
Dipeptidyl Peptidase Inhibition Enhances CD8 T Cell Recruitment and Activates Intrahepatic Inflammasome in a Murine Model of Hepatocellular Carcinoma

 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
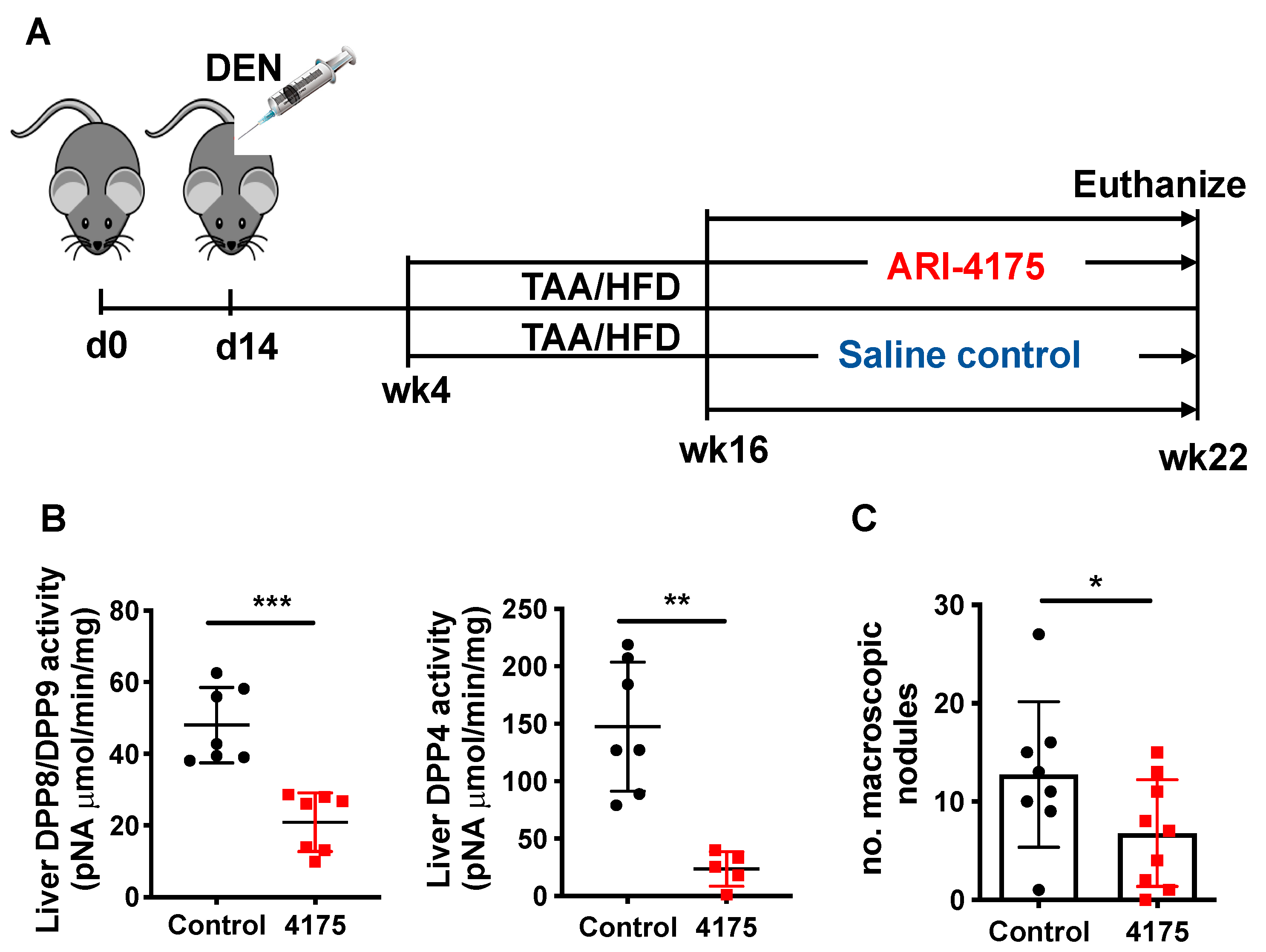
2.1. Mouse Model and Experimental Design
2.2. Enzyme Assay
2.3. Liver Lysate IL-1β Measurement
2.4. Western Blotting (WB)
2.5. Histology, Immunohistochemistry (IHC) and Image Analysis
2.6. Flow Cytometry
2.7. Real-Time Quantitative PCR (qPCR)
2.8. Data Analysis
3. Results
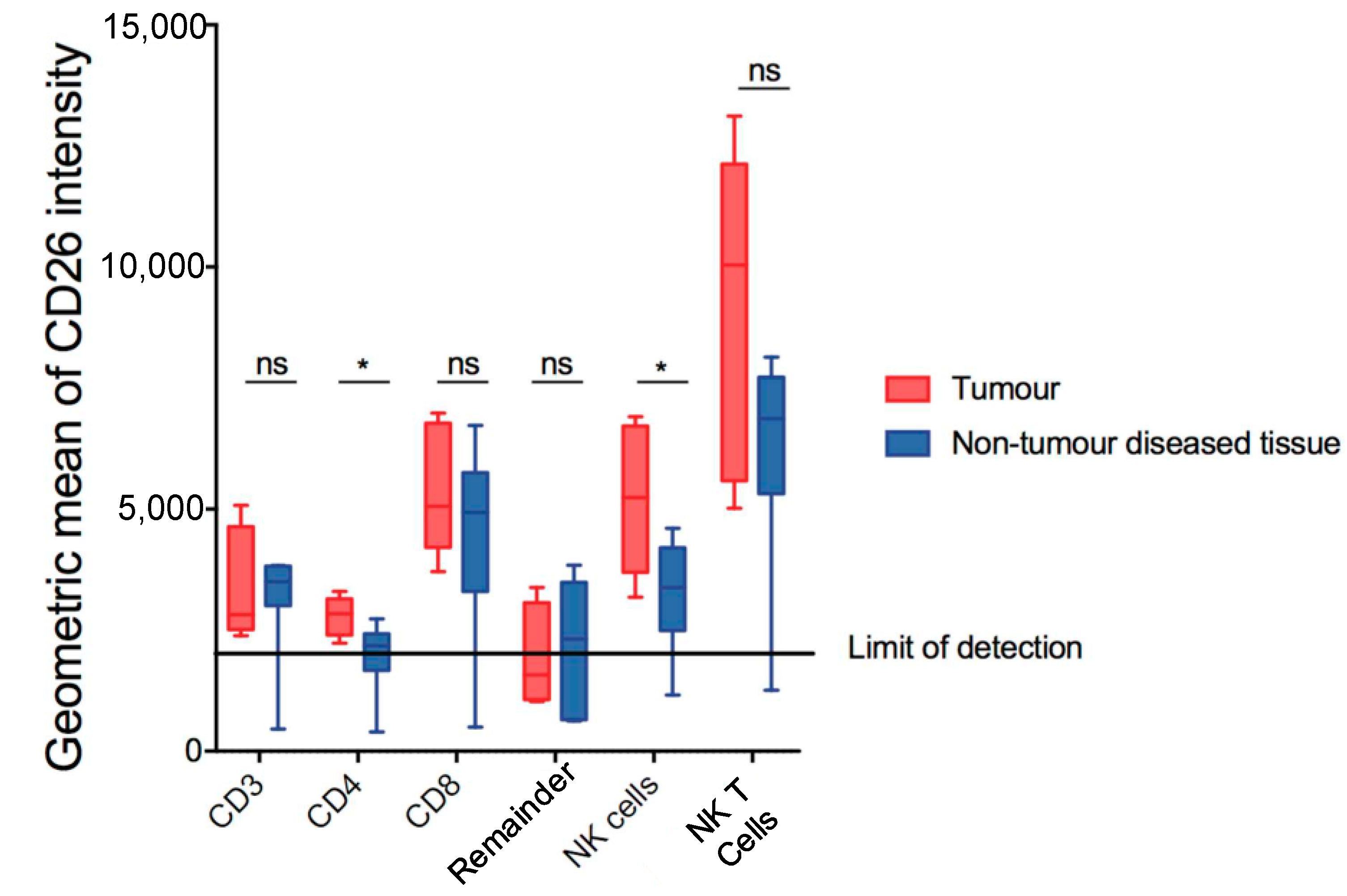
3.1. Dipeptidyl Peptidase in Mouse HCC
3.2. ARI-4175 Decreased Liver Nodule Abundance and Increased Inflammatory Cell Infiltration in the Liver
3.3. ARI-4175 Increased Intrahepatic CD8+ T Cell Density
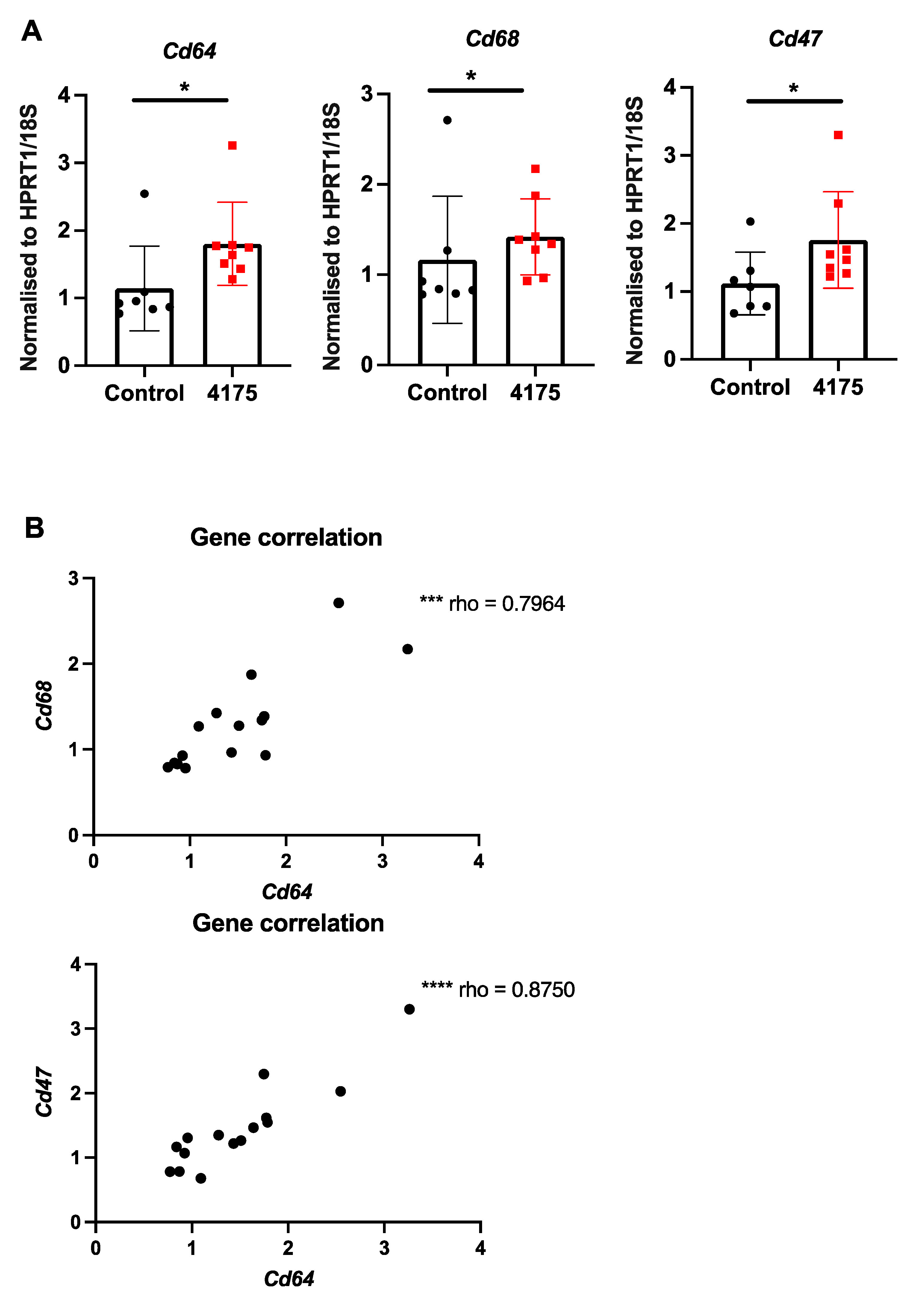
3.4. ARI-4175 Increased Intrahepatic Gene Expression of Macrophage Markers
3.5. ARI-4175 Activation of A Caspase-1 Mediated Inflammasone Pathway
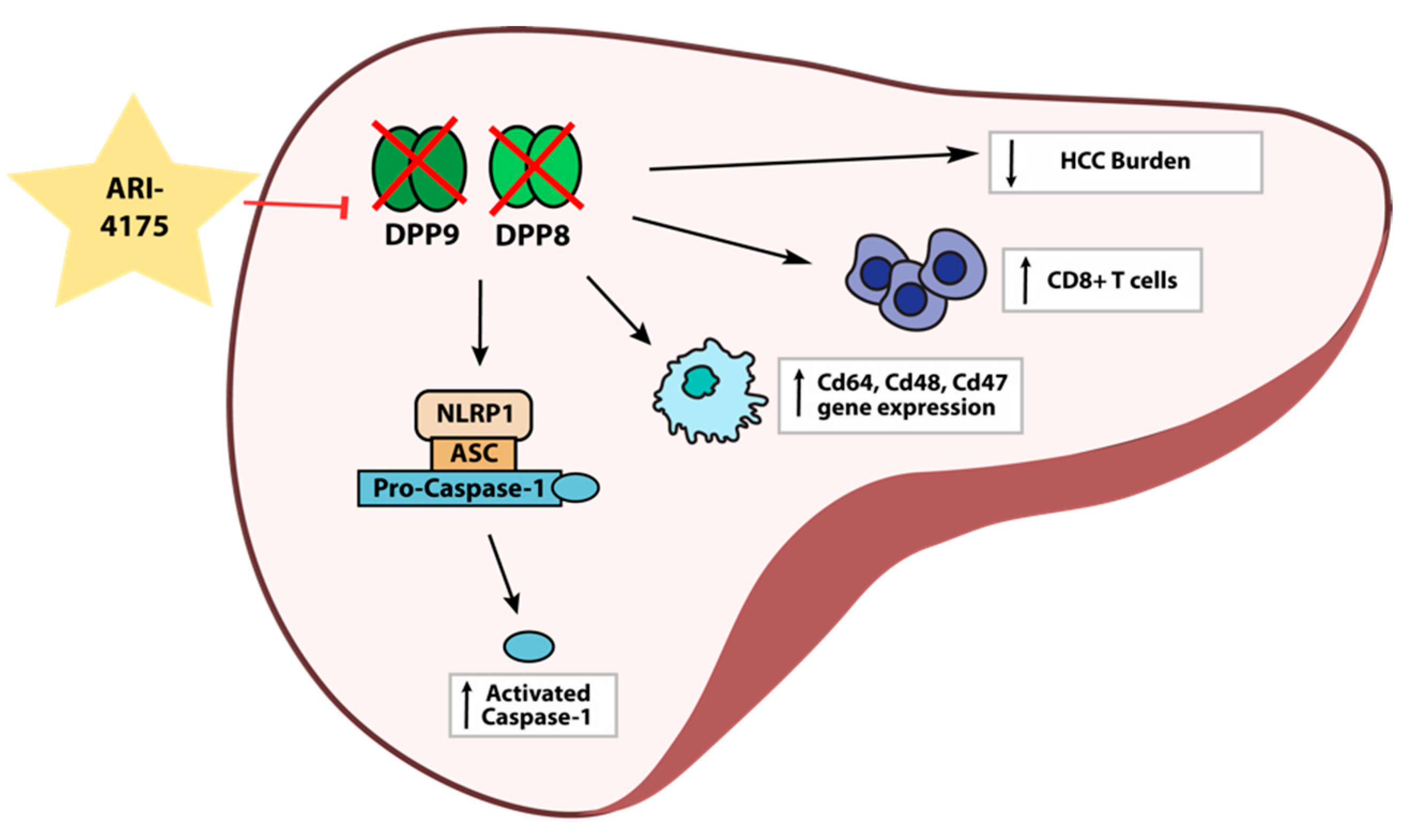
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Jiang, Y.; Yuan, H.; Fang, Q.; Cai, N.; Suo, C.; Jin, L.; Zhang, T.; Chen, X. The trends in incidence of primary liver cancer caused by specific etiologies: Results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J. Hepatol. 2018, 70, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yu, D.M.T.; Yao, T.-W.; Chowdhury, S.; Nadvi, N.A.; Osborne, B.; Church, W.B.; McCaughan, G.W.; Gorrell, M.D. The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J. 2010, 277, 1126–1144. [Google Scholar] [CrossRef]
- Dunaevsky, Y.E.; Tereshchenkova, V.F.; Oppert, B.; Belozersky, M.A.; Filippova, I.Y.; Elpidina, E.N. Human proline specific peptidases: A comprehensive analysis. Biochim. Biophys. Acta (BBA) Gen. Subj. 2020, 1864, 129636. [Google Scholar] [CrossRef]
- Huang, J.; Emran, A.; Endaya, J.; McCaughan, G.; Gorrell, M.; Zhang, H. DPP9: Comprehensive in Silico Analyses of Loss of Function Gene Variants and Associated Gene Expression Signatures in Human Hepatocellular Carcinoma. Cancers 2021, 13, 1637. [Google Scholar] [CrossRef] [PubMed]
- Duncan, B.B.; Highfill, S.L.; Qin, H.; Bouchkouj, N.; Larabee, S.; Zhao, P.; Woznica, I.; Liu, Y.; Li, Y.; Wu, W.; et al. A Pan-Inhibitor of DASH Family Enzymes Induces Immune-mediated Regression of Murine Sarcoma and Is a Potent Adjuvant to Dendritic Cell Vaccination and Adoptive T-cell Therapy. J. Immunother. 