
Autophagy in Cisplatin Nephrotoxicity during Cancer Therapy



Abstract
:Simple Summary
Abstract
1. Introduction
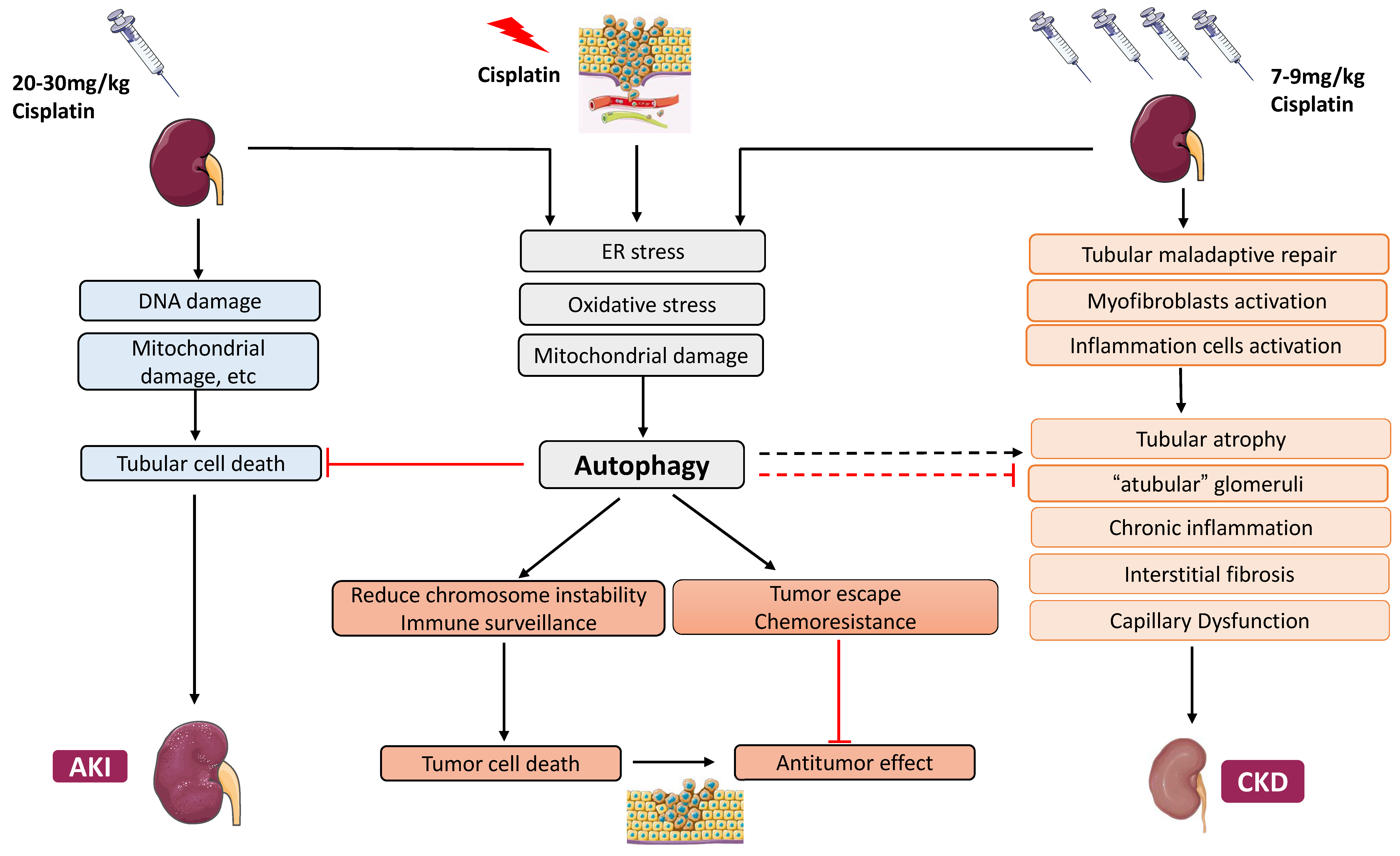
2. Cisplatin Nephrotoxicity
2.1. Cisplatin-Induced Acute Kidney Injury
2.2. Cisplatin-Induced Chronic Effects in the Kidney
3. Autophagy in Cisplatin-Induced AKI
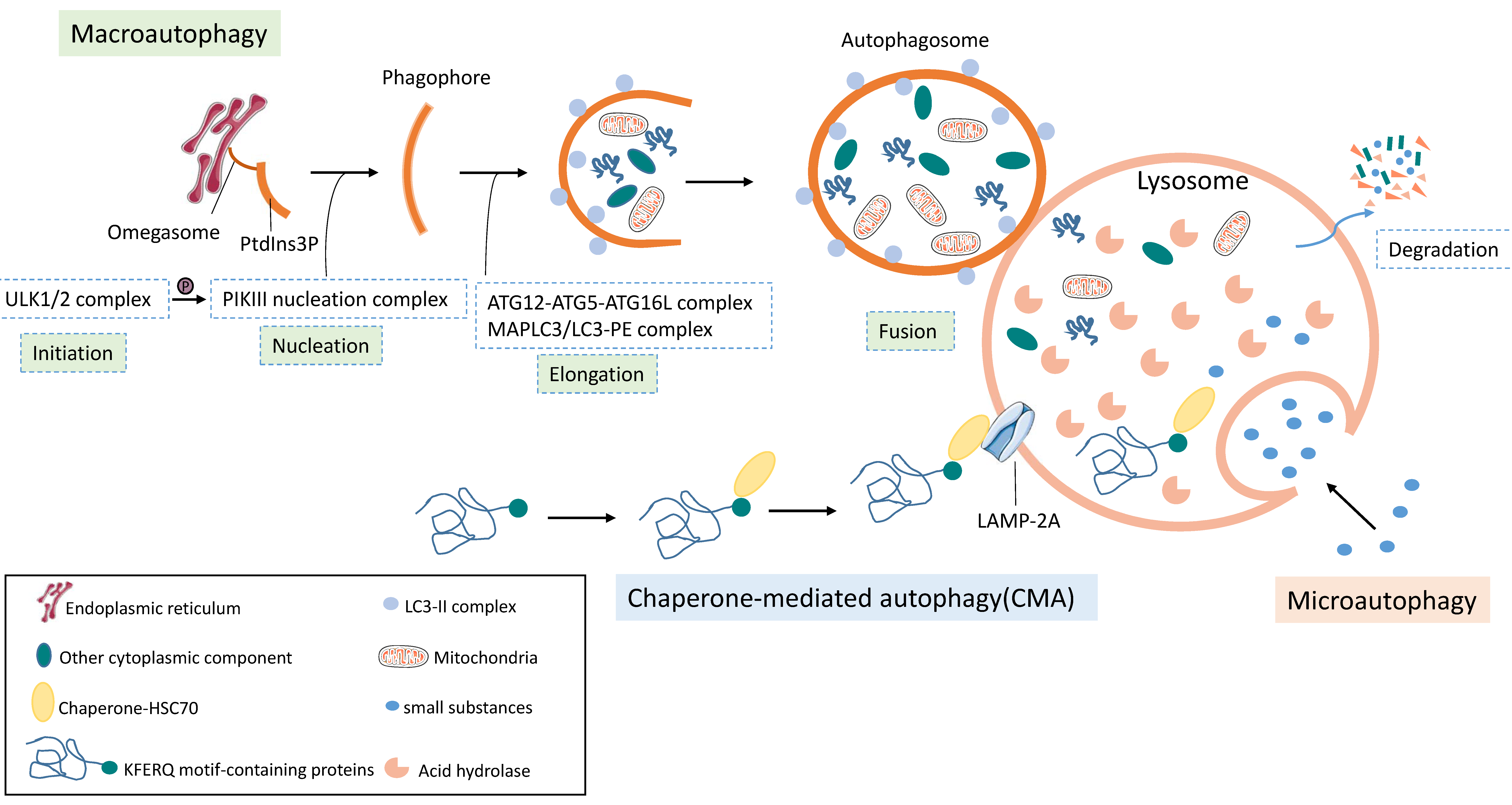
3.1. Basics of Autophagy
3.1.1. Three Forms of Autophagy
3.1.2. Overview of Autophagy
3.2. Induction of Autophagy in Cisplatin AKI
3.2.1. Oxidative Stress and Autophagy
3.2.2. Endoplasmic Reticulum Stress and Autophagy
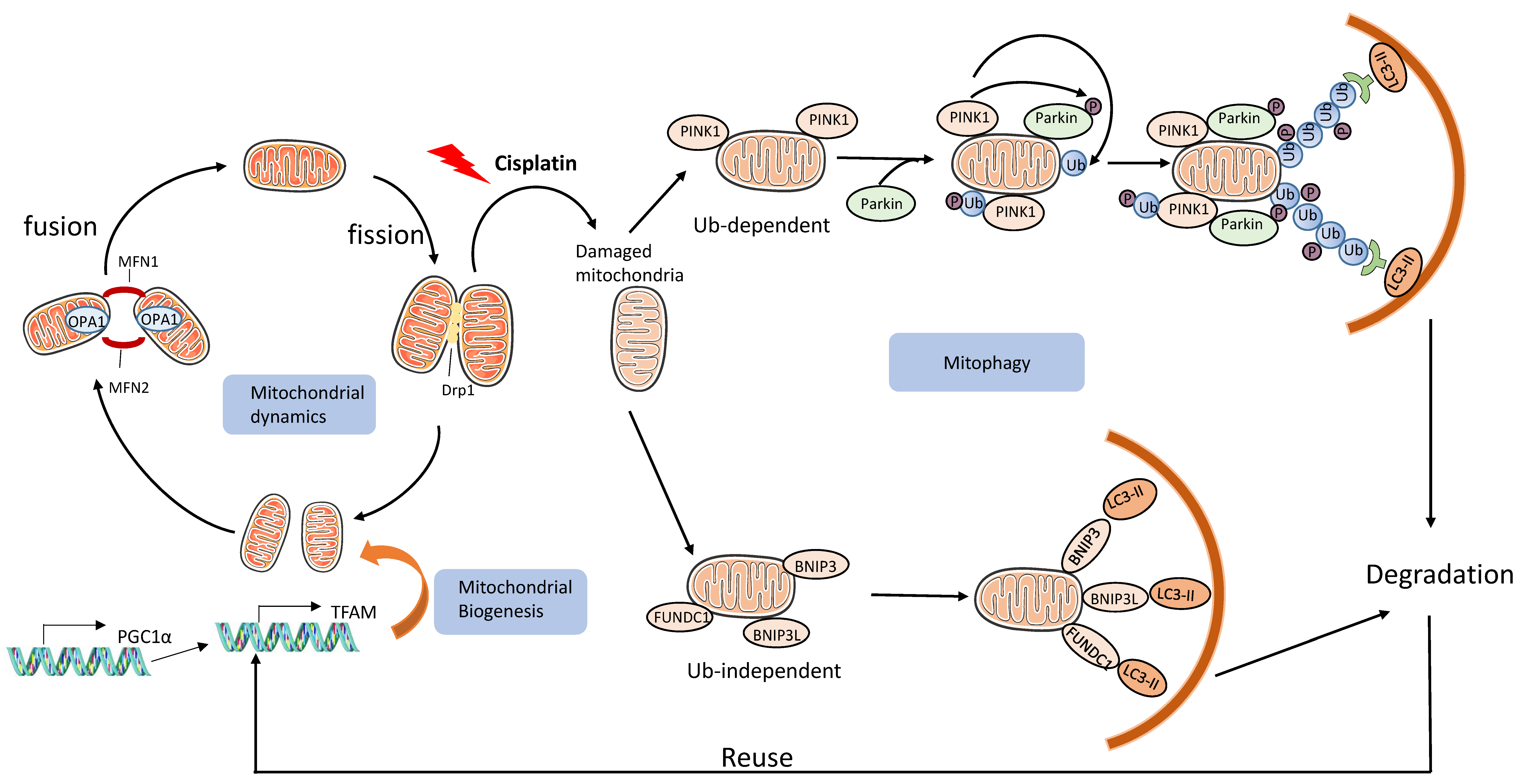
3.2.3. Mitochondrial Damage and Mitophagy
3.3. Roles of Autophagy in Cisplatin Nephrotoxicity and Underlying Mechanisms
3.3.1. Protective Role of Autophagy in Acute Cisplatin Nephrotoxicity
3.3.2. Mechanisms of the Renoprotective Effect of Autophagy
3.4. Regulation of Autophagy in Cisplatin AKI
3.4.1. Energy Signaling Pathway
3.4.2. Other Pathways
4. Autophagy in AKI–CKD Transition
5. Autophagy in Chronic Kidney Problems Following Cisplatin Treatment
5.1. Autophagy in Tubulointerstitial Fibrosis
5.2. Autophagy and Chronic Inflammation
6. Strategies Targeting Autophagy in Cisplatin Nephrotoxicity during Chemotherapy
6.1. Activation of Autophagy
6.2. Inhibition of Autophagy
6.3. Therapeutic Potential and Challenges
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar] [CrossRef]
- de Jonge, M.J.; Verweij, J. Renal Toxicities of Chemotherapy. Semin. Oncol. 2006, 33, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A. Acute Kidney Injury Network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latcha, S.; Jaimes, E.A.; Patil, S.; Glezerman, I.G.; Mehta, S.; Flombaum, C.D. Long–Term Renal Outcomes after Cisplatin Treatment. Clin. J. Am. Soc. Nephrol. 2016, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy-Thandavan, S.; Jiang, M.; Schoenlein, P.; Dong, Z. Autophagy: Molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Physiol. 2009, 297, F244–F256. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.A.; Kim, W.-Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.-S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Investig. 2019, 129, 2374–2389. [Google Scholar] [CrossRef] [Green Version]
- Livingston, M.J.; Ding, H.-F.; Huang, S.; Hill, J.A.; Yin, X.-M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sörensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy Induces Prosenescent Changes in Proximal Tubular S3 Segments. J. Am. Soc. Nephrol. 2015, 27, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Kim, S.L.; Lee, S.-Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy Regulates TGF-β Expression and Suppresses Kidney Fibrosis Induced by Unilateral Ureteral Obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [Green Version]
