
Autophagy in Cancer Therapy—Molecular Mechanisms and Current Clinical Advances


Abstract
:Simple Summary
Abstract
1. Autophagy in Cancer Treatment—A Molecular Introduction
2. Autophagy in Cancer Treatment—A Double-Edged Sword
2.1. Tumor-Suppressive Functions of Autophagy
2.2. Oncogenic Functions of Autophagy
2.3. Autophagy-Mediated Immune Evasion
2.4. Autophagy and Therapy Resistance
3. Autophagy in Cancer Treatment—Current Status and Perspectives
Clinical Trials
4. Conclusions and Outlook on Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Appelmans, F.; Wattiaux, R.; De Duve, C. Tissue fractionation studies. 5. The association of acid phosphatase with a special class of cytoplasmic granules in rat liver. Biochem. J. 1955, 59, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ishii, J.; Asai, E.; Ohsumi, Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 2012, 8, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.V.; Hefner-Gravink, A.; Morano, K.A.; Noda, T.; Ohsumi, Y.; Klionsky, D.J. Cytoplasm-to-vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc. Natl. Acad. Sci. USA 1996, 93, 12304–12308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, T.M.; Hefner-Gravink, A.; Thumm, M.; Klionsky, D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996, 271, 17621–17624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umekawa, M.; Klionsky, D.J. The Cytoplasm-to-Vacuole Targeting Pathway: A Historical Perspective. Int. J. Cell Biol. 2012, 2012, 142634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, S. Microautophagy-distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020. [Google Scholar] [CrossRef]
- Kissova, I.; Salin, B.; Schaeffer, J.; Bhatia, S.; Manon, S.; Camougrand, N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 2007, 3, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Choi, S.E.; Koh, H.C. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol. Lett. 2018, 284, 120–128. [Google Scholar] [CrossRef]
- Lu, W.; Karuppagounder, S.S.; Springer, D.A.; Allen, M.D.; Zheng, L.; Chao, B.; Zhang, Y.; Dawson, V.L.; Dawson, T.M.; Lenardo, M. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson’s-like movement disorder. Nat. Commun. 2014, 5, 4930. [Google Scholar] [CrossRef]
- Ganzleben, I.; He, G.W.; Gunther, C.; Prigge, E.S.; Richter, K.; Rieker, R.J.; Mougiakakos, D.; Neurath, M.F.; Becker, C. PGAM5 is a key driver of mitochondrial dysfunction in experimental lung fibrosis. Cell Mol. Life Sci. 2019, 76, 4783–4794. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef] [PubMed]
- Devenport, S.N.; Singhal, R.; Radyk, M.D.; Taranto, J.G.; Kerk, S.A.; Chen, B.; Goyert, J.W.; Jain, C.; Das, N.K.; Oravecz-Wilson, K.; et al. Colorectal cancer cells utilize autophagy to maintain mitochondrial metabolism for cell proliferation under nutrient stress. JCI Insight 2021. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; Pestell, R.G.; et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: A transcriptional informatics analysis with validation. Cell Cycle 2010, 9, 2201–2219. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Tsirigos, A.; Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010, 9, 3485–3505. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Perera, R.M.; Kimmelman, A.C. