostic factor in adrenocortical tumors of adults and children. Braz. J. Med. Biol. Res. 2003, 36, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sredni, S.T.; Alves, V.A.; Latorre Mdo, R.; Zerbini, M.C. Adrenocortical tumours in children and adults: A study of pathological and proliferation features. Pathology 2003, 35, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Boechat, G.A.; Stinghen, S.T.; Custódio, G.; Pianovski, M.A.; Figueiredo, F.R.; Jenkins, J.; Zambetti, G.P.; Ribeiro, R.C.; Figueiredo, B.C. Placental alkaline phosphatase in pediatric adrenocortical cancer. J. Pediatr. Hematol. Oncol. 2011, 33, e149–e153. [Google Scholar] [CrossRef]
- Rosati, R.; Cerrato, F.; Doghman, M.; Pianovski, M.A.; Parise, G.A.; Custódio, G.; Zambetti, G.P.; Ribeiro, R.C.; Riccio, A.; Figueiredo, B.C.; et al. High frequency of loss of heterozygosity at 11p15 and IGF2 overexpression are not related to clinical outcome in childhood adrenocortical tumors positive for the R337H TP53 mutation. Cancer Genet. Cytogenet. 2008, 186, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, B.M.; Trarbach, E.B.; Ribeiro, T.C.; Pereira, M.A.; Mendonca, B.B.; Fragoso, M.C. Genotype analysis of the human endostatin variant p.D104N in benign and malignant adrenocortical tumors. Clinics 2012, 67, 95–98. [Google Scholar] [CrossRef]
- Altieri, B.; Ronchi, C.L.; Kroiss, M.; Fassnacht, M. Next-generation therapies for adrenocortical carcinoma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101434. [Google Scholar] [CrossRef]
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): Interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 121–133. [Google Scholar] [CrossRef]
- Fragoso, M.C.; Almeida, M.Q.; Mazzuco, T.L.; Mariani, B.M.; Brito, L.P.; Gonçalves, T.C.; Alencar, G.A.; Lima Lde, O.; Faria, A.M.; Bourdeau, I.; et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: Validation in a Brazilian cohort of adult and pediatric patients. Eur. J. Endocrinol. 2012, 166, 61–67. [Google Scholar] [CrossRef]
- Kulshrestha, A.; Suman, S.; Ranjan, R. Network analysis reveals potential markers for pediatric adrenocortical carcinoma. OncoTargets Ther. 2016, 9, 4569–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.L.; Kim, E.S.; Nava-Parada, P.; Alam, S.; Johnson, F.M.; Stephens, A.W.; Simantov, R.; Poondru, S.; Gedrich, R.; Lippman, S.M.; et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X.; Krailo, M.; Villaluna, D.; Adamson, P.C.; Blaney, S.M. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Pianovski, M.; Blum, R.R.; Noronha, L.; Jung, J. Immunohistochemical expression of VEGFR1 her-2 and pten in pediatric adrenocortical carcinoma tumors. Pediatr. Blood Cancer 2013, 60, 104–105. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, B.; Bozilovic, J.; Diebold, M.; Schwarz, J.D.; Hofstetter, J.; Schroder, M.; Wanior, M.; Narain, A.; Vogt, M.; Dudvarski Stankovic, N.; et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat. Chem. Biol. 2020, 16, 1179–1188. [Google Scholar] [CrossRef]
- Pezzani, R.; Rubin, B.; Bertazza, L.; Redaelli, M.; Barollo, S.; Monticelli, H.; Baldini, E.; Mian, C.; Mucignat, C.; Scaroni, C.; et al. The aurora kinase inhibitor VX-680 shows anti-cancer effects in primary metastatic cells and the SW13 cell line. Investig. New Drugs 2016, 34, 531–540. [Google Scholar] [CrossRef]