2013, 36, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Miller, G.T.; Jesson, M.I.; Watanabe, T.; Jones, B.; Wallner, B.P. PT-100, a Small Molecule Dipeptidyl Peptidase Inhibitor, Has Potent Antitumor Effects and Augments Antibody-Mediated Cytotoxicity via a Novel Immune Mechanism. Cancer Res. 2004, 64, 5471–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.P.; Duncan, B.; Larabee, S.; Krauss, A.; Davis, J.P.; Cui, Y.; Kim, S.Y.; Guimond, M.; Bachovchin, W.; Fry, T.J. Val-BoroPro accelerates T cell priming via modulation of dendritic cell trafficking resulting in complete regression of established murine tumors. PLoS ONE 2013, 8, e58860. [Google Scholar] [CrossRef] [Green Version]
- Donahue, R.N.; Duncan, B.B.; Fry, T.J.; Jones, B.; Bachovchin, W.W.; Kiritsy, C.P.; Lai, J.H.; Wu, W.; Zhao, P.; Liu, Y.; et al. A pan inhibitor of DASH family enzymes induces immunogenic modulation and sensitizes murine and human carcinoma cells to antigen-specific cytotoxic T lymphocyte killing: Implications for combination therapy with cancer vaccines. Vaccine 2014, 32, 3223–3231. [Google Scholar] [CrossRef] [Green Version]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl Peptidase 4 Inhibitors Reduce Hepatocellular Carcinoma by Activating Lymphocyte Chemotaxis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2018, 7, 115–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacovicova, K.; Vinciguerra, M. Inhibition of dipeptidyl peptidase 4 (DPP4) activates immune cells chemotaxis in hepatocellular carcinoma. Oncol. Signal. 2019, 2, 1–3. [Google Scholar] [CrossRef]
- Wilson, A.; Moffitt, L.; Wilson, K.; Bilandzic, M.; Wright, M.; Gorrell, M.; Oehler, M.; Plebanski, M.; Stephens, A. DPP4 Inhibitor Sitagliptin Enhances Lymphocyte Recruitment and Prolongs Survival in a Syngeneic Ovarian Cancer Mouse Model. Cancers 2021, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, L.R.; Sharif, H.; Griswold, A.R.; Fontana, P.; Mintseris, J.; Dagbay, K.B.; Paulo, J.A.; Gygi, S.P.; Bachovchin, D.A.; Wu, H. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 2021, 592, 778–783. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, X.; Toh, G.A.; Gong, Q.; Wang, J.; Han, Z.; Wu, B.; Zhong, F.; Chai, J. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature 2021, 592, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Okondo, M.; Johnson, D.; Sridharan, R.; Bin Go, E.; Chui, A.; Wang, M.; Poplawski, S.E.; Wu, W.; Liu, Y.; Lai, J.H.; et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol. 2016, 13, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Okondo, M.C.; Rao, S.D.; Taabazuing, C.Y.; Chui, A.J.; Poplawski, S.E.; Johnson, D.C.; Bachovchin, D.A. Inhibition of DPP8/9 activates the NLRP1b inflammasome. Cell Chem. Biol. 2018, 25, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.; Taabazuing, C.Y.; Okondo, M.; Chui, A.; Rao, S.; Brown, F.C.; Reed, C.; Peguero, E.; De