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy 2020, 16, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Alissafi, T.; Hatzioannou, A.; Mintzas, K.; Barouni, R.M.; Banos, A.; Sormendi, S.; Polyzos, A.; Xilouri, M.; Wielockx, B.; Gogas, H.; et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Investig. 2018, 128, 3840–3852. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Avalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. BioMed Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, K.; Sundararajan, R.; Lindsten, T.; Thompson, C.; White, E. Bax and Bak independently promote cytochrome C release from mitochondria. J. Biol. Chem. 2002, 277, 14127–14134. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Mitsunaga, S.; Yamazaki, M.; Hasebe, T.; Ishii, G.; Kojima, M.; Kinoshita, T.; Ueno, T.; Esumi, H.; Ochiai, A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008, 99, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1alpha and HIF-2alpha target genes. Mol. Cancer 2010, 9, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchinetti, F.; Bluthgen, M.V.; Tergemina-Clain, G.; Faivre, L.; Pignon, J.P.; Planchard, D.; Remon, J.; Soria, J.C.; Lacroix, L.; Besse, B. LKB1/STK11 mutations in non-small cell lung cancer patients: Descriptive analysis and prognostic value. Lung Cancer 2017, 112, 62–68. [Google Scholar] [CrossRef]
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 2019, 33, 150–165. [Google Scholar] [CrossRef] [Green Version]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 2019, 29, 285–302.e287. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 is a critical determinant of autophagy, metabolic switching, and oxidative stress in vascular endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercier, I.; Casimiro, M.C.; Wang, C.; Rosenberg, A.L.; Quong, J.; Minkeu, A.; Allen, K.G.; Danilo, C.; Sotgia, F.; Bonuccelli, G.; et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol. Ther. 2008, 7, 1212–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Dasgupta, A.; Sammons, S.; Er, O.; Potoczek, M.B.; Guiles, F.; Sotgia, F.; Brody, J.R.; Mitchell, E.P.; Lisanti, M.P. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol. Ther. 2010, 10, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.; Janczara, J.; McCubrey, J.A.; Gizak, A.; Rakus, D. The Reverse Warburg Effect is Associated with Fbp2-Dependent Hif1alpha Regulation in Cancer Cells Stimulated by Fibroblasts. Cells 2020, 9, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, M.; Dai, D.; Vudem, A.; Varner, J.D.; Stroock, A.D. Multi-scale computational study of the Warburg effect, reverse Warburg effect and glutamine addiction in solid tumors. PLoS Comput. Biol. 2018, 14, e1006584. [Google Scholar] [CrossRef] [Green Version]
- DeVorkin, L.; Pavey, N.; Carleton, G.; Comber, A.; Ho, C.; Lim, J.; McNamara, E.; Huang, H.; Kim, P.; Zacharias, L.G.; et al. Autophagy Regulation of Metabolism Is Required for CD8(+) T Cell Anti-tumor Immunity. Cell Rep. 2019, 27, 502–513.e505. [Google Scholar] [CrossRef] [Green Version]