- Ikeya, A.; Nakashima, M.; Yamashita, M.; Kakizawa, K.; Okawa, Y.; Saitsu, H.; Sasaki, S.; Sasano, H.; Suda, T.; Oki, Y. CCNB2 and AURKA overexpression may cause atypical mitosis in Japanese cortisol-producing adrenocortical carcinoma with TP53 somatic variant. PLoS ONE 2020, 15, e0231665. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Edgerly, M.; Velarde, M.; Wilkerson, J.; Venkatesan, A.M.; Pittaluga, S.; Yang, S.X.; Nguyen, D.; Balasubramaniam, S.; Fojo, T. The VEGF inhibitor axitinib has limited effectiveness as a therapy for adrenocortical cancer. J. Clin. Endocrinol. Metab. 2014, 99, 1291–1297. [Google Scholar] [CrossRef] [Green Version]
- Fraenkel, M.; Gueorguiev, M.; Barak, D.; Salmon, A.; Grossman, A.B.; Gross, D.J. Everolimus therapy for progressive adrenocortical cancer. Endocrine 2013, 44, 187–192. [Google Scholar] [CrossRef]
- Faillot, S.; Assie, G. ENDOCRINE TUMOURS: The genomics of adrenocortical tumors. Eur. J. Endocrinol. 2016, 174, R249–R265. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.; Silva, R.; Friedrich-Medina, P.; Frazier, L.; Ribeiro, R.; Ribeiro, K.; Rodriguez-Galindo, C. Descriptive epidemiology ofadrenocortical carcinoma in the united states: Incidence, outcome, and risk of second malignant neoplasms. Pediatr. Blood Cancer 2013, 60, 105. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, R.C.; Figueiredo, B. Childhood adrenocortical tumours. Eur. J. Cancer 2004, 40, 1117–1126. [Google Scholar] [CrossRef]
- Bugg, M.F.; Ribeiro, R.C.; Roberson, P.K.; Lloyd, R.V.; Sandrini, R.; Silva, J.B.; Epelman, S.; Shapiro, D.N.; Parham, D.M. Correlation of pathologic features with clinical outcome in pediatric adrenocortical neoplasia. A study of a Brazilian population. Brazilian Group for Treatment of Childhood Adrenocortical Tumors. Am. J. Clin. Pathol. 1994, 101, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Passaia, B.D.S.; Dias, M.H.; Kremer, J.L.; Antonini, S.R.R.; de Almeida, M.Q.; Fragoso, M.; Lotfi, C.F.P. TCF21/POD-1, a Transcritional Regulator of SF-1/NR5A1, as a Potential Prognosis Marker in Adult and Pediatric Adrenocortical Tumors. Front. Endocrinol. 2018, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custódio, G.; Parise, G.A.; Filho, N.K.; Komechen, H.; Sabbaga, C.C.; Rosati, R.; Grisa, L.; Parise, I.Z.S.; Pianovski, M.A.D.; Fiori, C.M.C.M.; et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J. Clin. Oncol. 2013, 31, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambaiti, E.; Duci, M.; De Corti, F.; Gamba, P.; Dall’Igna, P.; Ghidini, F.; Virgone, C. Clinical prognostic factors in pediatric adrenocortical tumors: A meta-analysis. Pediatr. Blood Cancer 2021, 68, e28836. [Google Scholar] [CrossRef]
- Rodriguez-Galindo, C.; Krailo, M.D.; Pinto, E.M.; Pashankar, F.; Weldon, C.B.; Huang, L.; Caran, E.M.; Hicks, J.; McCarville, M.B.; Malkin, D.; et al. Treatment of Pediatric Adrenocortical Carcinoma With Surgery, Retroperitoneal Lymph Node Dissection, and Chemotherapy: The Children’s Oncology Group ARAR0332 Protocol. J. Clin. Oncol. 2021, 39, 2463–2473. [Google Scholar] [CrossRef]