- Sirichanchuen, B.; Pengsuparp, T.; Chanvorachote, P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol. Cell Biochem. 2012, 364, 11–18. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.S. Role of Autophagy in Cisplatin Resistance in Ovarian Cancer Cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Zhang, C.; Yu, B.; Chen, B.; Liu, Z.; Hou, C.; Wang, F.; Shen, H.; Chen, Z. Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am. J. Cancer Res. 2017, 7, 1407–1422. [Google Scholar] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, J.; Rick, O.; Weinknecht, S.; Kingreen, D.; Lenz, K.; Siegert, W. Nephrotoxicity after high-dose carboplatin, etoposide and ifosfamide in germ-cell tumors: Incidence and implications for hematologic recovery and clinical outcome. Bone Marrow Transplant. 1997, 20, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Campdera, F.J.G.; Gonzalez, P.; Carrillo, A.; Estelles, M.C.; Rengel, M. Cisplatin nephrotoxicity: Symptomatic hypomagnesemia and renal failure. Int. J. Pediatr. Nephrol. 1986, 7, 151–152. [Google Scholar] [PubMed]
- Brilleta, G.; Demya, G.; Jacquiauda, C.; Mignotb, L.; Bunkera, D.; Meilletc, D.; Raymond, F.; Jacobs, C.; Brillet, G.; Deray, G.; et al. Long-Term Renal Effect of Cisplatin in Man. Am. J. Nephrol. 1994, 14, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated Cell Death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Lieberthal, W.; Nigam, S.K. Acute Renal Failure. I. Relative importance of proximal vs. distal tubular injury. Am. J. Physiol. Physiol. 1998, 275, F623–F632. [Google Scholar] [CrossRef]
- Lieberthal, W.; Nigam, S.K. Acute Renal Failure. II. Experimental models of acute renal failure: Imperfect but indispensable. Am. J. Physiol. Physiol. 2000, 278, F1–F12. [Google Scholar] [CrossRef]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of Cisplatin-Induced Acute Kidney Injury. BioMed Res. Int. 2014, 2014, 1–17. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin Nephrotoxicity Is Critically Mediated via the Human Organic Cation Transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a Nephrotoxin in Proximal Tubule Cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Tang, C.; Ma, Z.; Huang, S.; Dong, Z. DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol. Appl. Pharmacol. 2016, 313, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Pabla, N.; Tang, C.; He, L.; Dong, Z. DNA damage response in cisplatin-induced nephrotoxicity. Arch. Toxicol. 2015, 89, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Ju, S.-M.; Kim, M.-S.; Jo, Y.-S.; Jeon, Y.-M.; Bae, J.-S.; Pae, H.-O.; Jeon, B.-H. Licorice and its active compound glycyrrhizic acid ameliorates cisplatin-induced nephrotoxicity through inactivation of p53 by scavenging ROS and overexpression of p21 in human renal proximal tubular epithelial cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 890–899. [Google Scholar]