- Cunha, L.D.; Yang, M.; Carter, R.; Guy, C.; Harris, L.; Crawford, J.C.; Quarato, G.; Boada-Romero, E.; Kalkavan, H.; Johnson, M.D.L.; et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018, 175, 429–441.e416. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.L.; Zhang, H.L.; Huang, Y.; Huang, J.H.; Sun, P.; Zhou, N.N.; Chen, Y.H.; Mai, J.; Wang, Y.; Yu, Y.; et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat. Commun. 2020, 11, 3806. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.X.; Xu, T.M.; Zhou, Z.L.; Lv, X.J.; Liu, J.; Zhang, W.J.; Cui, M.H. TRP14 promotes resistance to cisplatin by inducing autophagy in ovarian cancer. Oncol. Rep. 2019, 42, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 201. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, M.; Arias, A.; Martinez-Garcia, D.; Perez-Tomas, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Xu, T.; Xia, L.; Wang, X.; Zhang, X.; Zhang, X.; Zhu, Z.; Zhong, S.; Wang, C.; Shen, Z. Chloroquine enhances the efficacy of cisplatin by suppressing autophagy in human adrenocortical carcinoma treatment. Drug Des. Devel. Ther. 2016, 10, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Yoon, J.S.; Won, Y.W.; Park, B.B.; Lee, Y.Y. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. Acta Pathol. Microbiol. Immunol. Scand. 2012, 120, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Hosoda, R.; Akiyama, Y.; Sebori, R.; Wanibuchi, M.; Mikami, T.; Sugino, T.; Suzuki, K.; Maruyama, M.; Tsukamoto, M.; et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J. Neurooncol. 2015, 122, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyas, E.; Lopez-Bonet, E.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.Q.; Wang, S.B.; Shao, Y.F.; Shi, J.N.; Wang, W.; Chen, W.Y.; Ye, Z.Q.; Jiang, J.Y.; Fang, Q.X.; Zhang, G.B.; et al. Hydroxychloroquine potentiates the anti-cancer effect of bevacizumab on glioblastoma via the inhibition of autophagy. Biomed. Pharmacother. 2019, 118, 109339. [Google Scholar] [CrossRef]
- Zhao, Z.; Xia, G.; Li, N.; Su, R.; Chen, X.; Zhong, L. Autophagy Inhibition Promotes Bevacizumab-induced Apoptosis and Proliferation Inhibition in Colorectal Cancer Cells. J. Cancer 2018, 9, 3407–3416. [Google Scholar] [CrossRef] [Green Version]
- White, N.J. The treatment of malaria. N. Engl. J. Med. 1996, 335, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Moroni, G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin. Drug Saf. 2017, 16, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Axfors, C.; Schmitt, A.M.; Janiaud, P.; Van’t Hooft, J.; Abd-Elsalam, S.; Abdo, E.F.; Abella, B.S.; Akram, J.; Amaravadi, R.K.; Angus, D.C.; et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat. Commun. 2021, 12, 2349. [Google Scholar] [CrossRef]
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine 2018, 97, e12912. [Google Scholar] [CrossRef] [PubMed]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.B.; Rich, T.A.; Byrd, D.R.; Cleary, K.R.; Connelly, J.H.; Levin, B.; Charnsangavej, C.; Fenoglio, C.J.; Ames, F.C. Preoperative chemoradiation and pancreaticoduodenectomy for adenocarcinoma of the pancreas. Arch. Surg. 1992, 127, 1335–1339. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786. [Google Scholar] [CrossRef]
