
A Focus on Regulatory Networks Linking MicroRNAs, Transcription Factors and Target Genes in Neuroblastoma

 , ,
, ,  ,
, 

Abstract
:Simple Summary
Abstract
1. Neuroblastoma: Genetic Determinants and Developmental Origin
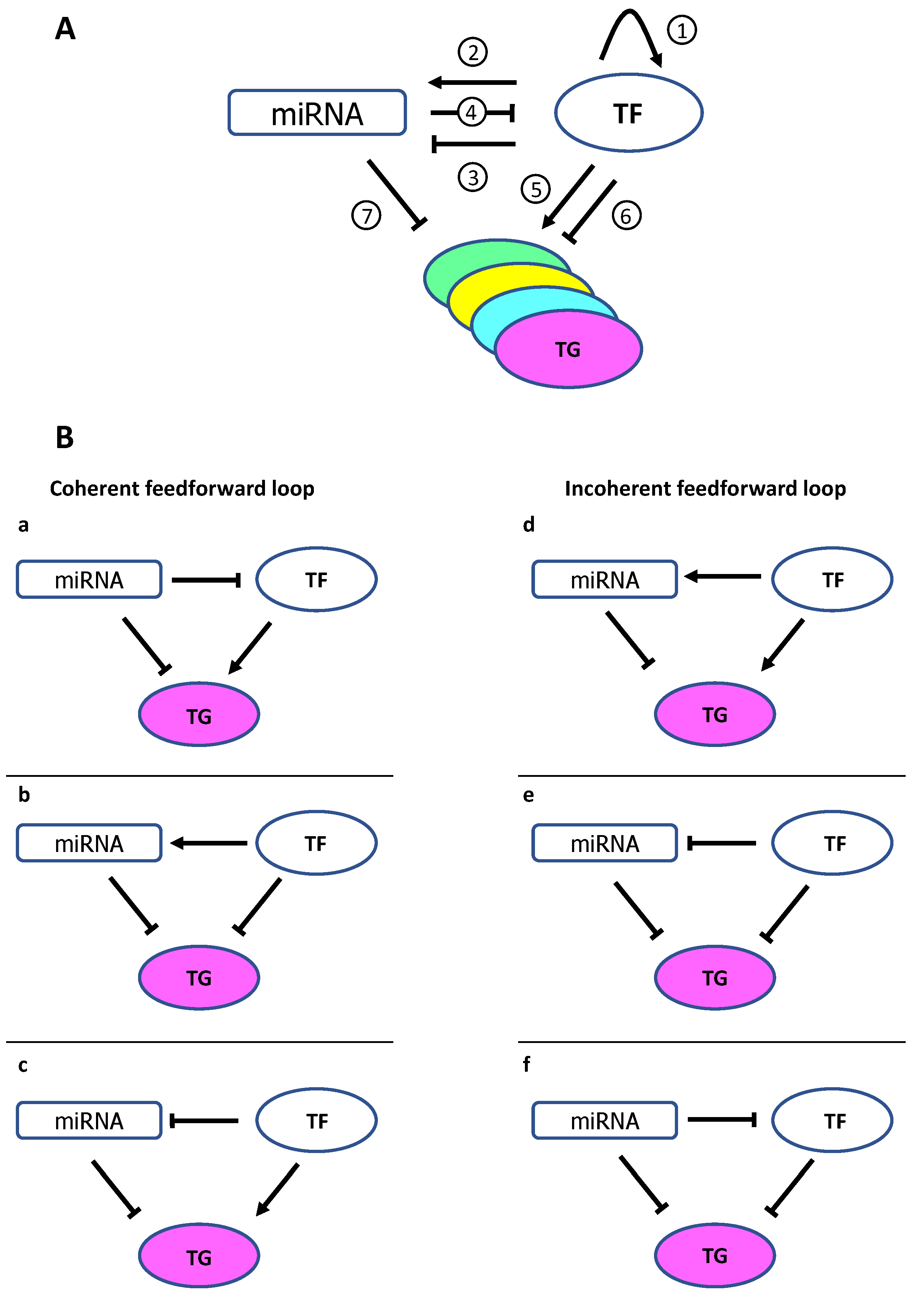
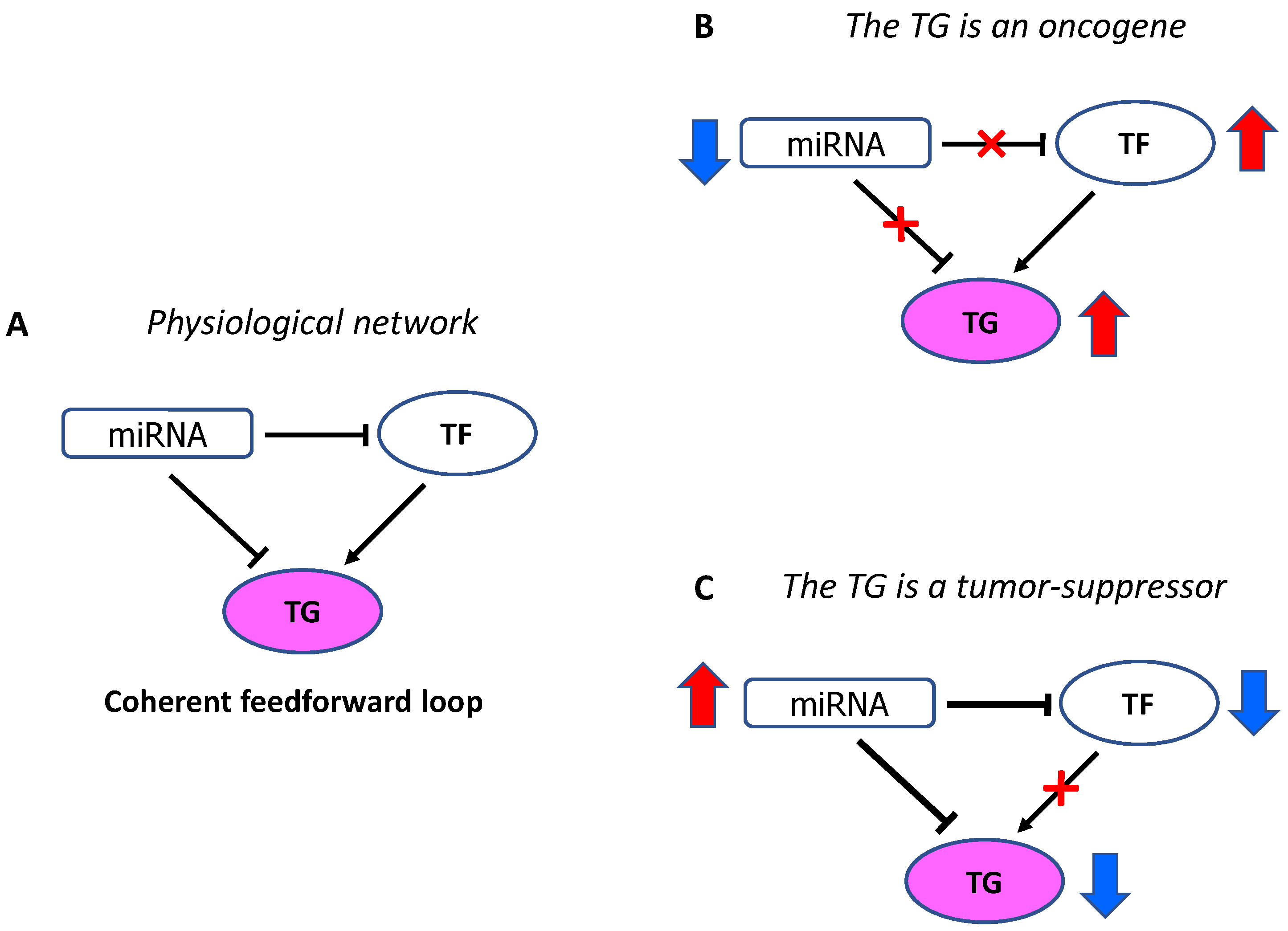
2. MicroRNA and Transcription Factor Co-Regulation
3. MicroRNAs and Transcription Factors in Neuronal and Neural Crest Development
4. MicroRNAs and Cancer
5. Tumor Suppressor miRNAs in Neuroblastoma
5.1. miR-34
5.2. Let-7
5.3. miR-204
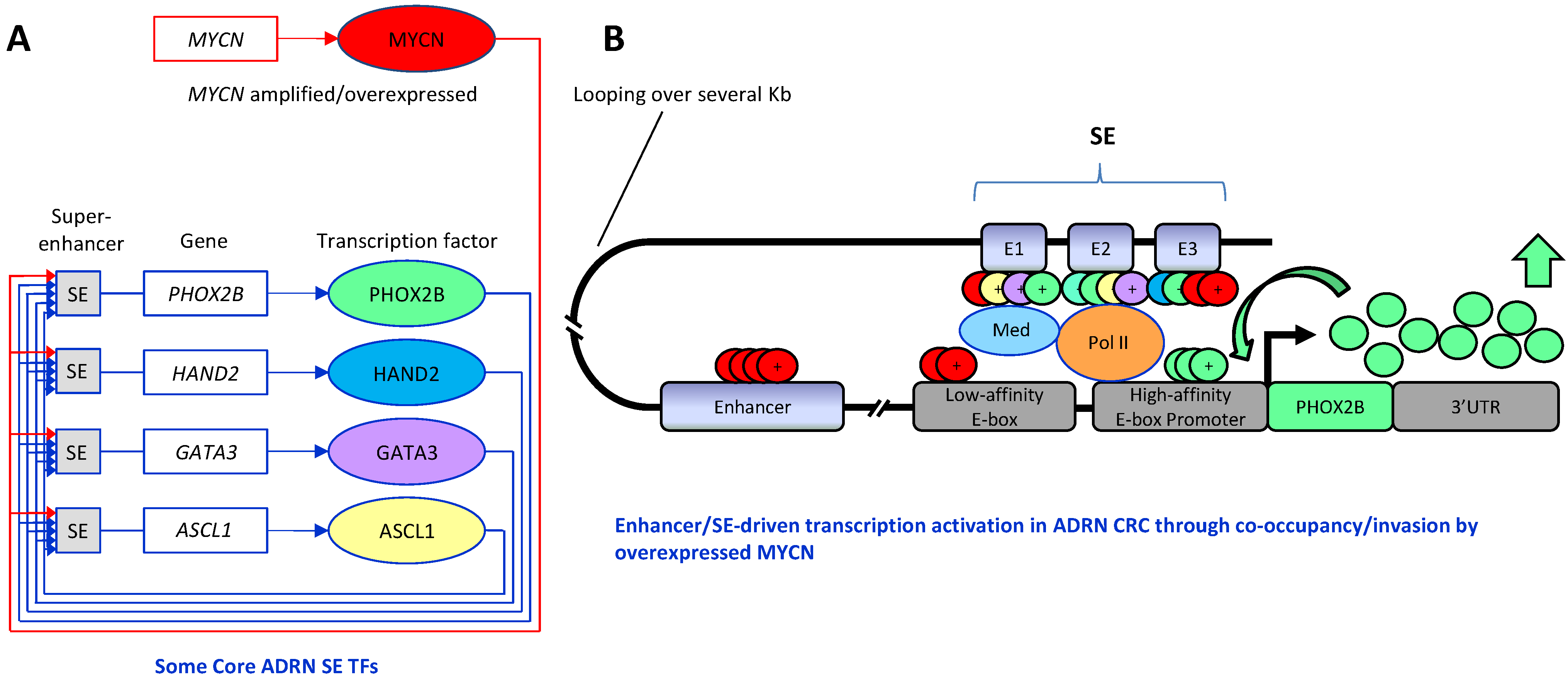
6. Key Transcription Factors and Target Genes in Neuroblastoma
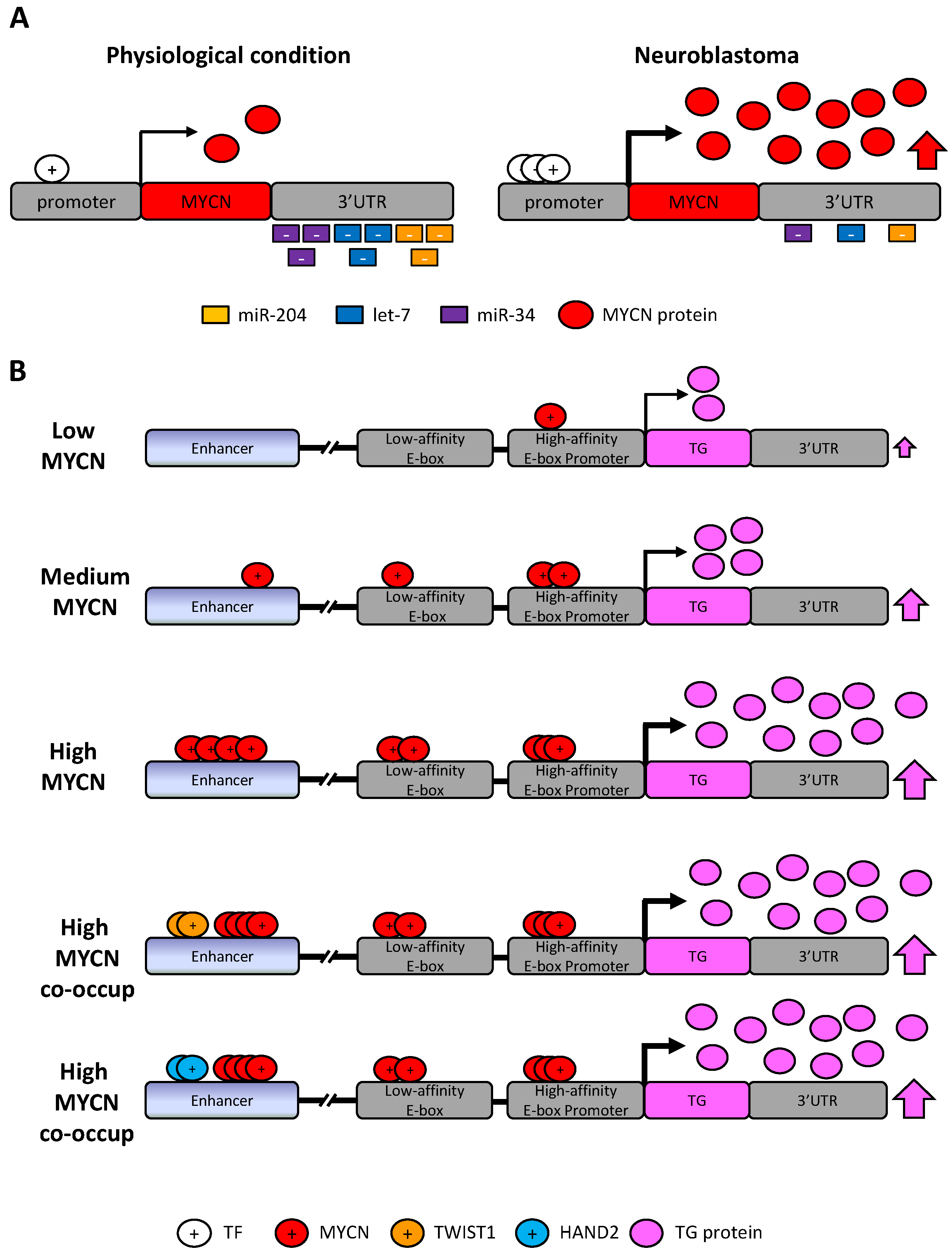
6.1. MYCN and c-MYC
6.2. PHOX2B
6.3. ALK
6.4. LIN28B
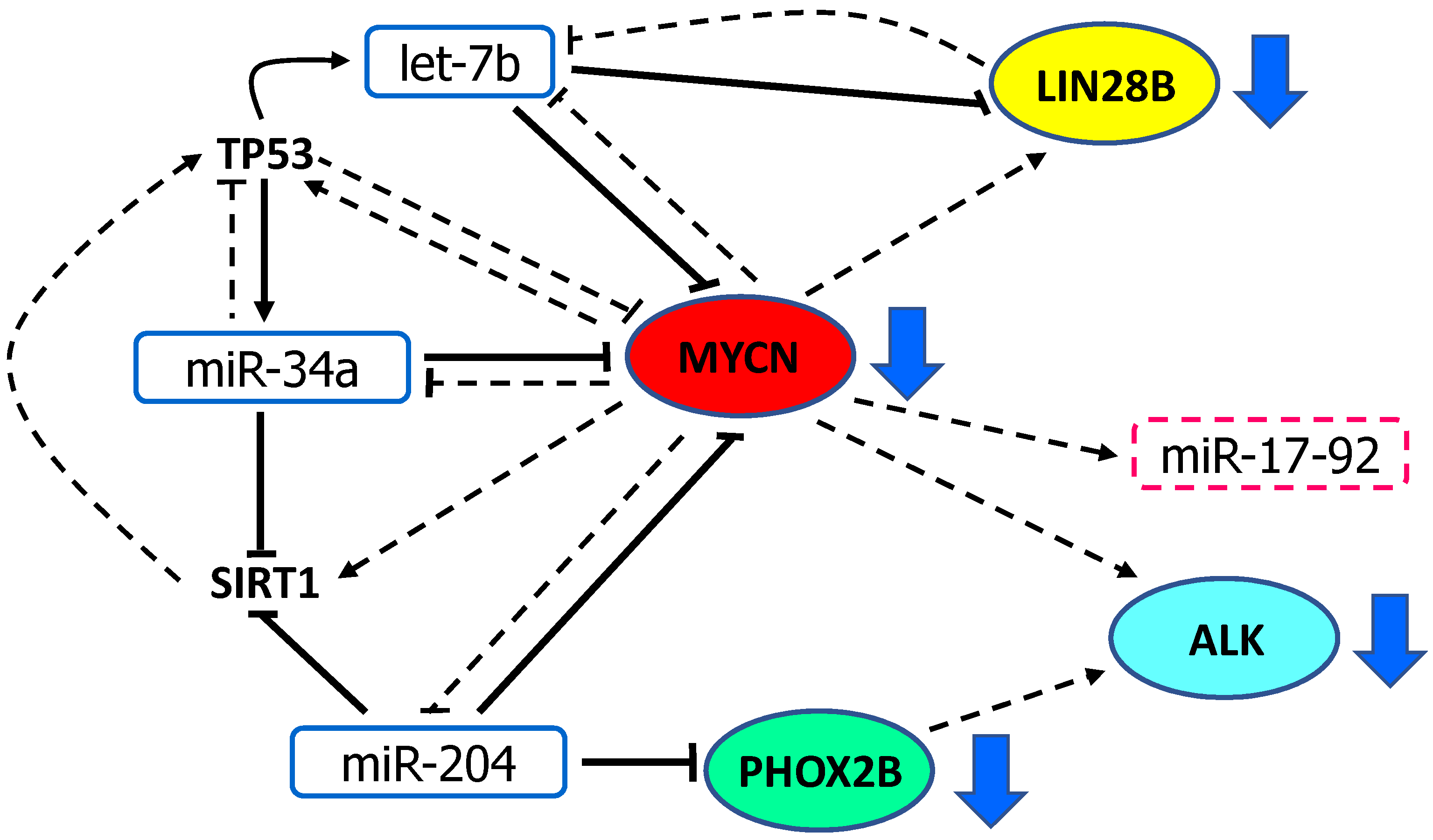
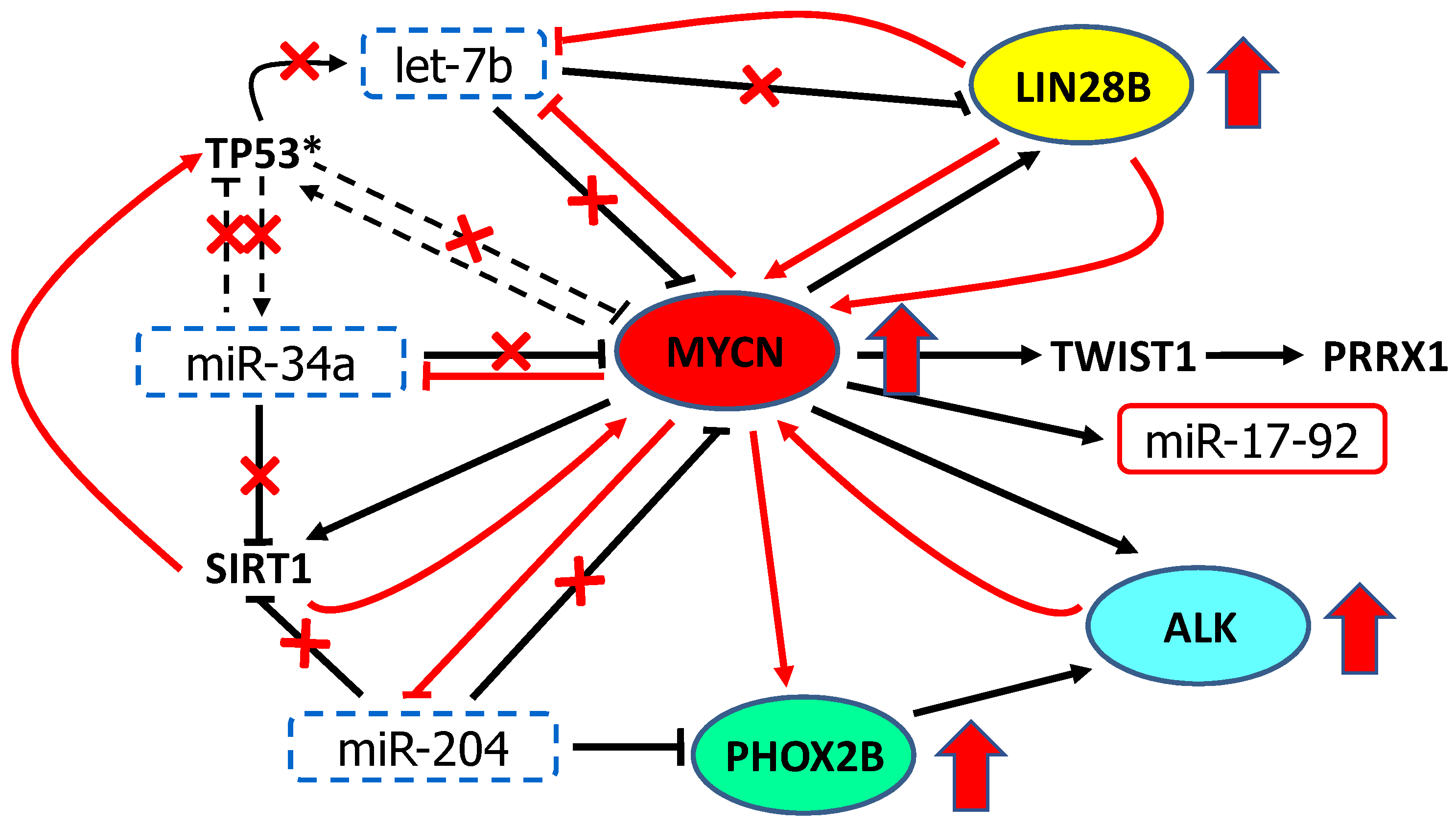
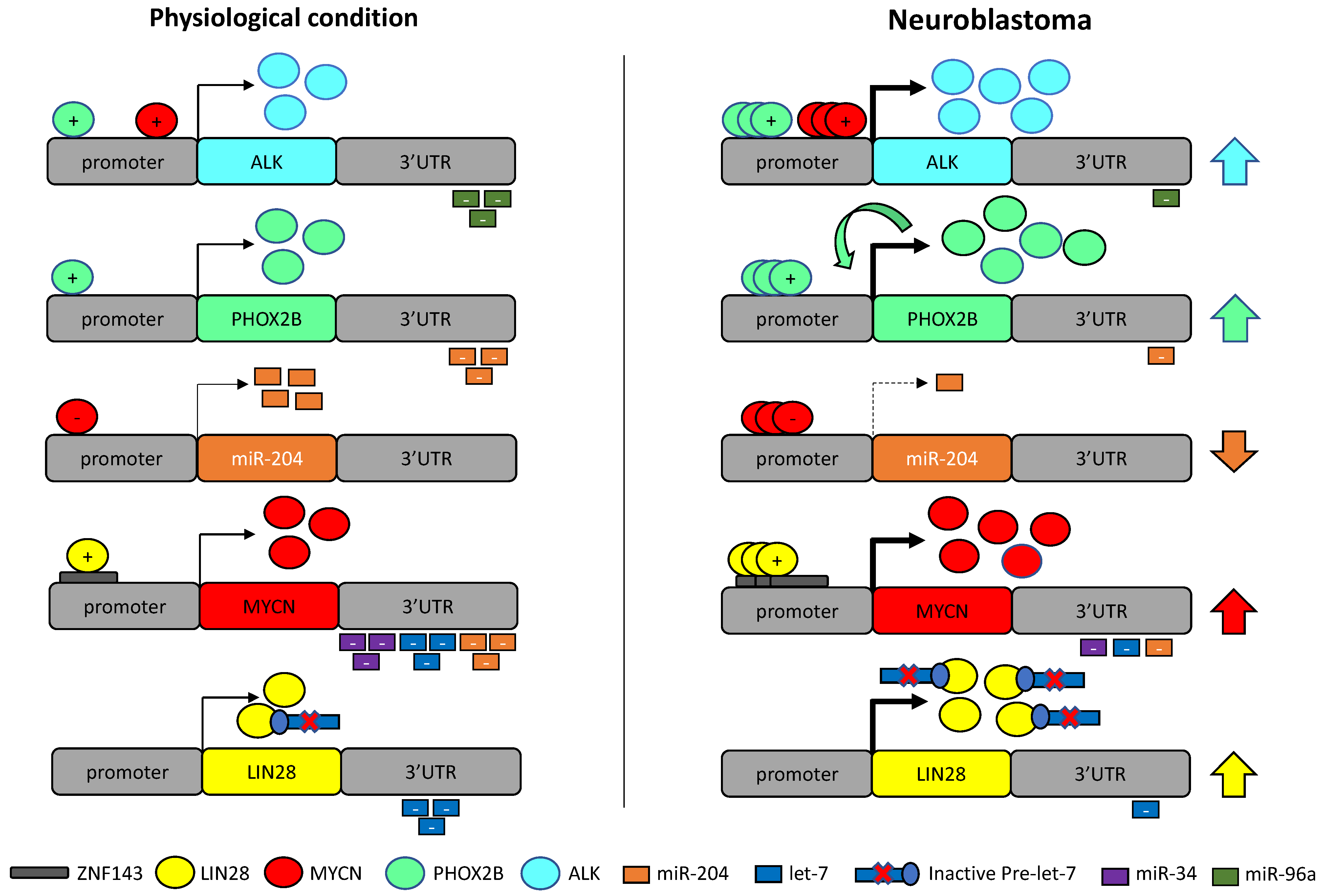
7. Discussion: Pathologic Dysregulation of miR-34a, let-7b, miR-204 and MYCN, PHOX2B, ALK and LIN28B in Neuroblastoma
8. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCHS | Congenital Central Hypoventilation Syndrome |
| ChIP-seq | Chromatin Immunoprecipitation Sequencing |
| E10.5, E11.5 | Embryonic Developmental Mouse Timeline Expressed in Days |
| EMT | Epithelial-to-Mesenchymal Transition |
| ESCs | Embryonic Stem Cells |
| FS | Frameshift |
| HD | Homeodomain |
| HSCR | Hirschsprung Disease |
| iPSC/s | Induced Pluripotent Stem Cell/s |
| miRNA | microRNA |
| MRX34 | Liposomal Formulation of miR-34a Mimics |
| MS | Missense |
| NC | Neural Crest |
| NCCs | Neural Crest Cells |
| NICDs | Notch Intracellular Domains |
| NSCs | Neural Stem Cells |
| RNAi | RNA Interference |
| scRNA-seq | Single-Cell RNA Sequencing |
| shRNA | Short Hairpin RNA |
| siRNA | Small Interference RNA |
| SNPs | Single Nucleotide Polymorphisms |
| SNS | Sympathetic Nervous System |
| UTR | Untraslated Region |
| AGO2 | Argonaute RISC Catalytic Component 2 |
| AKT | Serine/Threonine Kinase |
| ALK/ALK | Anaplastic Lymphoma Kinase (receptor tyrosine kinase) |
| ALKAL2 | ALK Additionally, LTK Ligand 2 |
| AP-1 | Activator Protein-1 |
| Ascl1/ASCL1 | Achaete-Scute Family bHLH Transcription Factor 1 |
| BANCR | BRAF-activated non-protein coding RNA |
| bHLH | Basic Helix-Loop-Helix |
| BLIMP1 | B-Lymphocyte-Induced Maturation Protein 1 |
| BMI1 | BMI1 Polycomb Ring Finger Proto-Oncogene |
| BMP | Bone Morphogenetic Protein |
| CCL2 | C-C Motif Chemokine Ligand 2 |
| CCNA2 | Cyclin A2 |
| CCNG2 | Cyclin G2 |
| CDK4 | Cyclin dependent kinase 7 |
| CDKL5 | Cyclin Dependent Kinase Like 5 |
| CDKN1A | Cyclin Dependent Kinase Inhibitor 1A |
| CHK1 | Checkpoint Kinase 1 |
| c-Myc/c-Myc | c-MYC proto-oncogene; homolog of v-myc avian myelocytomatosis viral related oncogene (hematopoietic cancer-derived), bHLH Transcription Factor |
| CRMP1 | Collapsin Response Mediator Protein 1 |
| c-SRC | SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase |
| CYP26A1 | Cytochrome P450 Family 26 Subfamily A Member 1 |
| DBH | Dopamine Beta-hydroxylase |
| DDC | DCC Netrin 1 Receptor |
| DKC1 | Dyskerin Pseudouridine Synthase 1 |
| DKK1 | Dickkopf WNT Signaling Pathway Inhibitor 1 |
| DLL3 | Delta Like Canonical Notch Ligand 3 |
| E2F8 | E2F Transcription Factor 8 |
| EFNB3 | Ephrin-B3 |
| EGFR | Epidermal Growth Factor Receptor |
| ERK or MAPK1 | Mitogen-Activated Protein Kinase 1 |
| ERK3 or MAPK12 | Mitogen-Activated Protein Kinase 12 |
| ERK5 or MAPK7 | Mitogen-Activated Protein Kinase 7 |
| FAK | Focal Adhesion Kinase |
| FGF | Fibroblast Growth Factor |
| FOS | Fos Proto-Oncogene |
| Foxd3 | Forkhead Box D3 |
| FZD7 | Frizzled Class Receptor 7 |
| GAP43 | Growth Associated Protein 43 |
| Gata2/GATA2 | GATA Binding Protein 2 |
| Gata3/GATA3 | GATA Binding Protein 3 |
| Hand2/HAND2 | Heart and Neural Crest Derivatives Expressed 2 |
| HDAC1 | Histone Deacetylase 1 |
| HES | Hairy and Enhancer of Split |
| HMGA2 | High Mobility Group AT-Hook 2 |
| HRAS | Harvey Rat Sarcoma Virus Proto-Oncogene |
| HuC | RNA-Binding Protein HuC |
| HuD | RNA-Binding Protein HuD |
| ID2 | Inhibitor of DNA Binding 2 |
| IL-6 | Interleukin 6 |
| INHBA | Inhibitor of DNA Binding 2 |
| ISL1 | Islet LIM Homeobox 1 |
| JUN | Jun Proto-Oncogene |
| Klf2, KLF2 | Kruppel Like Factor 2 |
| Klf4, KLF4 | Kruppel Like Factor 4 |
| KRAS | Kirsten Rat Sarcoma Virus |
| Lif | LIF Interleukin 6 Family Cytokine |
| LIN28A | Lin-28 Homolog A |
| Lin28b/LIN28B | Lin-28 Homolog B |
| LMO1 | LIM Domain Only 1 |
| LMO4 | LIM Domain Only 4 |
| MAML3 | Mastermind Like Transcriptional Coactivator 3 |
| MAPK | Mitogen-Activated Protein Kinase |
| MAX | MYC Associated Factor X |
| MCM | Minichromosome Maintenance Protein Complex |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MDM2 | Murine Double Minute 2 |
| MEIS | Meis Homeobox |
| MEK3 or MAPK3 | Mitogen-Activated Protein Kinase 3 |
| MEK5 or MAPK5 | Mitogen-Activated Protein Kinase 5 |
| MET | MET Proto-Oncogene, Receptor Tyrosine Kinase |
| Miz-1/MIZ-1 | Zinc Finger MIZ-Type Containing 1 |
| MKP3 | Mitogen-Activated Protein Kinase Phosphatase 3 |
| MSX1 | Msh Homeobox 1 |
| MYBL2 | MYB Proto-Oncogene Like 2 |
| MYC | MYC Transcription factor family, bHLH |
| Myc-ERT | Tamoxifen-Inducible Myc |
| MYCL | MYCL Proto-Oncogene; homolog of v-myc avian myelocytomatosis viral related oncogene (lung cancer-derived), bHLH Transcription Factor |
| Mycn/MYCN | MYCN Proto-Oncogene; homolog of v-myc avian myelocytomatosis viral related oncogene (neuroblastoma-derived), bHLH Transcription Factor |
| Nanog | Nanog Homeobox |
| NeuroD1 | Neuronal Differentiation 1 |
| NFKB | Nuclear Factor Kappa-B |
| Notch/NOTCH | Notch Receptor 1 |
| NTRK2 | Neurotrophic Receptor Tyrosine Kinase 2 |
| Oct3/4 | Octamer-Binding Transcription Factor 3/4 |
| p21 | alias Cyclin Dependent Kinase Inhibitor 1A (CDKN1A) |
| P75NTR | p75 Neurotrophin Receptor |
| PC12 | Pheochromocytoma Cells 12 |
| PD-L1 | Programmed Cell Death 1 Ligand 1 |
| Phox2a/PHOX2A | Paired Like Homeobox 2A |
| Phox2b/PHOX2B | Paired Like Homeobox 2B |
| PI3K | Phosphatidylinositol 3-Kinase |
| PRRX1 | Paired Related Homeobox 1 |
| PUMA | p53 Upregulated Modulator of Apoptosis |
| RAN | RAN, Member RAS Oncogene Family |
| RARB | Retinoic Acid Receptor Beta |
| RAS | Rat Sarcoma Oncogene |
| RET | Rearranged During Transfection |
| RUNX1 | RUNX Family Transcription Factor 1 |
| SKP2 | S-Phase Kinase Associated Protein 2 |
| SNAI1 | Snail Family Transcriptional Repressor 1 |
| Snail2/SNAI2 | Snail Family Transcriptional Repressor 2 |
| Sox2 | SRY-Box Transcription Factor 2 |
| Sox 9 | SRY-Box Transcription Factor 9 |
| Sox10/SOX10 | SRY-Box Transcription Factor 10 |
| SP1 | Specificity Protein 1 |
| SSEA-1 | Stage-Specific Embryonic Antigen 1 |
| TBX2 | T-box Transcription Factor 2 |
| TG2 | Tissue Transglutaminase 2 |
| TGF-β1 (TGFB1) | Transforming Growth Factor Beta 1 |
| TH | Tyrosine Hydroxylase |
| TIMP2 | Tissue Inhibitor of Metalloproteinases 2 |
| TP53 | Tumor Protein P53 |
| TP53INP1 | Tumor Protein P53 Inducible Nuclear Protein 1 |
| Tra2β | Transformer 2 Beta Homolog |
| Trk | Tropomyosin Receptor Kinase |
| TrkB/TRKB | Tropomyosin-Related Kinase B |
| TWIST1 | Twist Family bHLH Transcription Factor 1 |
| VEGF | Vascular Endothelial Growth Factor |
| Wnt/WNT | Wingless-Type MMTV Integration Site Family |
| ZCCHC11 | Zinc Finger CCHC Domain-Containing Protein 11 |
| ZEB1 | Zinc finger E-box Binding homeobox 1 |
| ZNF143 | Zinc Finger Protein 143 |
References