- Bertoin, F.; Letouze, E.; Grignani, P.; Patey, M.; Rossignol, S.; Libe, R.; Pasqual, C.; Lardiere-Deguelte, S.; Hoeffel-Fornes, C.; Gaillard, D.; et al. Genome-wide paternal uniparental disomy as a cause of Beckwith-Wiedemann syndrome associated with recurrent virilizing adrenocortical tumors. Horm. Metab. Res. 2015, 47, 497–503. [Google Scholar] [CrossRef]
- El Wakil, A.; Doghman, M.; Latre De Late, P.; Zambetti, G.P.; Figueiredo, B.C.; Lalli, E. Genetics and genomics of childhood adrenocortical tumors. Mol. Cell Endocrinol. 2011, 336, 169–173. [Google Scholar] [CrossRef]
- Ebbehoj, A.; Li, D.; Kaur, R.J.; Zhang, C.; Singh, S.; Li, T.; Atkinson, E.; Achenbach, S.; Khosla, S.; Arlt, W.; et al. Epidemiology of adrenal tumours in Olmsted County, Minnesota, USA: A population-based cohort study. Lancet Diabetes Endocrinol. 2020, 8, 894–902. [Google Scholar] [CrossRef]
- Jouinot, A.; Bertherat, J. Diseases Predisposing to Adrenocortical Malignancy (Li-Fraumeni Syndrome, Beckwith-Wiedemann Syndrome, and Carney Complex). Genet. Endocr. Dis. Syndr. 2019, 111, 149–169. [Google Scholar] [CrossRef]
- Magnusson, S.; Gisselsson, D.; Wiebe, T.; Kristoffersson, U.; Borg, Å.; Olsson, H. Prevalence of germline TP53 mutations and history of Li-Fraumeni syndrome in families with childhood adrenocortical tumors, choroid plexus tumors, and rhabdomyosarcoma: A population-based survey. Pediatr. Blood Cancer 2012, 59, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Chen, X.; Rodriguez-Galindo, C.; Zhang, J.; Ribeiro, R.; Zambetti, G. Whole-genome sequencing identifies genetic alterations in pediatric adrenocortical tumors. Horm. Res. Paediatr. 2016, 86, 1. [Google Scholar] [CrossRef]
- Raymond, V.M.; Everett, J.N.; Hammer, G.D.; Gustafson, S.L.; Stoffel, E.M.; Gruber, S.B.; Else, T. Prevalence of Lynch syndrome in adrenocortical carcinoma is comparable to the prevalence among colorectal and endometrial cancers. Fam. Cancer 2013, 12, 805. [Google Scholar] [CrossRef]
- Seidinger, A.L.; Caminha, I.P.; Mastellaro, M.J.; Gabetta, C.S.; Nowill, A.E.; Pinheiro, V.R.P.; Yunes, J.A. TP53 p.Arg337His geographic distribution correlates with adrenocortical tumor occurrence. Mol. Genet. Genom. Med. 2020, 8, e1168. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, I.; Deutschbein, T.; Jurowich, C.; Kroiss, M.; Ronchi, C.; Quinkler, M.; Waldmann, J.; Willenberg, H.S.; Beuschlein, F.; Fottner, C.; et al. The role of surgery in the management of recurrent adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Reibetanz, J.; Rinn, B.; Kunz, A.S.; Flemming, S.; Ronchi, C.L.; Kroiss, M.; Deutschbein, T.; Pulzer, A.; Hahner, S.; Kocot, A.; et al. Patterns of Lymph Node Recurrence in Adrenocortical Carcinoma: Possible Implications for Primary Surgical Treatment. Ann. Surg. Oncol. 2019, 26, 531–538. [Google Scholar] [CrossRef]
- Reibetanz, J.; Jurowich, C.; Erdogan, I.; Nies, C.; Rayes, N.; Dralle, H.; Behrend, M.; Allolio, B.; Fassnacht, M.; German, A.C.C.s.g. Impact of lymphadenectomy on the oncologic outcome of patients with adrenocortical carcinoma. Ann. Surg. 2012, 255, 363–369. [Google Scholar] [CrossRef]