Stanchina, E.; Kentsis, A.; et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med. 2018, 24, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Polak, N.; Chen, J.; Roediger, B.; Weninger, W.; Kench, J.G.; McCaughan, G.; Zhang, H.E.; Gorrell, M.D. Multiple liver insults synergize to accelerate experimental hepatocellular carcinoma. Sci. Rep. 2018, 8, 10283. [Google Scholar] [CrossRef]
- Grisham, J.W. Interspecies comparison of liver carcinogenesis: Implications for cancer risk assessment. Carcinogenesis 1997, 18, 59–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libbrecht, L.; Desmet, V.; Roskams, T. Preneoplastic lesions in human hepatocarcinogenesis. Liver Int. 2005, 25, 16–27. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research 2016, 5, 879. [Google Scholar] [CrossRef] [PubMed]
- Schlageter, M.; Terracciano, L.M.; D’Angelo, S.; Sorrentino, P. Histopathology of hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 15955–15964. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.M.; Ajami, K.; Gall, M.G.; Park, J.; Lee, C.S.; Evans, K.A.; McLaughlin, E.A.; Pitman, M.R.; Abbott, C.A.; McCaughan, G.; et al. The In Vivo Expression of Dipeptidyl Peptidases 8 and 9. J. Histochem. Cytochem. 2009, 57, 1025–1040. [Google Scholar] [CrossRef]
- Gall, M.G.; Chen, Y.; Ribeiro, A.J.V.d.; Zhang, H.; Bailey, C.G.; Spielman, D.; Yu, D.M.; Gorrell, M.D. Targeted inactivation of Dipeptidyl Peptidase 9 enzyme activity causes mouse neonate lethality. PLoS ONE 2013, 8, e0078378. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, Y.; Wadham, C.; McCaughan, G.W.; Keane, F.M.; Gorrell, M.D. Dipeptidyl peptidase 9 subcellular localization and a role in cell adhesion involving focal adhesion kinase and paxillin. Biochim. Biophys. Acta (BBA) Molec. Cell Res. 2015, 1853, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; Hoek, A.M.V.D. Establishment of a General NAFLD Scoring System for Rodent Models and Comparison to Human Liver Pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.M.; Holz, L.; Chowdhury, S.; Cordoba, S.P.; Evans, K.; Gall, M.G.; Vieira de Ribeiro, A.J.; Zheng, Y.Z.; Levy, M.T.; Yu, D.M.; et al. The pro-fibrotic role of dipeptidyl peptidase 4 in carbon tetrachloride-induced experimental liver injury. Immunol. Cell Biol. 2017, 95, 443–453. [Google Scholar] [CrossRef]
- Bertolino, P.; Bowen, D.G.; McCaughan, G.; Fazekas de St. Groth, B. Antigen-Specific Primary Activation of CD8+T Cells Within the Liver. J. Immunol. 2001, 166, 5430–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierro, F.; Evrard, M.; Rizzetto, S.; Melino, M.; Mitchell, A.; Florido, M.; Beattie, L.; Walters, S.B.; Tay, S.S.; Lu, B.; et al. A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity 2017, 47, 374–388.e6. [Google Scholar] [CrossRef]