- Padovan-Merhar, O.M.; Raman, P.; Ostrovnaya, I.; Kalletla, K.; Rubnitz, K.R.; Sanford, E.M.; Ali, S.M.; Miller, V.A.; Mosse, Y.P.; Granger, M.P.; et al. Enrichment of targetable mutations in the relapsed neuroblastoma genome. PLoS Genet. 2016, 12, e1006501. [Google Scholar] [CrossRef]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Longo, L.; Tonini, G.P.; Ceccherini, I.; Perri, P. Oligogenic inheritance in neuroblastoma. Cancer Lett. 2005, 228, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Ambros, P.F.; Ambros, I.M.; Brodeur, G.M.; Haber, M.; Khan, J.; Nakagawara, A.; Schleiermacher, G.; Speleman, F.; Spitz, R.; London, W.B.; et al. International consensus for neuroblastoma molecular diagnostics: Report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br. J. Cancer 2009, 100, 1471–1482. [Google Scholar] [CrossRef] [Green Version]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Dzieran, J.; Rodriguez Garcia, A.; Westermark, U.K.; Henley, A.B.; Eyre Sanchez, E.; Trager, C.; Johansson, H.J.; Lehtio, J.; Arsenian-Henriksson, M. MYCN-amplified neuroblastoma maintains an aggressive and undifferentiated phenotype by deregulation of estrogen and NGF signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E1229–E1238. [Google Scholar] [CrossRef] [Green Version]
- Westermark, U.K.; Wilhelm, M.; Frenzel, A.; Henriksson, M.A. The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 2011, 21, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, D.; Ambrogio, C.; Pastorino, F.; Brignole, C.; Martinengo, C.; Carosio, R.; Loi, M.; Pagnan, G.; Emionite, L.; Cilli, M.; et al. Selective therapeutic targeting of the anaplastic lymphoma kinase with liposomal siRNA induces apoptosis and inhibits angiogenesis in neuroblastoma. Mol. Ther. 2011, 19, 2201–2212. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.K.; Nafady, A.; Takatori, A.; Kishida, S.; Ohira, M.; Suenaga, Y.; Hossain, S.; Akter, J.; Ogura, A.; Nakamura, Y.; et al. ALK is a MYCN target gene and regulates cell migration and invasion in neuroblastoma. Sci. Rep. 2013, 3, 3450. [Google Scholar] [CrossRef] [Green Version]
- Ueda, T.; Nakata, Y.; Yamasaki, N.; Oda, H.; Sentani, K.; Kanai, A.; Onishi, N.; Ikeda, K.; Sera, Y.; Honda, Z.I.; et al. ALK(R1275Q) perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene 2016, 35, 4447–4458. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Domingo-Fernandez, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 44, 1199–1206. [Google Scholar] [CrossRef]
- Schnepp, R.W.; Diskin, S.J. LIN28B: An orchestrator of oncogenic signaling in neuroblastoma. Cell Cycle 2016, 15, 772–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, A.; Van Peer, G.; Carter, D.R.; Gartlgruber, M.; Herrmann, C.; Agarwal, S.; Helsmoortel, H.H.; Althoff, K.; Molenaar, J.J.; Cheung, B.B.; et al. MYCN-driven regulatory mechanisms controlling LIN28B in neuroblastoma. Cancer Lett. 2015, 366, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Powers, J.T.; Tsanov, K.M.; Pearson, D.S.; Roels, F.; Spina, C.S.; Ebright, R.; Seligson, M.; de Soysa, Y.; Cahan, P.; Theissen, J.; et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature 2016, 535, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Zhang, L.; Cui, L.; Lou, W.; Wang, D.; Lu, W.; Jin, D.; Liu, T. LIN28B suppresses microRNA let-7b expression to promote CD44+/LIN28B+ human pancreatic cancer stem cell proliferation and invasion. Am. J. Cancer Res. 2015, 5, 2643–2659. [Google Scholar]
- Bachetti, T.; Ceccherini, I. Causative and common PHOX2B variants define a broad phenotypic spectrum. Clin. Genet. 2020, 97, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lascio, S.; Benfante, R.; Di Zanni, E.; Cardani, S.; Adamo, A.; Fornasari, D.; Ceccherini, I.; Bachetti, T. Structural and functional differences in PHOX2B frameshift mutations underlie isolated or syndromic congenital central hypoventilation syndrome. Hum. Mutat. 2018, 39, 219–236. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B mutation in hereditary neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raabe, E.H.; Laudenslager, M.; Winter, C.; Wasserman, N.; Cole, K.; LaQuaglia, M.; Maris, D.J.; Mosse, Y.P.; Maris, J.M. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 2008, 27, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Reiff, T.; Tsarovina, K.; Majdazari, A.; Schmidt, M.; del Pino, I.; Rohrer, H. Neuroblastoma phox2b variants stimulate proliferation and dedifferentiation of immature sympathetic neurons. J. Neurosci. 2010, 30, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frebourg, T.; Munnich, A.; et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Aravindan, N.; Subramanian, K.; Somasundaram, D.B.; Herman, T.S.; Aravindan, S. MicroRNAs in neuroblastoma tumorigenesis, therapy resistance, and disease evolution. Cancer Drug Resist. 2019, 2, 1086–1105. [Google Scholar] [CrossRef] [Green Version]
- Salomao, K.B.; Pezuk, J.A.; de Souza, G.R.; Chagas, P.; Pereira, T.C.; Valera, E.T.; Brassesco, M.S. MicroRNA dysregulation interplay with childhood abdominal tumors. Cancer Metastasis Rev. 2019, 38, 783–811. [Google Scholar] [CrossRef] [PubMed]
- Bray, I.; Bryan, K.; Prenter, S.; Buckley, P.G.; Foley, N.H.; Murphy, D.M.; Alcock, L.; Mestdagh, P.; Vandesompele, J.; Speleman, F.; et al. Widespread dysregulation of MiRNAs by MYCN amplification and chromosomal imbalances in neuroblastoma: Association of miRNA expression with survival. PLoS ONE 2009, 4, e7850. [Google Scholar] [CrossRef] [Green Version]
- Hennchen, M.; Stubbusch, J.; Abarchan-El Makhfi, I.; Kramer, M.; Deller, T.; Pierre-Eugene, C.; Janoueix-Lerosey, I.; Delattre, O.; Ernsberger, U.; Schulte, J.B.; et al. Lin28B and Let-7 in the Control of Sympathetic Neurogenesis and Neuroblastoma Development. J. Neurosci. 2015, 35, 16531–16544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buechner, J.; Tomte, E.; Haug, B.H.; Henriksen, J.R.; Lokke, C.; Flaegstad, T.; Einvik, C. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br. J. Cancer 2011, 105, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Tivnan, A.; Tracey, L.; Buckley, P.G.; Alcock, L.C.; Davidoff, A.M.; Stallings, R.L. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer 2011, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.S.; Song, Y.K.; Durinck, S.; Chen, Q.R.; Cheuk, A.T.; Tsang, P.; Zhang, Q.; Thiele, C.J.; Slack, A.; Shohet, J.; et al. The MYCN oncogene is a direct target of miR-34a. Oncogene 2008, 27, 5204–5213. [Google Scholar] [CrossRef] [Green Version]
- Welch, C.; Chen, Y.; Stallings, R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007, 26, 5017–5022. [Google Scholar] [CrossRef] [Green Version]
- Bachetti, T.; Di Zanni, E.; Ravazzolo, R.; Ceccherini, I. miR-204 mediates post-transcriptional down-regulation of PHOX2B gene expression in neuroblastoma cells. Biochim. Biophys. Acta 2015, 1849, 1057–1065. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Carter, D.R.; Liu, B.; Mayoh, C.; Beckers, A.; Lalwani, A.; Nagy, Z.; De Brouwer, S.; Decaesteker, B.; Hung, T.T.; et al. Network modeling of microRNA-mRNA interactions in neuroblastoma tumorigenesis identifies miR-204 as a direct inhibitor of MYCN. Cancer Res. 2018, 78, 3122–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.; Tivnan, A.; Fay, J.; Bryan, K.; Meehan, M.; Creevey, L.; Lynch, J.; Bray, I.M.; O’Meara, A.; Tracey, L.; et al. MicroRNA-204 increases sensitivity of neuroblastoma cells to cisplatin and is associated with a favourable clinical outcome. Br. J. Cancer 2012, 107, 967–976. [Google Scholar] [CrossRef] [Green Version]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.D.; Ye, C.J.; Yao, X.Y.; Luo, W.Q.; Cheng, X.M.; Miao, J.J.; Wang, J.F.; et al. Single-cell characterization of malignant phenotypes and developmental trajectories of adrenal neuroblastoma. Cancer Cell 2020, 38, 716–733. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T.; Akkuratova, N.; Usoskin, D.; Kamenev, D.; et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 2017, 357, eaal3753. [Google Scholar] [CrossRef] [Green Version]