- Langenhuijsen, J.; Birtle, A.; Klatte, T.; Porpiglia, F.; Timsit, M.O. Surgical Management of Adrenocortical Carcinoma: Impact of Laparoscopic Approach, Lymphadenectomy, and Surgical Volume on Outcomes-A Systematic Review and Meta-analysis of the Current Literature. Eur. Urol. Focus 2016, 1, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.C.; Miller, C.C., 3rd; Joseph, M.; Huh, W.W.; Hayes-Jordan, A.A.; Austin, M.T. Retroperitoneal lymph node staging in paratesticular rhabdomyosarcoma-are we meeting expectations? J. Surg. Res. 2018, 224, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, A.; Diers, J.; Baum, P.; Weibel, S.; Kastner, C.; Muller, S.; Lock, J.F.; Kohler, F.; Meybohm, P.; Kranke, P.; et al. Systematic review and meta-analysis on volume-outcome relationship of abdominal surgical procedures in Germany. Int. J. Surg. 2021, 86, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Diers, J.; Wagner, J.; Baum, P.; Lichthardt, S.; Kastner, C.; Matthes, N.; Lob, S.; Matthes, H.; Germer, C.T.; Wiegering, A. Nationwide in-hospital mortality following colonic cancer resection according to hospital volume in Germany. BJS Open 2019, 3, 672–677. [Google Scholar] [CrossRef] [Green Version]
- Birkmeyer, J.D.; Siewers, A.E.; Finlayson, E.V.; Stukel, T.A.; Lucas, F.L.; Batista, I.; Welch, H.G.; Wennberg, D.E. Hospital volume and surgical mortality in the United States. N. Engl. J. Med. 2002, 346, 1128–1137. [Google Scholar] [CrossRef]
- Diers, J.; Baum, P.; Wagner, J.C.; Matthes, H.; Pietryga, S.; Baumann, N.; Uttinger, K.; Germer, C.T.; Wiegering, A. Hospital volume following major surgery for gastric cancer determines in-hospital mortality rate and failure to rescue: A nation-wide study based on German billing data (2009–2017). Gastric Cancer 2021, 24, 959–969. [Google Scholar] [CrossRef]
- Fassnacht, M.; Hahner, S.; Polat, B.; Koschker, A.C.; Kenn, W.; Flentje, M.; Allolio, B. Efficacy of adjuvant radiotherapy of the tumor bed on local recurrence of adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2006, 91, 4501–4504. [Google Scholar] [CrossRef] [Green Version]
- Viani, G.A.; Viana, B.S. Adjuvant radiotherapy after surgical resection for adrenocortical carcinoma: A systematic review of observational studies and meta-analysis. J. Cancer Res. Ther. 2019, 15, S20–S26. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Z.; Shen, J.; Lian, X.; Miao, Z.; Shen, J.; Zhang, F. Efficacy of adjuvant radiotherapy for treatment of adrenocortical carcinoma: A retrospective study and an updated meta-analysis. Radiat. Oncol. 2020, 15, 118. [Google Scholar] [CrossRef]
- Jazmati, D.; Butzer, S.; Hero, B.; Ahmad Khalil, D.; Merta, J.; Baumer, C.; Plum, G.; Fuchs, J.; Koerber, F.; Steinmeier, T.; et al. Proton Beam Therapy for Children With Neuroblastoma: Experiences From the Prospective KiProReg Registry. Front. Oncol. 2020, 10, 617506. [Google Scholar] [CrossRef]
- Fassnacht, M.; Libe, R.; Kroiss, M.; Allolio, B. Adrenocortical carcinoma: A clinician’s update. Nat. Reviews. Endocrinol. 2011, 7, 323–335. [Google Scholar] [CrossRef]
- Crona, J.; Baudin, E.; Terzolo, M.; Chrisoulidou, A.; Angelousi, A.; Ronchi, C.L.; Oliveira, C.L.; Nieveen van Dijkum, E.J.M.; Ceccato, F.; Borson-Chazot, F.; et al. ENSAT registry-based randomized clinical trials for adrenocortical carcinoma. Eur. J. Endocrinol. 2021, 184, R51–R59. [Google Scholar] [CrossRef]