- Chowdhury, S.; Chen, Y.; Yao, T.-W.; Ajami, K.; Wang, X.M.; Popov, Y.V.; Schuppan, D.; Bertolino, P.; McCaughan, G.; Yu, D.M.; et al. Regulation of dipeptidyl peptidase 8 and 9 expression in activated lymphocytes and injured liver. World J. Gastroenterol. 2013, 19, 2883–2893. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Knott, H.M.; Nadvi, N.A.; Collyer, C.A.; Wang, X.M.; Church, W.; Gorrell, M.D. Reversible Inactivation of Human Dipeptidyl Peptidases 8 and 9 by Oxidation. Open Enzym. Inhib. J. 2008, 1, 52–60. [Google Scholar] [CrossRef]
- Itou, M.; Kawaguchi, T.; Taniguchi, E.; Sata, M. Dipeptidyl peptidase-4: A key player in chronic liver disease. World J. Gastroenterol. 2013, 19, 2298–2306. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, R.; Hong, Y.; Shah, C.; Ali, A.; Iv, W.P.S.; Huo, J.; Dang, N.H.; Dang, L.H. Dipeptidyl peptidase 4 inhibitors as novel agents in improving survival in diabetic patients with colorectal cancer and lung cancer: A Surveillance Epidemiology and Endpoint Research Medicare study. Cancer Med. 2019, 8, 3918–3927. [Google Scholar] [CrossRef] [Green Version]
- Ghorpade, D.S.; Ozcan, L.; Zheng, Z.; Nicoloro, S.M.; Shen, Y.; Chen, E.; Blüher, M.; Czech, M.P.; Tabas, I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nat. Cell Biol. 2018, 555, 673–677. [Google Scholar] [CrossRef]
- Matsumoto, Y.; McCaughan, G.W.; Painter, D.M.; Bishop, G.A. Evidence that portal tract microvascular destruction precedes bile duct loss in human liver allograft rejection. Transplantation 1993, 56, 69–74. [Google Scholar] [CrossRef]
- McCaughan, G.; Wickson, J.E.; Creswick, P.F.; Gorrell, M.D. Identification of the bile canalicular cell surface molecule GP110 as the ectopeptidase dipeptidyl peptidase IV: An analysis by tissue distribution, purification andN-terminal amino acid sequence. Hepatology 1990, 11, 534–544. [Google Scholar] [CrossRef]
- Waumans, Y.; Vliegen, G.; Maes, L.; Rombouts, M.; Declerck, K.; Veken, P.V.D.; Berghe, W.V.; Meyer, G.R.Y.D.; Schrijvers, D.; Meester, I.D. The dipeptidyl peptidases 4, 8, and 9 in mouse monocytes and macrophages: DPP8/9 inhibition attenuates M1 macrophage activation in mice. Inflammation 2016, 39, 413–424. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Genet. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vasconcelos, N.M.; Vliegen, G.; Gonçalves, A.; De Hert, E.; Martín-Pérez, R.; Van Opdenbosch, N.; Jallapally, A.; Geiss-Friedlander, R.; Lambeir, A.-M.; Augustyns, K.; et al. DPP8/DPP9 inhibition elicits canonical Nlrp1b inflammasome hallmarks in murine macrophages. Life Sci. Alliance 2019, 2, e201900313. [Google Scholar] [CrossRef] [Green Version]