- Hanemaaijer, E.S.; Margaritis, T.; Sanders, K.; Bos, F.L.; Candelli, T.; Al-Saati, H.; van Noesel, M.M.; Meyer-Wentrup, F.A.G.; van de Wetering, M.; Holstege, F.C.P.; et al. Single-cell atlas of developing murine adrenal gland reveals relation of Schwann cell precursor signature to neuroblastoma phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2022350118. [Google Scholar] [CrossRef] [PubMed]
- Jansky, S.; Sharma, A.K.; Korber, V.; Quintero, A.; Toprak, U.H.; Wecht, E.M.; Gartlgruber, M.; Greco, A.; Chomsky, E.; Grunewald, T.G.P.; et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat. Genet. 2021, 53, 683–693. [Google Scholar] [CrossRef]
- Kameneva, P.; Artemov, A.V.; Kastriti, M.E.; Faure, L.; Olsen, T.K.; Otte, J.; Erickson, A.; Semsch, B.; Andersson, E.R.; Ratz, M.; et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat. Genet. 2021, 53, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Kildisiute, G.; Kholosy, W.M.; Young, M.D.; Roberts, K.; Elmentaite, R.; van Hooff, S.R.; Pacyna, C.N.; Khabirova, E.; Piapi, A.; Thevanesan, C.; et al. Tumor to normal single-cell mRNA comparisons reveal a pan-neuroblastoma cancer cell. Sci. Adv. 2021, 7, eabd3311. [Google Scholar] [CrossRef] [PubMed]
- Soldatov, R.; Kaucka, M.; Kastriti, M.E.; Petersen, J.; Chontorotzea, T.; Englmaier, L.; Akkuratova, N.; Yang, Y.; Haring, M.; Dyachuk, V.; et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 2019, 364, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugene, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Gartlgruber, M.; Sharma, A.K.; Quintero, A.; Dreidax, D.; Jansky, S.; Park, Y.G.; Kreth, S.; Meder, J.; Doncevic, D.; Saary, P.; et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat. Cancer 2021, 2, 114. [Google Scholar] [CrossRef]
- Oldridge, D.A.; Truong, B.; Russ, D.; DuBois, S.G.; Vaksman, Z.; Mosse, Y.P.; Diskin, S.J.; Maris, J.M.; Matthay, K.K. Differences in genomic profiles and outcomes between Thoracic and Adrenal Neuroblastoma. J. Natl. Cancer Inst. 2019, 111, 1192–1201. [Google Scholar] [CrossRef]
- Rohrer, H. Linking human sympathoadrenal development and neuroblastoma. Nat. Genet. 2021, 53, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tan, T.K.; Durbin, A.D.; Zimmerman, M.W.; Abraham, B.J.; Tan, S.H.; Ngoc, P.C.T.; Weichert-Leahey, N.; Akahane, K.; Lawton, L.N.; et al. ASCL1 is a MYCN- and LMO1-dependent member of the adrenergic neuroblastoma core regulatory circuitry. Nat. Commun. 2019, 10, 5622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.P.; Slack, F.J. microRNA-mediated silencing inside P-bodies. RNA Biol. 2006, 3, 97–100. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015, 25, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wolkenhauer, O.; Vera, J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 2016, 44, 6019–6035. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ferguson, J.; Chang, J.T.; Kluger, Y. Inter- and intra-combinatorial regulation by transcription factors and microRNAs. BMC Genom. 2007, 8, 396. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; George, R.E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowski, T.J.; Rani, N.; Golkaram, M.; Zhou, H.R.; Alvarado, B.; Huch, K.; West, J.A.; Leyrat, A.; Pollen, A.A.; Kriegstein, A.R.; et al. Regulation of cell-type-specific transcriptomes by microRNA networks during human brain development. Nat. Neurosci. 2018, 21, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Zolboot, N.; Du, J.X.; Zampa, F.; Lippi, G. MicroRNAs instruct and Maintain Cell type diversity in the nervous system. Front. Mol. Neurosci. 2021, 14, 646072. [Google Scholar] [CrossRef]
- Weiner, A.M.J. MicroRNAs and the neural crest: From induction to differentiation. Mech. Dev. 2018, 154, 98–106. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Lopez, A.; Moreno-Bueno, G.; Cano, A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag. Res. 2014, 6, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Simoes-Costa, M.; Bronner, M.E. Establishing neural crest identity: A gene regulatory recipe. Development 2015, 142, 242–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krispin, S.; Nitzan, E.; Kassem, Y.; Kalcheim, C. Evidence for a dynamic spatiotemporal fate map and early fate restrictions of premigratory avian neural crest. Development 2010, 137, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, R.R.; Otero, J.H.; Garcia-Lopez, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.D.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef] [Green Version]
- Saito, D.; Takase, Y.; Murai, H.; Takahashi, Y. The dorsal aorta initiates a molecular cascade that instructs sympatho-adrenal specification. Science 2012, 336, 1578–1581. [Google Scholar] [CrossRef]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 1999, 399, 366–370. [Google Scholar] [CrossRef]
- Wakamatsu, Y.; Watanabe, Y.; Nakamura, H.; Kondoh, H. Regulation of the neural crest cell fate by N-myc: Promotion of ventral migration and neuronal differentiation. Development 1997, 124, 1953–1962. [Google Scholar] [CrossRef]
- Knoepfler, P.S.; Cheng, P.F.; Eisenman, R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002, 16, 2699–2712. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, Y.; Zehir, A.; Maska, E.; Deng, C.; Schneider, M.D.; Mishina, Y.; Cserjesi, P. BMP signaling regulates sympathetic nervous system development through Smad4-dependent and -independent pathways. Development 2009, 136, 3575–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, G.; Cui, H.; Shi, H.; Yang, L.; Ding, J.; Mao, L.; Maltese, W.A.; Ding, H.F. MYCN promotes the expansion of Phox2B-positive neuronal progenitors to drive neuroblastoma development. Am. J. Pathol. 2009, 175, 856–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, M.; Sakai, K.; Fumino, S.; Aoi, S.; Furukawa, T.; Tajiri, T. The roles played by the MYCN, Trk, and ALK genes in neuroblastoma and neural development. Surg. Today 2019, 49, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, M.R.; Tiveron, M.C.; Guillemot, F.; Brunet, J.F.; Goridis, C. Control of noradrenergic differentiation and Phox2a expression by MASH1 in the central and peripheral nervous system. Development 1998, 125, 599–608. [Google Scholar] [CrossRef]
- Rohrer, H. Transcriptional control of differentiation and neurogenesis in autonomic ganglia. Eur. J. Neurosci. 2011, 34, 1563–1573. [Google Scholar] [CrossRef]
- Vincentz, J.W.; VanDusen, N.J.; Fleming, A.B.; Rubart, M.; Firulli, B.A.; Howard, M.J.; Firulli, A.B. A Phox2- and Hand2-dependent Hand1 cis-regulatory element reveals a unique gene dosage requirement for Hand2 during sympathetic neurogenesis. J. Neurosci. 2012, 32, 2110–2120. [Google Scholar] [CrossRef]
- Moore, R.; Alexandre, P. Delta-notch signaling: The long and the short of a neuron’s influence on progenitor fates. J. Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Tsarovina, K.; Schellenberger, J.; Schneider, C.; Rohrer, H. Progenitor cell maintenance and neurogenesis in sympathetic ganglia involves Notch signaling. Mol. Cell. Neurosci. 2008, 37, 20–31. [Google Scholar] [CrossRef]