- Melo-Leite, A.F.; Elias, P.C.; Teixeira, S.R.; Tucci, S.; Barros, G.E.; Antonini, S.R.; Muglia, V.F.; Elias, J. Adrenocortical neoplasms in adulthood and childhood: Distinct presentation. Review of the clinical, pathological and imaging characteristics. J. Pediatr. Endocrinol. Metab. 2017, 30, 253–276. [Google Scholar] [CrossRef]
- Almeida, M.Q.; Latronico, A.C. The molecular pathogenesis of childhood adrenocortical tumors. Horm. Metab. Res. 2007, 39, 461–466. [Google Scholar] [CrossRef]
- Blavier, L.; Yang, R.M.; DeClerck, Y.A. The Tumor Microenvironment in Neuroblastoma: New Players, New Mechanisms of Interaction and New Perspectives. Cancers 2020, 12, 2912. [Google Scholar] [CrossRef]
- Virgone, C.; Roganovic, J.; Vorwerk, P.; Redlich, A.; Schneider, D.T.; Janic, D.; Bien, E.; Lopez-Almaraz, R.; Godzinski, J.; Osterlundh, G.; et al. Adrenocortical tumours in children and adolescents: The EXPeRT/PARTNER diagnostic and therapeutic recommendations. Pediatr. Blood Cancer 2021, 68 (Suppl. 4), e29025. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Terzolo, M.; Allolio, B.; Baudin, E.; Haak, H.; Berruti, A.; Welin, S.; Schade-Brittinger, C.; Lacroix, A.; Jarzab, B.; et al. Combination chemotherapy in advanced adrenocortical carcinoma. N. Engl. J. Med. 2012, 366, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Allolio, B. What is the best approach to an apparently nonmetastatic adrenocortical carcinoma? Clin. Endocrinol. 2010, 73, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Kratz, C.P.; Achatz, M.I.; Brugieres, L.; Frebourg, T.; Garber, J.E.; Greer, M.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Aspect | Number of Identified Articles |
|---|---|
| Clinical characteristics, relapses, follow-up | 94 |
| Age-dependent clinical characteristics | 40 |
| Tumor stage at diagnosis | 43 |
| Metastasis | 69 |
| (Histo-)pathological characteristics | 31 |
| Druggable targets | 11 |
| Prognostic factors | 65 |
| Treatment modalities | 65 |
| Cohort | Clinical Characteristics | Number of Included Studies | ||||||||||
| n | Years of age | % | % | % | % | % | % | Time | Time (m) | n Relapses | ||
| ACT | Female Patients | Hormonally Active Tumors | Mixed | Androgens | Glucocorticoids | DOD | Interval | Symptoms–Diagnosis | ||||
| Brazilian cohort | 1761 | 3.3 | 70 | 97 | 30 | 55 | 3 | 19 | 1950–2019 | 6.8 | 144 | 37 |
| Non-Brazilian cohort | 1919 | 5.05 | 64 | 86 | 26 | 50 | 14 | 25 | 1950–2020 | 6.00 | 190 | 57 |
| Treatment Details | ||||||||||||
| Number of Patients | Surgery | R0 | >R0 | No Information on the Extent of Surgery | Tumor Spillage | Biopsy | Chemotherapy + Surgery | Only Chemo-Therapy | Radiotherapy | Mitotane | 59 | |
| Combined cohort (n) | 2221 | 2036 | 976 | 235 | 1009 | 69 | 69 | 510 | 18 | 74 | 360 | |
| (%) | 100 | 92 | 72 | 17 | 50 | 3 | 3 | 24 | 3 | 3 | 16 | |
| Tumor Stage Distribution | ||||||||||||
| Number of Patients | I | II | III | IV | 48 | |||||||
| Combined cohort (n) | 2238 | 985 | 568 | 287 | 371 | |||||||
| (%) | 100 | 44 | 25 | 13 | 17 | |||||||
| Characteristics | Age | |||||||
| 0–4 years | 4–14 years | >14 years | ||||||