- Francescato, H.D.; Costa, R.S.; Silva, C.G.; Coimbra, T.M. Treatment with a p38 MAPK inhibitor attenuates cisplatin nephrotoxicity starting after the beginning of renal damage. Life Sci. 2009, 84, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Dong, Z. Regulation and Pathological Role of p53 in Cisplatin Nephrotoxicity. J. Pharmacol. Exp. Ther. 2008, 327, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Yang, T.; Megyesi, J.; Price, P.M.; Dong, Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Physiol. 2007, 293, F1282–F1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, F.; Sharma, I.; Dai, Y.; Yang, M.; Kanwar, Y.S. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J. Clin. Investig. 2019, 129, 5033–5049. [Google Scholar] [CrossRef] [Green Version]
- Takai, N.; Abe, K.; Tonomura, M.; Imamoto, N.; Fukumoto, K.; Ito, M.; Momosaki, S.; Fujisawa, K.; Morimoto, K.; Takasu, N.; et al. Imaging of reactive oxygen species using [3H]hydromethidine in mice with cisplatin-induced nephrotoxicity. EJNMMI Res. 2015, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef]
- Zsengellér, Z.K.; Ellezian, L.; Brown, D.; Horváth, B.; Mukhopadhyay, P.; Kalyanaraman, B.; Parikh, S.M.; Karumanchi, S.A.; Stillman, I.E.; Pacher, P. Cisplatin Nephrotoxicity Involves Mitochondrial Injury with Impaired Tubular Mitochondrial Enzyme Activity. J. Histochem. Cytochem. 2012, 60, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.; Dong, Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef] [Green Version]
- Landau, S.I.; Guo, X.; Velazquez, H.; Torres, R.; Olson, E.; Garcia-Milian, R.; Moeckel, G.; Desir, G.V.; Safirstein, R. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int. 2019, 95, 797–814. [Google Scholar] [CrossRef]
- Fu, Y.; Cai, J.; Li, F.; Liu, Z.; Shu, S.; Wang, Y.; Liu, Y.; Tang, C.; Dong, Z. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am. J. Physiol. Physiol. 2019, 317, F1582–F1592. [Google Scholar] [CrossRef]
- Black, L.M.; Lever, J.M.; Traylor, A.M.; Chen, B.; Yang, Z.; Esman, S.K.; Jiang, Y.; Cutter, G.R.; Boddu, R.; George, J.F.; et al. Divergent effects of AKI to CKD models on inflammation and fibrosis. Am. J. Physiol. Physiol. 2018, 315, F1107–F1118. [Google Scholar] [CrossRef]
- Torres, R.; Velazquez, H.; Chang, J.J.; Levene, M.J.; Moeckel, G.; Desir, G.V.; Safirstein, R. Three-Dimensional Morphology by Multiphoton Microscopy with Clearing in a Model of Cisplatin-Induced CKD. J. Am. Soc. Nephrol. 2015, 27, 1102–1112. [Google Scholar] [CrossRef]