- Kirino, K.; Nakahata, T.; Taguchi, T.; Saito, M.K. Efficient derivation of sympathetic neurons from human pluripotent stem cells with a defined condition. Sci. Rep. 2018, 8, 12865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, B.D.; Wali, V.B.; Cheng, C.J.; Inukai, S.; Booth, C.J.; Agarwal, S.; Rimm, D.L.; Gyorffy, B.; Santarpia, L.; Pusztai, L.; et al. miR-34a silences c-SRC to attenuate tumor growth in triple-negative breast cancer. Cancer Res. 2016, 76, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Cole, K.A.; Attiyeh, E.F.; Mosse, Y.P.; Laquaglia, M.J.; Diskin, S.J.; Brodeur, G.M.; Maris, J.M. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res. 2008, 6, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective blockade of microRNA processing by Lin28. Science 2008, 320, 97–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afanasyeva, E.A.; Mestdagh, P.; Kumps, C.; Vandesompele, J.; Ehemann, V.; Theissen, J.; Fischer, M.; Zapatka, M.; Brors, B.; Savelyeva, L.; et al. MicroRNA miR-885-5p targets CDK2 and MCM5, activates p53 and inhibits proliferation and survival. Cell Death Differ. 2011, 18, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Althoff, K.; Lindner, S.; Odersky, A.; Mestdagh, P.; Beckers, A.; Karczewski, S.; Molenaar, J.J.; Bohrer, A.; Knauer, S.; Speleman, F.; et al. miR-542-3p exerts tumor suppressive functions in neuroblastoma by downregulating Survivin. Int. J. Cancer 2015, 136, 1308–1320. [Google Scholar] [CrossRef]
- Chen, Y.; Stallings, R.L. Differential patterns of microRNA expression in neuroblastoma are correlated with prognosis, differentiation, and apoptosis. Cancer Res. 2007, 67, 976–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Bryan, K.; Buckley, P.G.; Piskareva, O.; Bray, I.M.; Foley, N.; Ryan, J.; Lynch, J.; Creevey, L.; Fay, J.; et al. Modulation of neuroblastoma disease pathogenesis by an extensive network of epigenetically regulated microRNAs. Oncogene 2013, 32, 2927–2936. [Google Scholar] [CrossRef] [Green Version]
- Schulte, J.H.; Marschall, T.; Martin, M.; Rosenstiel, P.; Mestdagh, P.; Schlierf, S.; Thor, T.; Vandesompele, J.; Eggert, A.; Schreiber, S.; et al. Deep sequencing reveals differential expression of microRNAs in favorable versus unfavorable neuroblastoma. Nucleic Acids Res. 2010, 38, 5919–5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivnan, A.; Foley, N.H.; Tracey, L.; Davidoff, A.M.; Stallings, R.L. MicroRNA-184-mediated inhibition of tumour growth in an orthotopic murine model of neuroblastoma. Anticancer Res. 2010, 30, 4391–4395. [Google Scholar] [PubMed]
- Vishwamitra, D.; Li, Y.; Wilson, D.; Manshouri, R.; Curry, C.V.; Shi, B.; Tang, X.M.; Sheehan, A.M.; Wistuba, I.I.; Shi, P.; et al. MicroRNA 96 is a post-transcriptional suppressor of anaplastic lymphoma kinase expression. Am. J. Pathol. 2012, 180, 1772–1780. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zhang, J.; Jin, Y.; Yang, Y.; Shi, J.; Chen, F.; Han, S.; Chu, P.; Lu, J.; Wang, H.; et al. MiR-20a-5p suppresses tumor proliferation by targeting autophagy-related gene 7 in neuroblastoma. Cancer Cell Int. 2018, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Parodi, F.; Carosio, R.; Ragusa, M.; Di Pietro, C.; Maugeri, M.; Barbagallo, D.; Sallustio, F.; Allemanni, G.; Pistillo, M.P.; Casciano, I.; et al. Epigenetic dysregulation in neuroblastoma: A tale of miRNAs and DNA methylation. Biochim. Biophys. Acta 2016, 1859, 1502–1514. [Google Scholar] [CrossRef]
- Kaller, M.; Hermeking, H. Interplay between transcription factors and micrornas regulating epithelial-mesenchymal transitions in colorectal cancer. Adv. Exp. Med. Biol. 2016, 937, 71–92. [Google Scholar] [CrossRef]
- Mollinari, C.; Racaniello, M.; Berry, A.; Pieri, M.; de Stefano, M.C.; Cardinale, A.; Zona, C.; Cirulli, F.; Garaci, E.; Merlo, D. miR-34a regulates cell proliferation, morphology and function of newborn neurons resulting in improved behavioural outcomes. Cell Death Dis. 2015, 6, e1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, C.E.L.; Tang, B.L. miR-34a in Neurophysiology and Neuropathology. J. Mol. Neurosci. 2019, 67, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [Green Version]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1 and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef]
- Marshall, G.M.; Liu, P.Y.; Gherardi, S.; Scarlett, C.J.; Bedalov, A.; Xu, N.; Iraci, N.; Valli, E.; Ling, D.; Thomas, W.; et al. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genet. 2011, 7, e1002135. [Google Scholar] [CrossRef] [Green Version]
- Jauhari, A.; Singh, T.; Singh, P.; Parmar, D.; Yadav, S. Regulation of miR-34 Family in Neuronal Development. Mol. Neurobiol. 2018, 55, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Fineberg, S.K.; Datta, P.; Stein, C.S.; Davidson, B.L. MiR-34a represses Numbl in murine neural progenitor cells and antagonizes neuronal differentiation. PLoS ONE 2012, 7, e38562. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 2010, 9, 1031–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silber, J.; Jacobsen, A.; Ozawa, T.; Harinath, G.; Pedraza, A.; Sander, C.; Holland, E.C.; Huse, J.T. miR-34a repression in proneural malignant gliomas upregulates expression of its target PDGFRA and promotes tumorigenesis. PLoS ONE 2012, 7, e33844. [Google Scholar] [CrossRef]
- De Antonellis, P.; Medaglia, C.; Cusanelli, E.; Andolfo, I.; Liguori, L.; De Vita, G.; Carotenuto, M.; Bello, A.; Formiggini, F.; Galeone, A.; et al. MiR-34a targeting of Notch ligand delta-like 1 impairs CD15+/CD133+ tumor-propagating cells and supports neural differentiation in medulloblastoma. PLoS ONE 2011, 6, e24584. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Ma, X.; Shelton, S.D.; Sung, D.C.; Li, M.; Hernandez, D.; Zhang, M.; Losiewicz, M.D.; Chen, Y.; Pertsemlidis, A.; et al. A combined gene expression and functional study reveals the crosstalk between N-Myc and differentiation-inducing microRNAs in neuroblastoma cells. Oncotarget 2016, 7, 79372–79387. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Kelnar, K.; Stahlhut, C.; Orellana, E.; Zhao, J.; Shimer, E.; Dysart, S.; Chen, X.; Bader, A.G.; Slack, F.J. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene 2015, 34, 3547–3555. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef] [Green Version]
- Daige, C.L.; Wiggins, J.F.; Priddy, L.; Nelligan-Davis, T.; Zhao, J.; Brown, D. Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther. 2014, 13, 2352–2360. [Google Scholar] [CrossRef] [Green Version]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef]
- Bader, A.G. miR-34—A microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivnan, A.; Orr, W.S.; Gubala, V.; Nooney, R.; Williams, D.E.; McDonagh, C.; Prenter, S.; Harvey, H.; Domingo-Fernandez, R.; Bray, I.M.; et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles. PLoS ONE 2012, 7, e38129. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, D.; Pastorino, F.; Brignole, C.; Corrias, M.V.; Emionite, L.; Cilli, M.; Tamma, R.; Priddy, L.; Amaro, A.; Ferrari, D.; et al. Combined replenishment of miR-34a and let-7b by targeted nanoparticles inhibits tumor growth in neuroblastoma preclinical models. Small 2020, 16, e1906426. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, C.; Slack, F.J. Combinatorial Action of MicroRNAs let-7 and miR-34 Effectively Synergizes with Erlotinib to Suppress Non-small Cell Lung Cancer Cell Proliferation. Cell Cycle 2015, 14, 2171–2180. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bader, A.G. Evaluating Synergistic Effects of miR-34a Mimics in combination with other therapeutic agents in cultured non-small cell lung cancer cells. Methods Mol. Biol. 2017, 1517, 115–126. [Google Scholar] [CrossRef]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; LaPierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xie, C.; Zheng, X.; Nie, X.; Wang, Z.; Liu, H.; Zhao, Y. LIN28/let-7/PD-L1 Pathway as a Target for Cancer Immunotherapy. Cancer Immunol. Res. 2019, 7, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef]