| N | % | n | % | n | % | p | ||
| Gender | Female | 285 | 65.22% | 98 | 60.12% | 91 | 70.00% | |
| Male | 152 | 34.78% | 65 | 39.88% | 39 | 30.00% | 0.21 | |
| Hormonal activity | No | 23 | 6.93% | 12 | 9.45% | 18 | 23.08% | |
| Mixed | 113 | 34.04% | 34 | 26.77% | 31 | 39.74% | ||
| androgens | 180 | 54.22% | 68 | 53.54% | 13 | 16.67% | ||
| glucocorticoids | 16 | 4.82% | 13 | 10.24% | 16 | 20.51% | <0.00000005 *** | |
| DOD | Yes | 53 | 13.98% | 73 | 45.06% | 82 | 52.23% | |
| No | 326 | 86.02% | 89 | 54.94% | 75 | 47.77% | <0.00000005 *** | |
| Stage | I | 209 | 58.87% | 53 | 37.06% | 31 | 25.00% | |
| II | 78 | 21.97% | 32 | 22.38% | 23 | 18.55% | ||
| III | 44 | 12.39% | 21 | 14.69% | 14 | 11.29% | ||
| IV | 24 | 6.76% | 37 | 25.87% | 56 | 45.16% | <0.00000005 *** | |
| P53/CPS | Yes | 98 | 82.35% | 26 | 72.22% | 5 | 35.71% | |
| No | 21 | 17.65% | 10 | 27.78% | 9 | 64.29% | 0.0013 ** | |
| Chemotherapy | Yes | 53 | 24.54% | 38 | 55.07% | 30 | 55.56% | |
| No | 163 | 75.46% | 31 | 44.93% | 24 | 44.44% | 0.0000001 *** | |
| n = 1312, 37 patients excluded because of inexact information on age (>4 years) | ||||||||
| Characteristics | Age | |||||||
| 0–4 years | >4 years | |||||||
| N | % | n | % | p | ||||
| Gender | Female | 285 | 65.22% | 193 | 64.77% | |||
| Male | 152 | 34.78% | 105 | 35.23% | 0.90 | |||
| Hormonal activity | No | 23 | 6.93% | 31 | 14.76% | |||
| Mixed | 113 | 34.04% | 68 | 32.38% | ||||
| Androgens | 180 | 54.22% | 82 | 39.05% | ||||
| glucocorticoids | 16 | 4.82% | 29 | 13.81% | 0.0000088 *** | |||
| DOD | Yes | 53 | 13.98% | 160 | 49.38% | |||
| No | 326 | 86.02% | 164 | 50.62% | <0.00000005 *** | |||
| Stage | I | 209 | 58.87% | 84 | 30.88% | |||
| II | 78 | 21.97% | 56 | 20.59% | ||||
| III | 44 | 12.39% | 36 | 13.24% | ||||
| IV | 24 | 6.76% | 96 | 35.29% | <0.00000005 *** | |||
| P53/CPS | Yes | 98 | 82.35% | 31 | 62.00% | |||
| No | 21 | 17.65% | 19 | 38.00% | 0.0057 ** | |||
| Chemotherapy | Yes | 53 | 24.54% | 68 | 55.28% | |||
| No | 163 | 75.46% | 55 | 44.72% | <0.00000005 *** | |||
| n = 1349 | ||||||||
| Stage | UICC/WHO 2003 | ENSAT 2008 | UICC 2020 (since 2010) | AJCC, 8th Edition |
|---|---|---|---|---|
| I | T1, N0, M0 | T1, N0, M0 | T1, N0, M0 | T1, N0, M0 |
| II | T2, N0, M0 | T2, N0, M0 | T2, N0, M0 | T2, N0, M0 |
| III | T1–2, N1, M0 T3, N0, M0 | T1–2, N1, M0 T3–4, N0–1, M0 | T1–2, N1, M0 T3–4, N0–1, M0 | T3, N0, M0 T1/2, N1, M0 T4, N0, M0 T3/4, N1, M0 |
| IV | T1–4, N0–1, M1 T3, N1, M0 T4, N0–1, M0 | T1–4, N0–1, M1 | T1–4, N0–1, M1 | T1–4, N0–1, M1 |
| Factors of Poor Survival | Number of Patients | Number of Articles | Description | |
|---|---|---|---|---|
| Advanced stage | 1149 | 23 | A higher tumor stage is associated with poor survival | |
| Pathological grading: Weiss > 3, Wieneke > 3, ENSAT3/4 | 658 | 16 | High pathological tumor score is associated with poor survival | |
| Tumor size >100 g | 1083 | 24 | Large tumor mass is associated with poor survival | |
| Tumor volume >200 cm3 | 1289 | 27 | Large tumor volume is associated with poor survival | |
| >4 years old | 1260 | 24 | Age >4 years is associated with worse outcomes | |
| Metastasis | Distant metastases | 627 | 8 | Existence of metastases (lymph nodes and distant metastases) and tumor relapse are described as negative prognosis parameters |