- Malhotra, J.; Jabbour, S.; Orlick, M.; Riedlinger, G.; Guo, Y.; White, E.; Aisner, J. Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat. Res. Commun. 2019, 21, 100158. [Google Scholar] [CrossRef] [PubMed]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy Inhibition to Augment mTOR Inhibition: A Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Chen, G.; Ding, X.F.; Bouamar, H.; Pressley, K.; Sun, L.Z. Everolimus induces G1 cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells. Am. J. Physiol. Cell Physiol. 2019, 317, C244–C252. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Z.; Yan, L.; Li, X.; Zhang, J.; Zhang, X.; Zhu, D.; Sun, Y.; Jiang, Q. Everolimus reduces postoperative arthrofibrosis in rabbits by inducing autophagy-mediated fibroblast apoptosis by PI3K/Akt/mTOR signaling pathway. Biochem. Biophys. Res. Commun. 2020, 533, 1–8. [Google Scholar] [CrossRef]
- Lee, S.C.; Kim, K.H.; Kim, O.H.; Lee, S.K.; Kim, S.J. Activation of Autophagy by Everolimus Confers Hepatoprotection Against Ischemia-Reperfusion Injury. Am. J. Transplant. 2016, 16, 2042–2054. [Google Scholar] [CrossRef]
- Liu, W.; Huang, S.; Chen, Z.; Wang, H.; Wu, H.; Zhang, D. Temsirolimus, the mTOR inhibitor, induces autophagy in adenoid cystic carcinoma: In vitro and in vivo. Pathol. Res. Pract. 2014, 210, 764–769. [Google Scholar] [CrossRef]
- Inamura, S.O.; Ito, H.; Taga, M.; Tsuchiyama, K.; Hoshino, H.; Kobayashi, M.; Yokoyama, O. Low-dose Docetaxel Enhanced the Anticancer Effect of Temsirolimus by Overcoming Autophagy in Prostate Cancer Cells. Anticancer Res. 2019, 39, 5417–5425. [Google Scholar] [CrossRef]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saladini, S.; Aventaggiato, M.; Barreca, F.; Morgante, E.; Sansone, L.; Russo, M.A.; Tafani, M. Metformin Impairs Glutamine Metabolism and Autophagy in Tumour Cells. Cells 2019, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Hu, H.; Ye, S.; Wang, H.; Cui, R.; Yi, L. The effect of metformin therapy on incidence and prognosis in prostate cancer: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaout, A.; Robertson, S.J.; Pond, G.R.; Lee, H.; Jeong, A.; Ianni, L.; Kroeger, L.; Hilton, J.; Coupland, S.; Gottlieb, C.; et al. A randomized, double-blind, window of opportunity trial evaluating the effects of chloroquine in breast cancer patients. Breast Cancer Res. Treat. 2019, 178, 327–335. [Google Scholar] [CrossRef]
- Rojas-Puentes, L.L.; Gonzalez-Pinedo, M.; Crismatt, A.; Ortega-Gomez, A.; Gamboa-Vignolle, C.; Nunez-Gomez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 2013, 8, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- El-Chemaly, S.; Taveira-Dasilva, A.; Goldberg, H.J.; Peters, E.; Haughey, M.; Bienfang, D.; Jones, A.M.; Julien-Williams, P.; Cui, Y.; Villalba, J.A.; et al. Sirolimus and Autophagy Inhibition in Lymphangioleiomyomatosis: Results of a Phase I Clinical Trial. Chest 2017, 151, 1302–1310. [Google Scholar] [CrossRef]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehnert, J.M.; Kaveney, A.D.; Malhotra, J.; Spencer, K.; Portal, D.; Goodin, S.; Tan, A.R.; Aisner, J.; Moss, R.A.; Lin, H.; et al. A phase I trial of MK-2206 and hydroxychloroquine in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2019, 84, 899–907. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, S.B.; Supko, J.G.; Neal, J.W.; Muzikansky, A.; Digumarthy, S.; Fidias, P.; Temel, J.S.; Heist, R.S.; Shaw, A.T.; McCarthy, P.O.; et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1602–1608. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Garrido-Laguna, I.; Naing, A.; Fu, S.; Falchook, G.S.; Piha-Paul, S.A.; Wheler, J.J.; Hong, D.S.; Tsimberidou, A.M.; Subbiah, V.; et al. Phase I dose-escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget 2016, 7, 67521–67531. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gu, C.; Zhong, D.; Shi, L.; Kong, Y.; Zhou, Z.; Liu, S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014, 355, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Cai, M.; Zhang, Y.; Tao, L.; Guo, R. miR-29c-3p inhibits autophagy and cisplatin resistance in ovarian cancer by regulating FOXP1/ATG14 pathway. Cell Cycle 2020, 19, 193–206. [Google Scholar] [CrossRef]
- Zhang, S.F.; Wang, X.Y.; Fu, Z.Q.; Peng, Q.H.; Zhang, J.Y.; Ye, F.; Fu, Y.F.; Zhou, C.Y.; Lu, W.G.; Cheng, X.D.; et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy 2015, 11, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Kang, H.; Zhao, Y.; Min, I.; Wyrwas, B.; Moore, M.; Teng, L.; Zarnegar, R.; Jiang, X.; Fahey, T.J., 3rd. Targeting Autophagy Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. J. Clin. Endocrinol. Metab. 2017, 102, 634–643. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Gremke, N.; Polo, P.; Dort, A.; Schneikert, J.; Elmshauser, S.; Brehm, C.; Klingmuller, U.; Schmitt, A.; Reinhardt, H.C.; Timofeev, O.; et al. mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nat. Commun. 2020, 11, 4684. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Nicastri, M.C.; Fennelly, C.; Chude, C.I.; Barber-Rotenberg, J.S.; Ronghe, A.; McAfee, Q.; McLaughlin, N.P.; Zhang, G.; Goldman, A.R.; et al. PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov. 2019, 9, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Ojha, R.; Noguera-Ortega, E.; Rebecca, V.W.; Attanasio, J.; Liu, S.; Piao, S.; Lee, J.J.; Nicastri, M.C.; Harper, S.L.; et al. PPT1 inhibition enhances the antitumor activity of anti-PD-1 antibody in melanoma. JCI Insight 2020. [Google Scholar] [CrossRef] [PubMed]
- Brun, S.; Bassissi, F.; Serdjebi, C.; Novello, M.; Tracz, J.; Autelitano, F.; Guillemot, M.; Fabre, P.; Courcambeck, J.; Ansaldi, C.; et al. GNS561, a new lysosomotropic small molecule, for the treatment of intrahepatic cholangiocarcinoma. Investig. New Drugs 2019, 37, 1135–1145. [Google Scholar] [CrossRef] [PubMed]


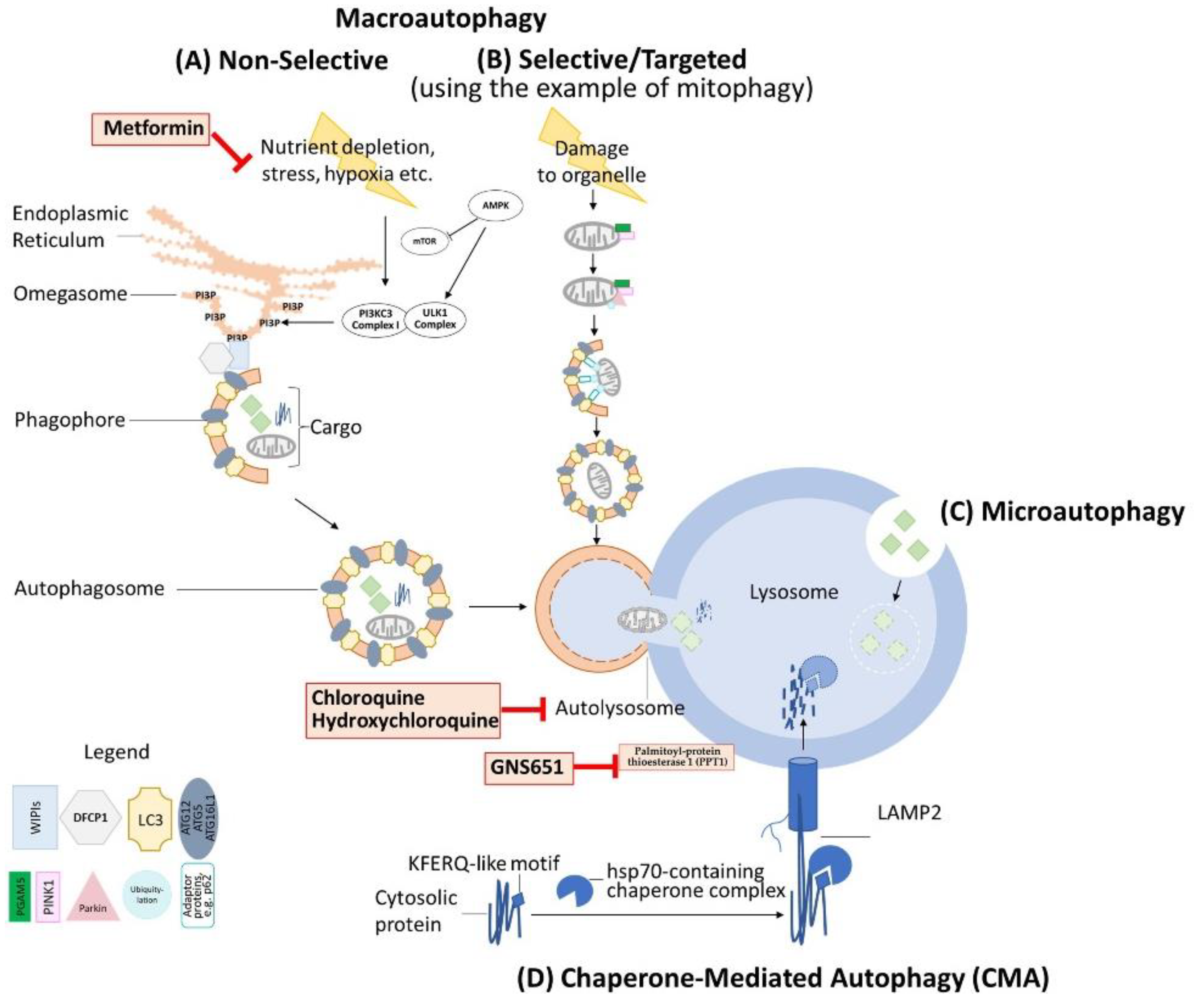{kind=link}
| Experimental Context | Mode of Action | Involved Components/ Regulators | Outcome | Conclusion | Reference |
|---|---|---|---|---|---|
| Sporadic breast, ovarian, and prostate cancer | Beclin-1 facilitates autophagy | Beclin 1 (beclin1+/−) heterozygous knock-out leads to reduction of autophagy (mice) | Disruption of beclin 1
| Beclin1 mediated autophagy acts tumor-suppressive | [39] |
| Liver neoplasms | Autophagy is required for detoxification of oxidative stress and prevention of associated damage | Mosaic deletion of Atg5 OR liver-specific Atg7−/− (mice) | Lack of Atg7 in hepatocytes causes:
| Autophagy acts tumor-suppressive | [40] |
| BrafV600E-induced lung cancer model | Autophagy causes prevention of ROS accumulation (from damaged mitochondria); autophagic recycling supports mitochondrial tricarboxylic acid (TCA) cycle/oxidative phosphorylation | Atg7 knock-out (mice) | Atg7 knock-out:
| Autophagy in BrafV600E tumors initially acts oncogenic and subsequently tumor-suppressive | [41] |
| Immortalized baby mouse kidney epithelial cells [BAX/BAK (W2) or deficient BAX/BAK (D3)] | Autophagy in apoptosis-defective cells prevents necrosis | Constitutive expression of AKT (myr-AKT) OR RAS (H-rasV12) (murine model) | Blocking autophagy causes necrosis and inflammation | Autophagy acts oncogenic regarding tumor cell survival but its inhibition leads to overall progression based on necrosis-triggered inflammation | [42] |
| Pancreatic ductal adenocarcinoma (PDAC) | Autophagic recycling is required to supply the tricarboxylic acid (TCA) cycle and oxidative phosphorylation | RNA interference of Atg5 OR chloroquine (CQ) (cell lines, murine model) | Blocking autophagy inhibits PDAC tumor growth both in vitro and in vivo | Autophagy acts oncogenic in RAS-dependent PDAC | [43] |
| K-Ras induced lung tumors OR liver kinase B1 (LKB1) knock-out (mice) | Autophagy provides e.g., glutamine for the TCA cycle | Atg7 knock-out (murine model) | Blocking autophagy leads to depleted energy metabolism with non-sustainable increased β-oxidation | Autophagy acts oncogenic in K-Ras-dependent lung tumors and liver kinase B1 mutated tumors | [44] |
| Acute myeloid leukemia (AML) | Feedback loop:
| Mitochondria-endoplasmic reticulum contact sites (MERCs) modulate autophagy (cell lines) | Mitochondria-endoplasmic reticulum contact sites (MERCs) modulate autoph-agy (cell lines) | Autophagy acts oncogenic | [45] |
| Colorectal cancer | Autophagy supplies metabolic intermediates for mitochondria. PINK1-mediated mitophagy causes mitochondrial recycling | Atg5 knock-out; RNA interference PINK1 (cell lines and murine model) | Blocking autophagy/mitophagy reduces tumor growth | Autophagy and mitophagy act oncogenic | [46] |
| Breast cancer | Caveolin-1 downregulation by ROS-induced autophagy in cancer associated fibroblasts (CAFs) | Caveolin-1 knock-out (mice) | Autophagy in CAFs leads to catabolism and supplies e.g., glutamine to adjacent cancer cells | Autophagy acts oncogenic | [47,48,49] |
| Pancreatic ductal adenocarcinoma (PDAC) | Autophagy degrades MHC class I molecules | Atg4B OR Atg7 RNA interference | Blocking autophagy leads to MHC class I molecule reappearance leading to increased immune detection of the tumor | Autophagy acts oncogenic | [50,51] |
| Melanoma | Autophagy degrades MHC class II molecules leading to myeloid-derived suppressor cells (MDSCs) blocking anti-cancer immune response | Atg5 knock-out (mice) | Blocking autophagy leads to MHC class II molecule reappearance and subsequent priming of anti-cancer leukocytes | Autophagy acts oncogenic | [52] |
| Autophagy-Inhibitor | Combination with | Tumor Entity | Outcome | Clinical Phase | Number of Patients | Reference |
|---|---|---|---|---|---|---|
| HCQ | Gemcitabine, nab-Paclitaxel | Pancreatic cancer (metastatic or advanced) | Primary endpoint: 12-month overall survival not improved. Improvement in overall response rate. | II | 112 | [97] |
| HCQ | Gemcitabine, nab-Paclitaxel | Pancreatic cancer (potentially resectable) | Primary endpoint: histological response at resection improved. HCQ led to increased autophagy-inhibition and immune activity in the tumor. | II | 64 | [98] |