- Sharp, C.N.; Doll, M.A.; Dupre, T.; Shah, P.P.; Subathra, M.; Siow, D.; Arteel, G.E.; Megyesi, J.; Beverly, L.J.; Siskind, L.J. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am. J. Physiol. Physiol. 2016, 310, F560–F568. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Negishi, K.; Sugaya, T.; Nangaku, M.; Noiri, E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016, 89, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Sharp, C.N.; Doll, M.; Dupre, T.V.; Beverly, L.J.; Siskind, L.J. Moderate aging does not exacerbate cisplatin-induced kidney injury or fibrosis despite altered inflammatory cytokine expression and immune cell infiltration. Am. J. Physiol. Physiol. 2019, 316, F162–F172. [Google Scholar] [CrossRef]
- Sears, S.M.; Sharp, C.N.; Krueger, A.; Oropilla, G.B.; Saforo, D.; Doll, M.A.; Megyesi, J.; Beverly, L.J.; Siskind, L.J. C57BL/6 mice require a higher dose of cisplatin to induce renal fibrosis and CCL2 correlates with cisplatin-induced kidney injury. Am. J. Physiol. Physiol. 2020, 319, F674–F685. [Google Scholar] [CrossRef] [PubMed]
- Menshikh, A.; Scarfe, L.; Delgado, R.; Finney, C.; Zhu, Y.; Yang, H.; De Caestecker, M.P. Capillary rarefaction is more closely associated with CKD progression after cisplatin, rhabdomyolysis, and ischemia-reperfusion-induced AKI than renal fibrosis. Am. J. Physiol. Physiol. 2019, 317, F1383–F1397. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.N.; Doll, M.A.; Megyesi, J.; Oropilla, G.B.; Beverly, L.J.; Siskind, L.J. Subclinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am. J. Physiol. Physiol. 2018, 315, F161–F172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, Y.; Satoh, T.; Hibi, D.; Ohno, Y.; Kohda, Y.; Miura, K.; Gemba, M. The effect of antioxidant on development of fibrosis by cisplatin in rats. J. Pharmacol. Sci. 2009, 111, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Shen, Y.; Huang, L.; Liu, C.; Wang, J. Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J. 2020, 35, e21229. [Google Scholar] [CrossRef]
- Li, C.; Xie, N.; Li, Y.; Liu, C.; Hou, F.F.; Wang, J. N-acetylcysteine ameliorates cisplatin-induced renal senescence and renal interstitial fibrosis through sirtuin1 activation and p53 deacetylation. Free Radic. Biol. Med. 2018, 130, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Biazik-Richmond, J.; Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nature 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12–Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.-M.; Dong, Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008, 74, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Baliga, R.; Ueda, N.; Walker, P.D.; Shah, S.V. Oxidant mechanisms in toxic acute renal failure. Am. J. Kidney Dis. 1997, 29, 465–477. [Google Scholar] [CrossRef]