- Kumar, M.S.; Erkeland, S.J.; Pester, R.E.; Chen, C.Y.; Ebert, M.S.; Sharp, P.A.; Jacks, T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl. Acad. Sci. USA 2008, 105, 3903–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.C.; Zeitels, L.R.; Hwang, H.W.; Chivukula, R.R.; Wentzel, E.A.; Dews, M.; Jung, J.; Gao, P.; Dang, C.V.; Beer, M.A.; et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 3384–3389. [Google Scholar] [CrossRef] [Green Version]
- Esquela-Kerscher, A.; Trang, P.; Wiggins, J.F.; Patrawala, L.; Cheng, A.; Ford, L.; Weidhaas, J.B.; Brown, D.; Bader, A.G.; Slack, F.J. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 2008, 7, 759–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trang, P.; Medina, P.P.; Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Homer, R.; Brown, D.; Bader, A.G.; Weidhaas, J.B.; et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene 2010, 29, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Rybak, A.; Fuchs, H.; Smirnova, L.; Brandt, C.; Pohl, E.E.; Nitsch, R.; Wulczyn, F.G. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat. Cell Biol. 2008, 10, 987–993. [Google Scholar] [CrossRef]
- Schultz, J.; Lorenz, P.; Gross, G.; Ibrahim, S.; Kunz, M. MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008, 18, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangi-Garimella, S.; Yun, J.; Eves, E.M.; Newman, M.; Erkeland, S.J.; Hammond, S.M.; Minn, A.J.; Rosner, M.R. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009, 28, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barh, D.; Malhotra, R.; Ravi, B.; Sindhurani, P. MicroRNA let-7: An emerging next-generation cancer therapeutic. Curr. Oncol. 2010, 17, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Krell, J.; Stebbing, J.; Carissimi, C.; Dabrowska, A.F.; de Giorgio, A.; Frampton, A.E.; Harding, V.; Fulci, V.; Macino, G.; Colombo, T.; et al. TP53 regulates miRNA association with AGO2 to remodel the miRNA-mRNA interaction network. Genome Res. 2016, 26, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Jiang, Y.; Tan, C. Let-7 Sensitizes KRAS Mutant Tumor Cells to Chemotherapy. PLoS ONE 2015, 10, e0126653. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Koster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef]
- Conte, I.; Merella, S.; Garcia-Manteiga, J.M.; Migliore, C.; Lazarevic, D.; Carrella, S.; Marco-Ferreres, R.; Avellino, R.; Davidson, N.P.; Emmett, W.; et al. The combination of transcriptomics and informatics identifies pathways targeted by miR-204 during neurogenesis and axon guidance. Nucleic Acids Res. 2014, 42, 7793–7806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittstatt, J.; Weider, M.; Wegner, M.; Reiprich, S. MicroRNA miR-204 regulates proliferation and differentiation of oligodendroglia in culture. Glia 2020, 68, 2015–2027. [Google Scholar] [CrossRef]
- Chiu, C.C.; Yeh, T.H.; Chen, R.S.; Chen, H.C.; Huang, Y.Z.; Weng, Y.H.; Cheng, Y.C.; Liu, Y.C.; Cheng, A.J.; Lu, Y.C.; et al. Upregulated Expression of MicroRNA-204-5p Leads to the Death of Dopaminergic Cells by Targeting DYRK1A-Mediated Apoptotic Signaling Cascade. Front. Cell. Neurosci. 2019, 13, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepko, T.; Pusch, M.; Muller, T.; Schulte, D.; Ehses, J.; Kiebler, M.; Hasler, J.; Huttner, H.B.; Vandenbroucke, R.E.; Vandendriessche, C.; et al. Choroid plexus-derived miR-204 regulates the number of quiescent neural stem cells in the adult brain. EMBO J. 2019, 38, e100481. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Wang, Y.; Xu, Y.; Zhang, S.; Sun, X.; Guan, H.; Zhao, X.; Wang, Y.; Li, Y.; Zhao, G. Long non-coding RNA TCONS_00041960 enhances osteogenesis and inhibits adipogenesis of rat bone marrow mesenchymal stem cell by targeting miR-204-5p and miR-125a-3p. J. Cell. Physiol. 2018, 233, 6041–6051. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Pan, H.; Li, R. The dual regulatory role of miR-204 in cancer. Tumor Biol. 2016, 37, 11667–11677. [Google Scholar] [CrossRef] [Green Version]
- Saunders, L.R.; Sharma, A.D.; Tawney, J.; Nakagawa, M.; Okita, K.; Yamanaka, S.; Willenbring, H.; Verdin, E. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging 2010, 2, 415–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, X.; Chen, P. MiR-204 down regulates SIRT1 and reverts SIRT1-induced epithelial-mesenchymal transition, anoikis resistance and invasion in gastric cancer cells. BMC Cancer 2013, 13, 290. [Google Scholar] [CrossRef] [Green Version]
- Sacconi, A.; Biagioni, F.; Canu, V.; Mori, F.; Di Benedetto, A.; Lorenzon, L.; Ercolani, C.; Di Agostino, S.; Cambria, A.M.; Germoni, S.; et al. miR-204 targets Bcl-2 expression and enhances responsiveness of gastric cancer. Cell Death Dis. 2012, 3, e423. [Google Scholar] [CrossRef]
- Kuwano, Y.; Nishida, K.; Kajita, K.; Satake, Y.; Akaike, Y.; Fujita, K.; Kano, S.; Masuda, K.; Rokutan, K. Transformer 2beta and miR-204 regulate apoptosis through competitive binding to 3′ UTR of BCL2 mRNA. Cell Death Differ. 2015, 22, 815–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoehner, J.C.; Hedborg, F.; Wiklund, H.J.; Olsen, L.; Pahlman, S. Cellular death in neuroblastoma: In situ correlation of apoptosis and bcl-2 expression. Int. J. Cancer 1995, 62, 19–24. [Google Scholar] [CrossRef]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef]
- Zhang, B.; Cui, H.; Sun, Y.; Wang, X.; Jia, Q.; Li, J.; Yin, Y.; Sun, X.; Xu, H.; Li, H.; et al. Up-regulation of miR-204 inhibits proliferation, invasion and apoptosis of gallbladder cancer cells by targeting Notch2. Aging 2021, 13, 2941–2958. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Zheng, Y.; Ma, S.; Xing, Q.; Wang, X.; Yang, B.; Yin, G.; Guan, F. BANCR contributes to the growth and invasion of melanoma by functioning as a competing endogenous RNA to upregulate Notch2 expression by sponging miR204. Int. J. Oncol. 2017, 51, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Varmus, H.E.; Bishop, J.M. Human N-myc gene contributes to neoplastic transformation of mammalian cells in culture. Nature 1985, 316, 160–162. [Google Scholar] [CrossRef]
- Breit, S.; Schwab, M. Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J. Neurosci. Res. 1989, 24, 21–28. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Charron, J.; Malynn, B.A.; Fisher, P.; Stewart, V.; Jeannotte, L.; Goff, S.P.; Robertson, E.J.; Alt, F.W. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992, 6, 2248–2257. [Google Scholar] [CrossRef]
- Malynn, B.A.; de Alboran, I.M.; O’Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef]
- Grady, E.F.; Schwab, M.; Rosenau, W. Expression of N-myc and c-src during the development of fetal human brain. Cancer Res. 1987, 47, 2931–2936. [Google Scholar]