| HCQ | None | Pancreatic cancer (previously treated and metastatic) | Inconsistent autophagy inhibition. No survival benefits. | II | 20 | [99] |
| HCQ | Gemcitabine | Pancreatic cancer (adenocarcinoma, preoperative) | Patients with >51% reduction of autophagy (surrogate: LC3-II in circulating PMNs) had significant (p < 0.05) improvement in disease-free survival (15.03 vs. 6.9 months) and median overall survival (34.83 vs. 10.83 months). | I/II | 35 | [100] |
| HCQ | Imatinib | Chronic myeloid leukemia (major cytogenetic response with residual disease) | 12 months: ’Success’ rate not improved. Major Molecular Remission (MMR): 80% (Imatinib) compared to 92% (Imatinib/HCQ) (n.s.). 24 months: ’Success’ rate increased 20.8% for Imatinib/HCQ vs. Imatinib (n.s.). | II | 62 | [102] |
| CQ | None | Breast cancer (preoperative) | No effect on cancer cell proliferation (n.s.). | II | 70 | [114] |
| HCQ | Everolimus | Clear-cell renal cell carcinoma (previously treated) | Longer stable disease in some patients, inconsistent autophagy inhibition. | I/II | 38 | [104] |
| CQ | Whole-brain irradiation | Brain metastases | Overall response rate (ORR): CQ 54% vs. Control 55% (n.s.). Progression-free survival: CQ 83.9% (95% CI 69.4–98.4) control 55.1% (95% CI 33.6–77.6). CQ significantly improves PFS: RR 0.31 (95% CI [0.1–0.9]). No difference in response rate or overall survival. | II | 73 | [115] |
| HCQ | Carboplatin, Paclitaxel, Bevacizumab (if criteria met) | NSCLC (metastatic and untreated) | Progression-free survival longer than expected. | Ib/II | 40 | [103] |
| HCQ | Radiation therapy; concurrent, adjuvant Temozolomide | Glioblastoma multiforme (newly diagnosed) | No significant improvement in overall survival. Significant but inconsistent autophagy inhibition. | I/II | 92 | [116] |
| HCQ | Temsirolimus | Advanced solid tumors and melanoma | No significant improvements | I | 40 | [105] |
| HCQ | Sirolimus | Lymphangioleiomyomatosis | No improvement of lung function | I | 14 | [117] |
| HCQ | Bortezomib | Multiple Myeloma (relapsed, refractory) | Very good partial responses (14%), minor response (14%), temporary stable disease (45%) | I | 25 | [118] |
| HCQ | MK-2206 (AKT inhibitor) | Advanced solid tumors | Stable disease 15%. No significant antineoplastic activity. | I | 35 | [119] |
| HCQ | Temozolomide | Advanced solid tumors and melanoma | Metastatic melanoma: Partial response 14%, stable disease 27%. Subgroup analysis refractory BRAF wild-type melanoma: 2/6 patients almost complete response, prolonged stable disease. Significant inhibition of autophagy. | I | 40 | [120] |
| HCQ | Erlotinib | NSCLC (advanced, prior clinical response to EGFR-TKI) | No relevant toxicities. | I | 27 | [121] |
| HCQ | Sirolimus, Vorinostat | Advanced Cancers | Partial response: Refractory Hodgkin lymphoma; perivascular epithelioid tumor. Stable disease: Hepatocellular carcinoma, fibromyxoid sarcoma. | I | 70 | [122] |
| NCT Number | Tumor Entity | Autophagy Modulator | Combination with… | Clinical Phase | Enrollment | Registration |
|---|---|---|---|---|---|---|
| NCT03037437 | Hepatocellular cancer | HCQ | Sorafenib | II | 68 ** | https://ClinicalTrials.gov/show/NCT03037437, accessed on 1 October 2021 |
| NCT04214418 | Gastrointestinal cancer | HCQ | Cobimetinib, Atezolizumab | I/II | 175 ** | https://ClinicalTrials.gov/show/NCT04214418, accessed on 1 October 2021 |