- Bolisetty, S.; Traylor, A.M.; Kim, J.; Joseph, R.; Ricart, K.; Landar, A.; Agarwal, A. Heme Oxygenase-1 Inhibits Renal Tubular Macroautophagy in Acute Kidney Injury. J. Am. Soc. Nephrol. 2010, 21, 1702–1712. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-W.; Kim, Y.-J.; Kim, H.-T.; Park, S.-R.; Lee, M.-Y.; Park, Y.-D.; Lee, C.-H.; Jung, J.-Y. NQO1 Deficiency Leads Enhanced Autophagy in Cisplatin-Induced Acute Kidney Injury Through the AMPK/TSC2/mTOR Signaling Pathway. Antioxid. Redox Signal. 2016, 24, 867–883. [Google Scholar] [CrossRef]
- Rovetta, F.; Stacchiotti, A.; Consiglio, A.; Cadei, M.; Grigolato, P.G.; Lavazza, A.; Rezzani, R.; Aleo, M.F. ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Exp. Cell Res. 2012, 318, 238–250. [Google Scholar] [CrossRef]
- Pallet, N.; Bouvier, N.; Legendre, C.; Gilleron, J.; Codogno, P.; Beaune, P.; Thervet, E.; Anglicheau, D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 2008, 4, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Chandrika, B.B.; Yang, C.; Ou, Y.; Feng, X.; Muhoza, D.; Holmes, A.F.; Theus, S.; Deshmukh, S.; Haun, R.S.; Kaushal, G.P. Endoplasmic Reticulum Stress-Induced Autophagy Provides Cytoprotection from Chemical Hypoxia and Oxidant Injury and Ameliorates Renal Ischemia-Reperfusion Injury. PLoS ONE 2015, 10, e0140025. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Inagi, R.; Takano, H.; Sato, S.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol. Dial. Transplant. 2009, 24, 2665–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deegan, S.; Saveljeva, S.; Gorman, A.; Samali, A. Stress-induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress. Cell. Mol. Life Sci. 2012, 70, 2425–2441. [Google Scholar] [CrossRef]
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886. [Google Scholar] [CrossRef] [Green Version]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Cai, J.; Yin, X.-M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2020, 17, 299–318. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Schuh, C.-D. Mitochondria as therapeutic targets in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2016, 25, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Chen, Z.; Qi, J.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zeng, M.; Zhang, B.; Wang, N.; et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 2017, 8, 20988–21000. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Scorrano, L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1777, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.; Kher, A.; Zsengeller, Z.; Akhavan-Sharif, M.R.; Khankin, E.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, M.; Zsengeller, Z.; Berg, A.H.; Khankin, E.; Bhasin, M.; Kim, W.; Clish, C.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Fermín, L.M.; Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Tapia, E.; Rivero, I.; Pedraza-Chaverri, J. The Protective Effect of Alpha-Mangostin against Cisplatin-Induced Cell Death in LLC-PK1 Cells is Associated to Mitochondrial Function Preservation. Antioxidants 2019, 8, 133. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.R.; Tran, M.T.; Ralto, K.M.; Zsengeller, Z.K.; Raman, V.; Bhasin, S.S.; Sun, N.; Chen, X.; Brown, D.; Rovira, I.I.; et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 2019, 4, e126749. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Herzog, C.; Yang, C.; Holmes, A.; Kaushal, G.P. zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am. J. Physiol. Physiol. 2012, 303, F1239–F1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tang, Y.; Wen, L.; Kong, X.; Chen, X.; Liu, P.; Zhou, Z.; Chen, W.; Xiao, C.; Xiao, P.; et al. Neferine reduces cisplatin-induced nephrotoxicity by enhancing autophagy via the AMPK/mTOR signaling pathway. Biochem. Biophys. Res. Commun. 2017, 484, 694–701. [Google Scholar] [CrossRef]
- Liu, H.; Gu, L.-B.; Tu, Y.; Hu, H.; Huang, Y.-R.; Sun, W. Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro by activating autophagy. Acta Pharmacol. Sin. 2016, 37, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Hou, J.; Ma, Z.; Wang, Z.; Ren, S.; Wang, Y.; Liu, W.; Chen, C.; Li, W. Ginsenoside Rb3 provides protective effects against cisplatin-induced nephrotoxicity via regulation of AMPK-/mTOR-mediated autophagy and inhibition of apoptosis in vitro and in vivo. Cell Prolif. 2019, 52, e12627. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gao, Y.; Fan, X.; Liu, X.; Peng, L.; Ci, X. Oridonin Sensitizes Cisplatin-Induced Apoptosis via AMPK/Akt/mTOR-Dependent Autophagosome Accumulation in A549 Cells. Front. Oncol. 2019, 9, 769. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.P.; Chauhan, A.K.; Kang, S.C. Morin hydrate ameliorates cisplatin-induced ER stress, inflammation and autophagy in HEK-293 cells and mice kidney via PARP-1 regulation. Int. Immunopharmacol. 2018, 56, 156–167. [Google Scholar] [CrossRef]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy Guards Against Cisplatin-Induced Acute Kidney Injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kuwana, H.; Shimamura, Y.; Ogata, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T.; Morita, T.; Sasaki, S.; et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin. Exp. Nephrol. 2009, 14, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Hu, C.; Zhang, P.; Jiang, H.; Chen, J. Mesenchymal stem cell therapy targeting mitochondrial dysfunction in acute kidney injury. J. Transl. Med. 2019, 17, 142. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, Z.; Xu, X.; An, X.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zhang, B.; Zhang, A.; et al. Pink1/Parkin-mediated mitophagy play a protective role in cisplatin induced renal tubular epithelial cells injury. Exp. Cell Res. 2017, 350, 390–397. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, L.; Zhang, Y.; Yu, X.; Sun, X.; Zhu, T.; Li, X.; Liang, W.; Han, Y.; Qin, C. PINK1 Deficiency Ameliorates Cisplatin-Induced Acute Kidney Injury in Rats. Front. Physiol. 2019, 10, 1225. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia–reperfusion injury. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yang, Y.; Huang, Z.; Zhou, J.; Li, Y.; Zhong, X. Panax notoginseng saponins mitigate cisplatin induced nephrotoxicity by inducing mitophagy via HIF-1α. Oncotarget 2017, 8, 102989–103003. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Wei, L.; Chen, W.; Zou, Y.; Huang, H.; Pan, B.; Jin, S.; Huang, R.; Nie, S.; Kong, G. AMP-activated protein kinase regulates autophagic protection against cisplatin-induced tissue injury in the kidney. Genet. Mol. Res. 2015, 14, 12006–12015. [Google Scholar] [CrossRef]
- Yang, A.; Liu, F.; Guan, B.; Luo, Z.; Lin, J.; Fang, W.; Liu, L.; Zuo, W. p53 induces miR-199a-3p to suppress mechanistic target of rapamycin activation in cisplatin-induced acute kidney injury. J. Cell. Biochem. 2019, 120, 17625–17634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pan, J.; Xiang, X.; Liu, Y.; Dong, G.; Livingston, M.J.; Chen, J.-K.; Yin, X.-M.; Dong, Z. Protein Kinase Cδ Suppresses Autophagy to Induce Kidney Cell Apoptosis in Cisplatin Nephrotoxicity. J. Am. Soc. Nephrol. 2016, 28, 1131–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.; Luo, J.; Kumar, V.; Dong, Z. Inhibitors of histone deacetylases suppress cisplatin-induced p53 activation and apoptosis in renal tubular cells. Am. J. Physiol. Physiol. 2010, 298, F293–F300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.-M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Mehr, A.P.; Tran, M.; Ralto, K.M.; Leaf, D.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Wang, S.-R.; Huang, X.-Z.; Xie, Q.-H.; Xu, Y.-Y.; Shang, D.; Hao, C.-M. Nicotinamide Mononucleotide, an NAD+ Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1–Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morevati, M.; Egstrand, S.; Nordholm, A.; Mace, M.L.; Andersen, C.B.; Salmani, R.; Olgaard, K.; Lewin, E. Effect of NAD+ boosting on kidney ischemia-reperfusion injury. PLoS ONE 2021, 16, e0252554. [Google Scholar] [CrossRef]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of Nuclear LC3 Drives Autophagy Initiation under Starvation. Mol. Cell 2015, 57, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; DePinho, R.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- B’Chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2009, 29, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia-Arencibia, M.; Vetrini, F.; Erdin, S.; Huynh, T.; Medina, D.L.; Colella, P.; Sardiello, M.