- Wakamatsu, Y.; Watanabe, Y.; Shimono, A.; Kondoh, H. Transition of localization of the N-Myc protein from nucleus to cytoplasm in differentiating neurons. Neuron 1993, 10, 1–9. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotterman, R.; Knoepfler, P.S. N-Myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PLoS ONE 2009, 4, e5799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinatl, J.; Cinatl, J.; Mainke, M.; Weissflog, A.; Rabenau, H.; Kornhuber, B.; Doerr, H.W. In vitro differentiation of human neuroblastoma cells induced by sodium phenylacetate. Cancer Lett. 1993, 70, 15–24. [Google Scholar] [CrossRef]
- Thiele, C.J.; Reynolds, C.P.; Israel, M.A. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 1985, 313, 404–406. [Google Scholar] [CrossRef]
- Reynolds, C.P.; Matthay, K.K.; Villablanca, J.G.; Maurer, B.J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003, 197, 185–192. [Google Scholar] [CrossRef]
- Duffy, D.J.; Krstic, A.; Halasz, M.; Schwarzl, T.; Konietzny, A.; Iljin, K.; Higgins, D.G.; Kolch, W. Retinoic acid and TGF-beta signalling cooperate to overcome MYCN-induced retinoid resistance. Genome Med. 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeshima, R.; Moulding, D.; Stoker, A.W.; Hart, S.L. MYCN Silencing by RNAi Induces Neurogenesis and Suppresses Proliferation in Models of Neuroblastoma with Resistance to Retinoic Acid. Nucleic Acid Ther. 2020, 30, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Westermann, F.; Muth, D.; Benner, A.; Bauer, T.; Henrich, K.O.; Oberthuer, A.; Brors, B.; Beissbarth, T.; Vandesompele, J.; Pattyn, F.; et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 2008, 9, R150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmsauer, K.; Valieva, M.E.; Ali, S.; Chamorro Gonzalez, R.; Schopflin, R.; Roefzaad, C.; Bei, Y.; Dorado Garcia, H.; Rodriguez-Fos, E.; Puiggros, M.; et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat. Commun. 2020, 11, 5823. [Google Scholar] [CrossRef]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X.; et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov. 2018, 8, 320–335. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M. Amplification of the MYCN oncogene and deletion of putative tumour suppressor gene in human neuroblastomas. Brain Pathol. 1990, 1, 41–46. [Google Scholar] [CrossRef]
- Bordow, S.B.; Norris, M.D.; Haber, P.S.; Marshall, G.M.; Haber, M. Prognostic significance of MYCN oncogene expression in childhood neuroblastoma. J. Clin. Oncol. 1998, 16, 3286–3294. [Google Scholar] [CrossRef]
- Chan, H.S.; Gallie, B.L.; DeBoer, G.; Haddad, G.; Ikegaki, N.; Dimitroulakos, J.; Yeger, H.; Ling, V. MYCN protein expression as a predictor of neuroblastoma prognosis. Clin. Cancer Res. 1997, 3, 1699–1706. [Google Scholar] [PubMed]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Althoff, K.; Beckers, A.; Bell, E.; Nortmeyer, M.; Thor, T.; Sprussel, A.; Lindner, S.; De Preter, K.; Florin, A.; Heukamp, L.C.; et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene 2015, 34, 3357–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Zhang, X.; Weichert-Leahey, N.; Dong, Z.; Zhang, C.; Lopez, G.; Tao, T.; He, S.; Wood, A.C.; Oldridge, D.; et al. LMO1 Synergizes with MYCN to Promote Neuroblastoma Initiation and Metastasis. Cancer Cell 2017, 32, 310–323.e315. [Google Scholar] [CrossRef] [Green Version]
- Niemas-Teshiba, R.; Matsuno, R.; Wang, L.L.; Tang, X.X.; Chiu, B.; Zeki, J.; Coburn, J.; Ornell, K.; Naranjo, A.; Van Ryn, C.; et al. MYC-family protein overexpression and prominent nucleolar formation represent prognostic indicators and potential therapeutic targets for aggressive high-MKI neuroblastomas: A report from the children’s oncology group. Oncotarget 2018, 9, 6416–6432. [Google Scholar] [CrossRef]
- Wang, L.L.; Suganuma, R.; Ikegaki, N.; Tang, X.; Naranjo, A.; McGrady, P.; London, W.B.; Hogarty, M.D.; Gastier-Foster, J.M.; Look, A.T.; et al. Neuroblastoma of undifferentiated subtype, prognostic significance of prominent nucleolar formation, and MYC/MYCN protein expression: A report from the Children’s Oncology Group. Cancer 2013, 119, 3718–3726. [Google Scholar] [CrossRef]
- Wang, L.L.; Teshiba, R.; Ikegaki, N.; Tang, X.X.; Naranjo, A.; London, W.B.; Hogarty, M.D.; Gastier-Foster, J.M.; Look, A.T.; Park, J.R.; et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: A Children’s Oncology Group study. Br. J. Cancer 2015, 113, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Prochownik, E.V.; VanAntwerp, M.E. Differential patterns of DNA binding by myc and max proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 960–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlstrom, T.; Henriksson, M.A. Impact of MYC in regulation of tumor cell metabolism. Biochim. Biophys. Acta 2015, 1849, 563–569. [Google Scholar] [CrossRef]
- Wenzel, A.; Cziepluch, C.; Hamann, U.; Schurmann, J.; Schwab, M. The N-Myc oncoprotein is associated in vivo with the phosphoprotein Max(p20/22) in human neuroblastoma cells. EMBO J. 1991, 10, 3703–3712. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Lutz, W.; Schwab, M.; Debatin, K.M. MycN sensitizes neuroblastoma cells for drug-induced apoptosis. Oncogene 1999, 18, 1479–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, J.H.; Horn, S.; Otto, T.; Samans, B.; Heukamp, L.C.; Eilers, U.C.; Krause, M.; Astrahantseff, K.; Klein-Hitpass, L.; Buettner, R.; et al. MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int. J. Cancer 2008, 122, 699–704. [Google Scholar] [CrossRef]
- Buechner, J.; Einvik, C. N-myc and noncoding RNAs in neuroblastoma. Mol. Cancer Res. 2012, 10, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Gogolin, S.; Dreidax, D.; Becker, G.; Ehemann, V.; Schwab, M.; Westermann, F. MYCN/MYC-mediated drug resistance mechanisms in neuroblastoma. Int. J. Clin. Pharmacol. Ther. 2010, 48, 489–491. [Google Scholar] [CrossRef]
- Haber, M.; Bordow, S.B.; Gilbert, J.; Madafiglio, J.; Kavallaris, M.; Marshall, G.M.; Mechetner, E.B.; Fruehauf, J.P.; Tee, L.; Cohn, S.L.; et al. Altered expression of the MYCN oncogene modulates MRP gene expression and response to cytotoxic drugs in neuroblastoma cells. Oncogene 1999, 18, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Manohar, C.F.; Bray, J.A.; Salwen, H.R.; Madafiglio, J.; Cheng, A.; Flemming, C.; Marshall, G.M.; Norris, M.D.; Haber, M.; Cohn, S.L. MYCN-mediated regulation of the MRP1 promoter in human neuroblastoma. Oncogene 2004, 23, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafar, A.; Wang, W.; Liu, G.; Xian, W.; McKeon, F.; Zhou, J.; Zhang, R. Targeting the p53-MDM2 pathway for neuroblastoma therapy: Rays of hope. Cancer Lett. 2021, 496, 16–29. [Google Scholar] [CrossRef]
- Song, C.; Lu, P.; Sun, G.; Yang, L.; Wang, Z.; Wang, Z. miR-34a sensitizes lung cancer cells to cisplatin via p53/miR-34a/MYCN axis. Biochem. Biophys. Res. Commun. 2017, 482, 22–27. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.M.; Buckley, P.G.; Bryan, K.; Das, S.; Alcock, L.; Foley, N.H.; Prenter, S.; Bray, I.; Watters, K.M.; Higgins, D.; et al. Global MYCN transcription factor binding analysis in neuroblastoma reveals association with distinct E-box motifs and regions of DNA hypermethylation. PLoS ONE 2009, 4, e8154. [Google Scholar] [CrossRef] [Green Version]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Gherardi, S.; Valli, E.; Erriquez, D.; Perini, G. MYCN-mediated transcriptional repression in neuroblastoma: The other side of the coin. Front. Oncol. 2013, 3, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef]