- Bachovchin, D.A. NLRP1: A jack of all trades, or a master of one? Mol. Cell 2021, 81, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Ceccarelli, V.; Cimini, F.A.; Barone, E.; Sentinelli, F.; Coluzzi, M.; Chiappetta, C.; Bertoccini, L.; Tramutola, A.; Labbadia, G.; et al. Circulating dipeptidyl peptidase-4 is independently associated with the presence and severity of NAFLD/NASH in individuals with and without obesity and metabolic disease. J. Endocrinol. Investig. 2020, 44, 979–988. [Google Scholar] [CrossRef]
- Williams, K.; de Ribeiro, A.V.; Prakoso, E.; Veillard, A.; Shackel, N.; Bu, Y.; Brooks, B.; Cavanagh, E.; Raleigh, J.; McLennan, S.; et al. Lower serum fibroblast activation protein shows promise in the exclusion of clinically significant liver fibrosis due to non-alcoholic fatty liver disease in diabetes and obesity. Diabetes Res. Clin. Pr. 2015, 108, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.H.; De Ribeiro, A.J.V.; Prakoso, E.; Veillard, A.-S.; Shackel, N.A.; Brooks, B.; Bu, Y.; Cavanagh, E.; Raleigh, J.; McLennan, S.V.; et al. Circulating dipeptidyl peptidase-4 activity correlates with measures of hepatocyte apoptosis and fibrosis in non-alcoholic fatty liver disease in type 2 diabetes mellitus and obesity: A dual cohort cross-sectional study. J. Diabetes 2015, 7, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, R.; Stuhec, K.; Dietrich, R. Changes of dipeptidyl peptidase IV as a membrane marker of lymphocytes in acute and chronic liver diseases--biochemical and cytochemical investigations. Physiol. Res. 1991, 40, 95–102. [Google Scholar]
- Uitte de Willige, S.; Keane, F.M.; Bowen, D.G.; Malfliet, J.J.M.C.; Zhang, H.E.; Maneck, B.; McCaughan, G.W.; Leebeek, F.W.G.; Rijken, D.C.; Gorrell, M.D. Circulating fibroblast activation protein activity and antigen levels correlate strongly when measured in liver disease and coronary heart disease. PLoS ONE 2017, 12, e0178987. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; McCaughan, G.; Marinos, G.; Gorrell, M.D. Intrahepatic expression of the hepatic stellate cell marker fibroblast activation protein correlates with the degree of fibrosis in hepatitis C virus infection. Liver Int. 2002, 22, 93–101. [Google Scholar] [CrossRef]