| Lymph nodes | 416 | 5 | ||
| Relapses | 27 | 1 | ||
| Tumor extension, vascular invasion, and/or venous thrombosis | 779 | 13 | Tumor extension, vascular invasion, and venous thrombosis are associated with worse outcomes | |
| Surgical outcome: | Non-R0 | 636 | 12 | Non R0-resection, biopsy, and tumor spillage are described as negative prognostic markers |
| Biopsy | 154 | 2 | ||
| Tumor spillage | 245 | 6 | ||
| Hormone activity | 110 | 2 | Hormone activity was associated with both better and worse outcomes | |
| Immunohistochemistry | Atypical mitosis | 137 | 4 | Atypical mitosis, aneuploidy, tumor necrosis, high mitotic index with high Ki67 expression are associated with poor outcomes |
| Aneuploidy | 227 | 6 | ||
| Tumor necrosis | 360 | 8 | ||
| High mitotic index (MI), | 410 | 11 | ||
| Ki67 | 348 | 9 | ||
| Target | Intervention | In Vitro Data | Case Reports/Studies | References |
|---|---|---|---|---|
| Crosstalk between YAP1 and Wnt/beta-catenin | Hippo/YAP1 signaling inhibition | yes | none | Abduch et al., 2016 [23] |
| PDL1 expression | Checkpoint inhibition | yes | case reports/several trials | Altieri, 2020, Geoerger, 2020 [131,132] |
| Overexpression of Aurora kinases A | Aurora kinase inhibition | yes | none | Borges, 2013 [26] |
| CDK1, CCNB1, CDC20, and BUB1B | yes | none | Fragoso, 2012; Kulshrestha, 2016 [133,134] | |
| HMGCR-overexpressing tumors | Lovastatin | yes | none | Lin, 2010 [123] |
| Overexpression of IGF1R | IGF1R inhibition | yes | phase ½ | Lira, 2016; Jones, 2015; Weigel, 2014 [33,135,136] |
| Overexpression of VEGFR | VEGFR inhibition/tyrosine kinase inhibition | yes | case reports/several trials | Pianovski, 2013, Altieri, 2020 [131,137] |
| mTOR kinase activity | mTOR inhibition | yes | case reports/several trials | Pianovski, 2013, Altieri, 2020 |
| ATRX | yes | none | Pinto, 2015 [111,131,137] | |
| ZNRF3 | yes | none | Pinto, 2015 [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riedmeier, M.; Decarolis, B.; Haubitz, I.; Müller, S.; Uttinger, K.; Börner, K.; Reibetanz, J.; Wiegering, A.; Härtel, C.; Schlegel, P.-G.; et al. Adrenocortical Carcinoma in Childhood: A Systematic Review. Cancers 2021, 13, 5266. https://doi.org/10.3390/cancers13215266
Riedmeier M, Decarolis B, Haubitz I, Müller S, Uttinger K, Börner K, Reibetanz J, Wiegering A, Härtel C, Schlegel P-G, et al. Adrenocortical Carcinoma in Childhood: A Systematic Review. Cancers. 2021; 13(21):5266. https://doi.org/10.3390/cancers13215266
Chicago/Turabian StyleRiedmeier, Maria, Boris Decarolis, Imme Haubitz, Sophie Müller, Konstantin Uttinger, Kevin Börner, Joachim Reibetanz, Armin Wiegering, Christoph Härtel, Paul-Gerhardt Schlegel, and et al. 2021. "Adrenocortical Carcinoma in Childhood: A Systematic Review" Cancers 13, no. 21: 5266. https://doi.org/10.3390/cancers13215266
APA StyleRiedmeier, M., Decarolis, B., Haubitz, I., Müller, S., Uttinger, K., Börner, K., Reibetanz, J., Wiegering, A., Härtel, C., Schlegel, P. -G., Fassnacht, M., & Wiegering, V. (2021). Adrenocortical Carcinoma in Childhood: A Systematic Review. Cancers, 13(21), 5266. https://doi.org/10.3390/cancers13215266