- Morton, A.R.; Dogan-Artun, N.; Faber, Z.J.; MacLeod, G.; Bartels, C.F.; Piazza, M.S.; Allan, K.C.; Mack, S.C.; Wang, X.; Gimple, R.C.; et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 2019, 179, 1330–1341.e1313. [Google Scholar] [CrossRef]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selmi, A.; de Saint-Jean, M.; Jallas, A.C.; Garin, E.; Hogarty, M.D.; Benard, J.; Puisieux, A.; Marabelle, A.; Valsesia-Wittmann, S. TWIST1 is a direct transcriptional target of MYCN and MYC in neuroblastoma. Cancer Lett. 2015, 357, 412–418. [Google Scholar] [CrossRef]
- Visel, A.; Rubin, E.M.; Pennacchio, L.A. Genomic views of distant-acting enhancers. Nature 2009, 461, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Limpt, V.; Schramm, A.; van Lakeman, A.; Sluis, P.; Chan, A.; van Noesel, M.; Baas, F.; Caron, H.; Eggert, A.; Versteeg, R. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 2004, 23, 9280–9288. [Google Scholar] [CrossRef] [Green Version]
- Bachetti, T.; Matera, I.; Borghini, S.; Di Duca, M.; Ravazzolo, R.; Ceccherini, I. Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum. Mol. Genet. 2005, 14, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Di Lascio, S.; Bachetti, T.; Saba, E.; Ceccherini, I.; Benfante, R.; Fornasari, D. Transcriptional dysregulation and impairment of PHOX2B auto-regulatory mechanism induced by polyalanine expansion mutations associated with congenital central hypoventilation syndrome. Neurobiol. Dis. 2013, 50, 187–200. [Google Scholar] [CrossRef]
- Pei, D.; Luther, W.; Wang, W.; Paw, B.H.; Stewart, R.A.; George, R.E. Distinct neuroblastoma-associated alterations of PHOX2B impair sympathetic neuronal differentiation in zebrafish models. PLoS Genet. 2013, 9, e1003533. [Google Scholar] [CrossRef] [Green Version]
- Nagashimada, M.; Ohta, H.; Li, C.; Nakao, K.; Uesaka, T.; Brunet, J.F.; Amiel, J.; Trochet, D.; Wakayama, T.; Enomoto, H. Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. J. Clin. Investig. 2012, 122, 3145–3158. [Google Scholar] [CrossRef] [Green Version]
- Revet, I.; Huizenga, G.; Chan, A.; Koster, J.; Volckmann, R.; van Sluis, P.; Ora, I.; Versteeg, R.; Geerts, D. The MSX1 homeobox transcription factor is a downstream target of PHOX2B and activates the Delta-Notch pathway in neuroblastoma. Exp. Cell Res. 2008, 314, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Corrias, M.V.; Haupt, R.; Carlini, B.; Cappelli, E.; Giardino, S.; Tripodi, G.; Tonini, G.P.; Garaventa, A.; Pistoia, V.; Pistorio, A. Multiple target molecular monitoring of bone marrow and peripheral blood samples from patients with localized neuroblastoma and healthy donors. Pediatr. Blood Cancer 2012, 58, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.; Borghini, S.; Schena, F.; Parodi, S.; Albino, D.; Bachetti, T.; Da Prato, L.; Truini, M.; Gambini, C.; Tonini, G.P.; et al. PHOX2A and PHOX2B genes are highly co-expressed in human neuroblastoma. Int. J. Oncol. 2008, 33, 985–991. [Google Scholar] [PubMed]
- Thwin, K.K.M.; Ishida, T.; Uemura, S.; Yamamoto, N.; Lin, K.S.; Tamura, A.; Kozaki, A.; Saito, A.; Kishimoto, K.; Mori, T.; et al. Level of Seven Neuroblastoma-Associated mRNAs Detected by Droplet Digital PCR Is Associated with Tumor Relapse/Regrowth of High-Risk Neuroblastoma Patients. J. Mol. Diagn. 2020, 22, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Viprey, V.F.; Gregory, W.M.; Corrias, M.V.; Tchirkov, A.; Swerts, K.; Vicha, A.; Dallorso, S.; Brock, P.; Luksch, R.; Valteau-Couanet, D.; et al. Neuroblastoma mRNAs predict outcome in children with stage 4 neuroblastoma: A European HR-NBL1/SIOPEN study. J. Clin. Oncol. 2014, 32, 1074–1083. [Google Scholar] [CrossRef]
- Bachetti, T.; Di Paolo, D.; Di Lascio, S.; Mirisola, V.; Brignole, C.; Bellotti, M.; Caffa, I.; Ferraris, C.; Fiore, M.; Fornasari, D.; et al. PHOX2B-mediated regulation of ALK expression: In vitro identification of a functional relationship between two genes involved in neuroblastoma. PLoS ONE 2010, 5, e13108. [Google Scholar] [CrossRef] [PubMed]
- Cargnin, F.; Flora, A.; Di Lascio, S.; Battaglioli, E.; Longhi, R.; Clementi, F.; Fornasari, D. PHOX2B regulates its own expression by a transcriptional auto-regulatory mechanism. J. Biol. Chem. 2005, 280, 37439–37448. [Google Scholar] [CrossRef] [Green Version]
- Flora, A.; Lucchetti, H.; Benfante, R.; Goridis, C.; Clementi, F.; Fornasari, D. Sp proteins and Phox2b regulate the expression of the human Phox2a gene. J. Neurosci. 2001, 21, 7037–7045. [Google Scholar] [CrossRef] [Green Version]
- Di Zanni, E.; Fornasari, D.; Ravazzolo, R.; Ceccherini, I.; Bachetti, T. Identification of novel pathways and molecules able to down-regulate PHOX2B gene expression by in vitro drug screening approaches in neuroblastoma cells. Exp. Cell Res. 2015, 336, 43–57. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Yao, S.; Li, F.; Xin, L.; Lai, M.; Bracchi-Ricard, V.; Xu, H.; Yen, W.; Meng, W.; et al. Nuclear factor kappa B signaling initiates early differentiation of neural stem cells. Stem Cells 2012, 30, 510–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachetti, T.; Bagnasco, S.; Piumelli, R.; Palmieri, A.; Ceccherini, I. A Common 3′UTR Variant of the PHOX2B Gene Is Associated With Infant Life-Threatening and Sudden Death Events in the Italian Population. Front. Neurol. 2021, 12, 642735. [Google Scholar] [CrossRef] [PubMed]
- Pulford, K.; Lamant, L.; Morris, S.W.; Butler, L.H.; Wood, K.M.; Stroud, D.; Delsol, G.; Mason, D.Y. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997, 89, 1394–1404. [Google Scholar] [CrossRef]
- Hurley, S.P.; Clary, D.O.; Copie, V.; Lefcort, F. Anaplastic lymphoma kinase is dynamically expressed on subsets of motor neurons and in the peripheral nervous system. J. Comp. Neurol. 2006, 495, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Delisle, L.; Pierre-Eugene, C.; Bloch-Gallego, E.; Birling, M.C.; Duband, J.L.; Durand, E.; Bourgeois, T.; Matrot, B.; Sorg, T.; Huerre, M.; et al. Hyperactivation of Alk induces neonatal lethality in knock-in AlkF1178L mice. Oncotarget 2014, 5, 2703–2713. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.B.; Xue, C.; Benice, T.; Xue, L.; Morris, S.W.; Raber, J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: Functions and genetic interactions in learning, memory and adult neurogenesis. Pharm. Biochem. Behav. 2012, 100, 566–574. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Lopez-Delisle, L.; Delattre, O.; Rohrer, H. The ALK receptor in sympathetic neuron development and neuroblastoma. Cell Tissue Res. 2018, 372, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Cheng, M.; Zhang, Q.; Wasik, M.; Kelsh, R.; Winkler, C. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PLoS ONE 2013, 8, e63757. [Google Scholar] [CrossRef] [Green Version]