- Wang, X.M.; Yu, D.M.T.; McCaughan, G.W.; Gorrell, M.D. Fibroblast activation protein increases apoptosis, cell adhesion and migration by the LX-2 human stellate cell line. Hepatology 2005, 42, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Minoux, M.; Piaia, A.; Kueng, B.; Gapp, B.; Weber, D.; Haller, C.; Barbieri, S.; Namoto, K.; Lorenz, T.; et al. DPP9 enzyme activity controls survival of mouse migratory tongue muscle progenitors and its absence leads to neonatal lethality due to suckling defect. Dev. Biol. 2017, 431, 297–308. [Google Scholar] [CrossRef]
- Gall, M.G.; Zhang, H.E.; Lee, Q.; Jolly, C.; McCaughan, G.; Cook, A.; Roediger, B.; Gorrell, M.D. Immune regeneration in irradiated mice is not impaired by the absence of DPP9 enzymatic activity. Sci. Rep. 2019, 9, 7292. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, K.; Li, Y.; Zhao, W.; Wang, L.; Chen, Z.; Ma, X.; Yao, T.; Wang, J.; Dong, W.; et al. Profibrotic mechanisms of DPP8 and DPP9 highly expressed in the proximal renal tubule epithelial cells. Pharmacol. Res. 2021, 169, 105630. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G. Immune Therapy for Cancer. Annu. Rev. Immunol. 2009, 27, 83–117. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2017, 75, 689–713. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Hamson, E.J.; Keane, F.M.; Tholen, S.; Schilling, O.; Gorrell, M.D. Understanding fibroblast activation protein (FAP): Substrates, activities, expression and targeting for cancer therapy. Proteom. Clin. Appl. 2014, 8, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.E.; Hamson, E.J.; Koczorowska, M.M.; Tholen, S.; Chowdhury, S.; Bailey, C.G.; Lay, A.J.; Twigg, S.M.; Lee, Q.; Roediger, B.; et al. Identification of Novel Natural Substrates of Fibroblast Activation Protein-alpha by Differential Degradomics and Proteomics. Mol. Cell. Proteom. 2019, 18, 65–85. [Google Scholar] [CrossRef] [Green Version]
- Brennen, W.N.; Thorek, D.L.J.; Jiang, W.; E Krueger, T.; Antony, L.; Denmeade, S.R.; Isaacs, J.T. Overcoming stromal barriers to immuno-oncological responses via fibroblast activation protein-targeted therapy. Immunotherapy 2021, 13, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Nakano, D.; Koga, H.; Torimura, T. Effects of a DPP4 Inhibitor on Progression of NASH-related HCC and the p62/ Keap1/Nrf2-Pentose Phosphate Pathway in a Mouse Model. Liver Cancer 2019, 8, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Davoodi, J.; Kelly, J.; Gendron, N.H.; MacKenzie, A.E. The Simpson–Golabi–Behmel syndrome causative Glypican-3, binds to and inhibits the dipeptidyl peptidase activity of CD26. Proteomics 2007, 7, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Bachovchin, W.W.; Lai, H.-S.; Wu, W. Combination Therapies Using Immuno-DASH Inhibitors and PGE2 Antagonists; Tufts: Medford, MA, USA, 2017; Available online: https://patents.google.com/patent/WO2018049027A1/en?inventor=Wengen+Wu&page=1 (accessed on 9 July 2020).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Supplier | Catalogue | Dilution | Application |