| NCT04386057 | Pancreatic cancer (advanced) | HCQ | LY3214996 | II | 52 ** | https://ClinicalTrials.gov/show/NCT04386057, accessed on 1 October 2021 |
| NCT05036226 | Prostate Cancer (recurrent), solid tumors | HCQ | Metformin, Sirolimus, Nelfinavir, Dasatinib | I/II | 76 ** | https://ClinicalTrials.gov/show/NCT05036226, accessed on 1 October 2021 |
| NCT02339168 | Prostate cancer | Metformin | Enzalutamide | I | 24 * | https://ClinicalTrials.gov/show/NCT02339168, accessed on 1 October 2021 |
| NCT01506973 | Adenocarcinoma (advanced, metastatic) | HCQ | Gemcitabine, Abraxane | I/II | 119 * | https://ClinicalTrials.gov/show/NCT01506973, accessed on 1 October 2021 |
| NCT04566133 | Cholangiocarcinoma | HCQ | Trametinib | II | 30 ** | https://ClinicalTrials.gov/show/NCT04566133, accessed on 1 October 2021 |
| NCT02042989 | Advanced cancers | MLN9708, Vorinostat | I | 68 * | https://ClinicalTrials.gov/show/NCT02042989, accessed on 1 October 2021 | |
| NCT01023737 | Malignant solid tumors | HCQ | Vorinostat | I | 72 * | https://ClinicalTrials.gov/show/NCT01023737, accessed on 1 October 2021 |
| NCT04333914 | Hematological or solid tumor (advanced, metastatic) | Autophagy inhibitor (GNS651) | Standard of care, Avdoralimab, Monalizumab | II | 219 ** | https://ClinicalTrials.gov/show/NCT04333914, accessed on 1 October 2021 |
| NCT03774472 | Breast Cancer | HCQ | Letrozole, Palbociclib | I/II | 54 ** | https://ClinicalTrials.gov/show/NCT03774472, accessed on 1 October 2021 |
| NCT01480154 | Malignant solid neoplasms (advanced), cutaneous melanoma, prostate cancer, renal cell cancer | HCQ | Akt Inhibitor MK2206 | I | 62 * | https://ClinicalTrials.gov/show/NCT01480154, accessed on 1 October 2021 |
| NCT04527549 | Melanoma (advanced) | HCQ | Placebo, Trametinib, Dabrafenib | II | 84 ** | https://ClinicalTrials.gov/show/NCT04527549, accessed on 1 October 2021 |
| NCT04841148 | Breast cancer | HCQ | Avelumab, Palbociclib | II | 96 ** | https://ClinicalTrials.gov/show/NCT04841148, accessed on 1 October 2021 |
| NCT03979651 | Melanoma (metastatic NRAS) | HCQ | Trametinib | N/A | 29 ** | https://ClinicalTrials.gov/show/NCT03979651, accessed on 1 October 2021 |
| NCT03598595 | Osteosarcoma (recurrent, refractory) | HCQ | Docetaxel, Gemcitabine | I/II | 31 ** | https://ClinicalTrials.gov/show/NCT03598595, accessed on 1 October 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganzleben, I.; Neurath, M.F.; Becker, C. Autophagy in Cancer Therapy—Molecular Mechanisms and Current Clinical Advances. Cancers 2021, 13, 5575. https://doi.org/10.3390/cancers13215575
Ganzleben I, Neurath MF, Becker C. Autophagy in Cancer Therapy—Molecular Mechanisms and Current Clinical Advances. Cancers. 2021; 13(21):5575. https://doi.org/10.3390/cancers13215575
Chicago/Turabian StyleGanzleben, Ingo, Markus F. Neurath, and Christoph Becker. 2021. "Autophagy in Cancer Therapy—Molecular Mechanisms and Current Clinical Advances" Cancers 13, no. 21: 5575. https://doi.org/10.3390/cancers13215575
APA StyleGanzleben, I., Neurath, M. F., & Becker, C. (2021). Autophagy in Cancer Therapy—Molecular Mechanisms and Current Clinical Advances. Cancers, 13(21), 5575. https://doi.org/10.3390/cancers13215575