- Mao, R.; Zhang, X.; Kong, Y.; Wu, S.; Huo, H.Q.; Kong, Y.; Wang, Z.; Liu, Y.; Jia, Z.; Zhou, Z. Transcriptome Regulation by Oncogenic ALK Pathway in Mammalian Cortical Development Revealed by Single-Cell RNA Sequencing. Cereb. Cortex. 2021, 31, 3911–3924. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Wood, A.; Maris, J.M. Inhibition of ALK signaling for cancer therapy. Clin. Cancer Res. 2009, 15, 5609–5614. [Google Scholar] [CrossRef] [Green Version]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Javanmardi, N.; Fransson, S.; Djos, A.; Umapathy, G.; Ostensson, M.; Milosevic, J.; Borenas, M.; Hallberg, B.; Kogner, P.; Martinsson, T.; et al. Analysis of ALK, MYCN, and the ALK ligand ALKAL2 (FAM150B/AUGalpha) in neuroblastoma patient samples with chromosome arm 2p rearrangements. Genes Chromosomes Cancer 2019, 59, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Caren, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Bellini, A.; Potschger, U.; Bernard, V.; Lapouble, E.; Baulande, S.; Ambros, P.F.; Auger, N.; Beiske, K.; Bernkopf, M.; Betts, D.R.; et al. Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J. Clin. Oncol. 2021, 39, 3377–3390. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, G.; El Wakil, A.; Witek, B.; Chesler, L.; Danielson, L.; Deng, X.; Gray, N.S.; Johansson, M.; Kvarnbrink, S.; Ruuth, K.; et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci. Signal. 2014, 7, ra102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonherr, C.; Ruuth, K.; Kamaraj, S.; Wang, C.L.; Yang, H.L.; Combaret, V.; Djos, A.; Martinsson, T.; Christensen, J.G.; Palmer, R.H.; et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 2012, 31, 5193–5200. [Google Scholar] [CrossRef] [Green Version]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Schulte, J.H.; Lindner, S.; Bohrer, A.; Maurer, J.; De Preter, K.; Lefever, S.; Heukamp, L.; Schulte, S.; Molenaar, J.; Versteeg, R.; et al. MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 2013, 32, 1059–1065. [Google Scholar] [CrossRef]
- Montavon, G.; Jauquier, N.; Coulon, A.; Peuchmaur, M.; Flahaut, M.; Bourloud, K.B.; Yan, P.; Delattre, O.; Sommer, L.; Joseph, J.M.; et al. Wild-type ALK and activating ALK-R1275Q and ALK-F1174L mutations upregulate Myc and initiate tumor formation in murine neural crest progenitor cells. Oncotarget 2014, 5, 4452–4466. [Google Scholar] [CrossRef] [Green Version]
- Heukamp, L.C.; Thor, T.; Schramm, A.; De Preter, K.; Kumps, C.; De Wilde, B.; Odersky, A.; Peifer, M.; Lindner, S.; Spruessel, A.; et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci. Transl. Med. 2012, 4, 141ra191. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Ribeiro, D.; Arsenian-Henriksson, M.; Deller, T.; Rohrer, H. Proliferation and Survival of Embryonic Sympathetic Neuroblasts by MYCN and Activated ALK Signaling. J. Neurosci. 2016, 36, 10425–10439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954. [Google Scholar] [CrossRef]
- Sekimizu, M.; Osumi, T.; Fukano, R.; Koga, Y.; Kada, A.; Saito, A.M.; Mori, T. A Phase I/II Study of Crizotinib for Recurrent or Refractory Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma and a Phase I Study of Crizotinib for Recurrent or Refractory Neuroblastoma: Study Protocol for a Multicenter Single-arm Open-label Trial. Acta Med. Okayama. 2018, 72, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Fransson, S.; Siaw, J.T.; Treis, D.; Van den Eynden, J.; Chand, D.; Umapathy, G.; Ruuth, K.; Svenberg, P.; Wessman, S.; et al. Clinical response of the novel activating ALK-I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002550. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Halilovic, E.; Li, X.; Liang, J.; Cao, Y.; Rakiec, D.P.; Ruddy, D.A.; Jeay, S.; Wuerthner, J.U.; Timple, N.; et al. Combined ALK and MDM2 inhibition increases antitumor activity and overcomes resistance in human ALK mutant neuroblastoma cell lines and xenograft models. Elife 2017, 6, e17137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.Y.; et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin Cancer Res. 2020, 26, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paolo, D.; Brignole, C.; Pastorino, F.; Carosio, R.; Zorzoli, A.; Rossi, M.; Loi, M.; Pagnan, G.; Emionite, L.; Cilli, M.; et al. Neuroblastoma-targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol. Ther. 2011, 19, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, D.; Yang, D.; Pastorino, F.; Emionite, L.; Cilli, M.; Daga, A.; Destafanis, E.; Di Fiore, A.; Piaggio, F.; Brignole, C.; et al. New therapeutic strategies in neuroblastoma: Combined targeting of a novel tyrosine kinase inhibitor and liposomal siRNAs against ALK. Oncotarget 2015, 6, 28774–28789. [Google Scholar] [CrossRef] [Green Version]
- Diskin, S.J.; Capasso, M.; Schnepp, R.W.; Cole, K.A.; Attiyeh, E.F.; Hou, C.; Diamond, M.; Carpenter, E.L.; Winter, C.; Lee, H.; et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 2012, 44, 1126–1130. [Google Scholar] [CrossRef]
- Corallo, D.; Candiani, S.; Ori, M.; Aveic, S.; Tonini, G.P. The zebrafish as a model for studying neuroblastoma. Cancer Cell Int. 2016, 16, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corallo, D.; Donadon, M.; Pantile, M.; Sidarovich, V.; Cocchi, S.; Ori, M.; De Sarlo, M.; Candiani, S.; Frasson, C.; Distel, M.; et al. LIN28B increases neural crest cell migration and leads to transformation of trunk sympathoadrenal precursors. Cell Death Differ. 2020, 27, 1225–1242. [Google Scholar] [CrossRef]
- Tao, T.; Shi, H.; Mariani, L.; Abraham, B.J.; Durbin, A.D.; Zimmerman, M.W.; Powers, J.T.; Missios, P.; Ross, K.N.; Perez-Atayde, A.R.; et al. LIN28B regulates transcription and potentiates MYCN-induced neuroblastoma through binding to ZNF143 at target gene promotors. Proc. Natl. Acad. Sci. USA 2020, 117, 16516–16526. [Google Scholar] [CrossRef]
- Bresler, S.C.; Wood, A.C.; Haglund, E.A.; Courtright, J.; Belcastro, L.T.; Plegaria, J.S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter, E.L.; et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, S.S.; Clarke, S.; Veschi, V.; Thiele, C.J. Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 2020, 10, 623679. [Google Scholar] [CrossRef] [PubMed]
- Wolpaw, A.J.; Bayliss, R.; Buchel, G.; Dang, C.V.; Eilers, M.; Gustafson, W.C.; Hansen, G.H.; Jura, N.; Knapp, S.; Lemmon, M.A.; et al. Drugging the “Undruggable” MYCN Oncogenic Transcription Factor: Overcoming Previous Obstacles to Impact Childhood Cancers. Cancer Res. 2021, 81, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Uchiumi, T.; Niina, I.; Wakasugi, T.; Igarashi, T.; Momii, Y.; Yoshida, T.; Matsuo, K.; Miyamoto, N.; Izumi, H. Transcription factors and drug resistance. Eur. J. Cancer 2005, 41, 2577–2586. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TF/TG | MUTATIONS | OVEREXPRESSION | ||
|---|---|---|---|---|
| Transcriptional Activation | Gene Amplification | miRNA Downregulation | ||
| MYCN | rare | X | X | miR-34, let-7, miR-204 |
| ALK | X | X | X | miR-96 |
| PHOX2B | X | X | nd | miR-204 |
| LIN28 | rare | X | rare | let-7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perri, P.; Ponzoni, M.; Corrias, M.V.; Ceccherini, I.; Candiani, S.; Bachetti, T. A Focus on Regulatory Networks Linking MicroRNAs, Transcription Factors and Target Genes in Neuroblastoma. Cancers 2021, 13, 5528. https://doi.org/10.3390/cancers13215528
Perri P, Ponzoni M, Corrias MV, Ceccherini I, Candiani S, Bachetti T. A Focus on Regulatory Networks Linking MicroRNAs, Transcription Factors and Target Genes in Neuroblastoma. Cancers. 2021; 13(21):5528. https://doi.org/10.3390/cancers13215528
Chicago/Turabian StylePerri, Patrizia, Mirco Ponzoni, Maria Valeria Corrias, Isabella Ceccherini, Simona Candiani, and Tiziana Bachetti. 2021. "A Focus on Regulatory Networks Linking MicroRNAs, Transcription Factors and Target Genes in Neuroblastoma" Cancers 13, no. 21: 5528. https://doi.org/10.3390/cancers13215528
APA StylePerri, P., Ponzoni, M., Corrias, M. V., Ceccherini, I., Candiani, S., & Bachetti, T. (2021). A Focus on Regulatory Networks Linking MicroRNAs, Transcription Factors and Target Genes in Neuroblastoma. Cancers, 13(21), 5528. https://doi.org/10.3390/cancers13215528