|---|---|---|---|---|---|
| Activated caspase-1 (P20) | Mouse | AdipoGen Life Sciences, San Diego, CA, USA | AG-20B-oo48-C100 | 1:1000 | WB |
| Alpha-SMA | Rabbit | Abcam, Cambridge, UK | AB32575 | 1:100 | IHC |
| Anti-mouse-HRP | Rabbit | Dako, Glostrup, Denmark | P0161 | 1:5000 | WB |
| Anti-rabbit-HRP | Goat | Dako | P0448 | 1:5000 | WB |
| Anti-rat-HRP | Goat | Invitrogen, Carlsbad, CA | 31470 | 1:100 | IHC |
| β-actin | Mouse | Abcam | Ab49900 | 1:50,000 | WB |
| Beclin-1 | Mouse | Genetex, Irvine, CA, USA | GTX34055 | 1:1500 | WB |
| CD4 | Rabbit | Cell Signaling Technology, Danvers, MA, USA | 25229 | 1:50 | IHC |
| CD8 | Rabbit | Cell Signaling Technology | 98941 | 1:150 | IHC |
| Dipeptidyl peptidase 9 | Rabbit | Abcam | Ab42080 | 1:100 | IHC |
| F4/80 | Rat | Dr Patrick Bertolino, Centenary Institute, Sydney, Australia | Hybridoma A3-1 | neat | IHC |
| LC3B | Rabbit | Genetex | GTX127375 | 1:500–1:3000 | IHC 1:500 WB 1:3000 |
| Pro-caspase-1 | Mouse | AdipoGen Life Sciences | AG-20B-0042-C100 | 1:1000 | WB |
| Antibody; Conjugate | Origin | Clone | Company | Catalogue | Dilution |
|---|---|---|---|---|---|
| CD45 BUV395 | Rat (LOU) Anti-mouse IgG2b, κ | 30-F11 | BD Horizon | 564279 | 1/100 |
| CD3e BV421 | Hamster Anti-mouse IgG1k | 145-2C11 | BD Horizon | 562600 | 1/200 |
| NK1.1 PE | Mouse Anti-mouse IgG2a, κ | PK136 | BD Pharmingen | 557391 | 1/100 |
| CD26 PerCP-Cy5.5 | Rat Anti-mouse IgG2a, κ | H194-112 | eBioscience | 45-0261-82 | 1/100 |
| CD8a APC | Rat Anti-mouse IgG2a, κ | 53–6.7 | BD Pharmingen | 553035 | 1/100 |
| CD4 APC-Cy7 | Rat Anti-mouse IgG2b, κ | GK1.5 | BD Pharmingen | 552051 | 1/100 |
| Gene Name | Gene Symbol | Assay ID | Amplicon Length |
|---|---|---|---|
| Caspase-1 | Casp1 | Mm00438023_m1 | 99 |
| Caspase-3 | Casp3 | Mm01195085_m1 | 70 |
| CD47 antigen | Cd47 | Mm00495011_m1 | 77 |
| CD64 antigen | Cd64 | Mm00438874_m1 | 58 |
| CD68 antigen | Cd68 | Mm00839636_g1 | 86 |
| Chemokine (C-C motif) ligand 2 | Ccl2 | Mm99999056_m1 | 96 |
| Chemokine (C-C motif) ligand 5 | Ccl5 | Mm01302427_m1 | 103 |
| Chemokine (C-C motif) receptor 2 | Ccr2 | Mm99999051_gH | 60 |
| Chemokine (C-C motif) receptor 3 | Cxcr3 | Mm99999054_s1 | 57 |
| Chemokine (C-X-C motif) ligand 10 | Cxcl10 | Mm00445235_m1 | 59 |
| Chemokine (C-X3-X motif) receptor 1 | Cx3cr1 | Mm02620111_s1 | 107 |
| Collagen, type I, alpha 2 | Col1a2 | Mm00483937_m1 | 77 |
| Collagen, type III, alpha 1 | Col3a1 | Mm00802300_m1 | 88 |
| Dipeptidyl peptidase 4 | Dpp4 | Mm00494538_m1 | 88 |
| Dipeptidyl peptidase 8 | Dpp8 | Mm00547049_m1 | 95 |
| Dipeptidyl peptidase 9 | Dpp9 | Mm00841122_m1 | 61 |
| Eukaryotic 18S rRNA | 18S | Hs99999901_s1 | 187 |
| Fibroblast activation protein | Fap | Mm00484254_m1 | 107 |
| Glypican 3 | Gpc3 | Mm00516722_m1 | 91 |
| Hypoxanthine phosphoribosyltransferase 1 | Hprt1 | Mm00446968_m1 | 65 |
| Interleukin 1 beta | Il-1β | Mm00434228_m1 | 90 |
| Interleukin 18 | Il-18 | Mm00434226_m1 | 141 |
| NLR family, pyrin domain containing 1B | Nlrp1b | Mm01241387_m1 | 68 |
| NLR family, pyrin domain containing 3 | Nlrp3 | Mm00840904_m1 | 84 |
| Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha | Nfkbia | Mm00477798_m1 | 70 |
| Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, beta | Nfkbib | Mm00456853_m1 | 64 |
| Ribosomal protein L37a | Rpl37a | Mm01546394_s1 | 111 |
| Tumour necrosis factor | Tnf | Mm00443258_m1 | 81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henderson, J.M.; Xiang, M.S.W.; Huang, J.C.; Wetzel, S.; Jiang, L.; Lai, J.H.; Wu, W.; Kench, J.G.; Bachovchin, W.W.; Roediger, B.; et al. Dipeptidyl Peptidase Inhibition Enhances CD8 T Cell Recruitment and Activates Intrahepatic Inflammasome in a Murine Model of Hepatocellular Carcinoma. Cancers 2021, 13, 5495. https://doi.org/10.3390/cancers13215495
Henderson JM, Xiang MSW, Huang JC, Wetzel S, Jiang L, Lai JH, Wu W, Kench JG, Bachovchin WW, Roediger B, et al. Dipeptidyl Peptidase Inhibition Enhances CD8 T Cell Recruitment and Activates Intrahepatic Inflammasome in a Murine Model of Hepatocellular Carcinoma. Cancers. 2021; 13(21):5495. https://doi.org/10.3390/cancers13215495
Chicago/Turabian StyleHenderson, James M., Michelle S. W. Xiang, Jiali Carrie Huang, Stefanie Wetzel, Linxuan Jiang, Jack H. Lai, Wengen Wu, James G. Kench, William W. Bachovchin, Ben Roediger, and et al. 2021. "Dipeptidyl Peptidase Inhibition Enhances CD8 T Cell Recruitment and Activates Intrahepatic Inflammasome in a Murine Model of Hepatocellular Carcinoma" Cancers 13, no. 21: 5495. https://doi.org/10.3390/cancers13215495
APA StyleHenderson, J. M., Xiang, M. S. W., Huang, J. C., Wetzel, S., Jiang, L., Lai, J. H., Wu, W., Kench, J. G., Bachovchin, W. W., Roediger, B., McCaughan, G. W., Zhang, H. E., & Gorrell, M. D. (2021). Dipeptidyl Peptidase Inhibition Enhances CD8 T Cell Recruitment and Activates Intrahepatic Inflammasome in a Murine Model of Hepatocellular Carcinoma. Cancers, 13(21), 5495. https://doi.org/10.3390/cancers13215495