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 Is a Master Transcriptional Repressor of Autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yuan, Y.; Yuan, L.; Li, L.; Liu, F.; Liu, J.; Chen, Y.; Lu, Y.; Cheng, J. Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics 2020, 10, 5829–5844. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.L.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2015, 27, 687–697. [Google Scholar] [CrossRef]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Livingston, M.J.; Dong, Z. Autophagy in Acute Kidney Injury and Repair. Nephron 2014, 127, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Shi, Y.; Tao, M.; Liu, N.; Zhuang, S.; Yuan, W. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy. Clin. Sci. 2018, 132, 2299–2322. [Google Scholar] [CrossRef] [Green Version]
- Canaud, G.; Brooks, C.R.; Kishi, S.; Taguchi, K.; Nishimura, K.; Magassa, S.; Scott, A.; Hsiao, L.-L.; Ichimura, T.; Terzi, F.; et al. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci. Transl. Med. 2019, 11, eaav4754. [Google Scholar] [CrossRef]
- Choi, M.E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 2020, 82, 297–322. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy Is a Component of Epithelial Cell Fate in Obstructive Uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Zehender, A.; Li, Y.-N.; Lin, N.-Y.; Stefanica, A.; Nüchel, J.; Chen, C.-W.; Hsu, H.-H.; Zhu, H.; Ding, X.; Huang, J.; et al. TGFβ promotes fibrosis by MYST1-dependent epigenetic regulation of autophagy. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-A.; Kim, H.-J.; Gwon, M.-G.; Gu, H.; An, H.-J.; Bae, S.; Leem, J.; Jung, H.; Park, K.-K. Inhibitory Effects of STAT3 Transcription Factor by Synthetic Decoy ODNs on Autophagy in Renal Fibrosis. Biomedicines 2021, 9, 331. [Google Scholar] [CrossRef]
- Shi, Y.; Tao, M.; Ma, X.; Hu, Y.; Huang, G.; Qiu, A.; Zhuang, S.; Liu, N. Delayed treatment with an autophagy inhibitor 3-MA alleviates the progression of hyperuricemic nephropathy. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Na, H.-J.; Ding, Y.; Wang, Z.; Lee, S.-J.; Choi, M.E. Autophagy Promotes Intracellular Degradation of Type I Collagen Induced by Transforming Growth Factor (TGF)-β. J. Biol. Chem. 2012, 287, 11677–11688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Yue, F.; Huang, H.; He, Y.; Li, X.; Zhao, H.; Su, Z.; Jiang, X.; Li, W.; Zou, J.; et al. Defects in MAP1S-mediated autophagy turnover of fibronectin cause renal fibrosis. Aging 2016, 8, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.-Y.; Nam, S.A.; Song, H.C.; Ko, J.S.; Park, S.H.; Kim, H.L.; Choi, E.J.; Kim, Y.-S.; Kim, J. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology 2011, 17, 148–159. [Google Scholar] [CrossRef]
- Brooks, C.R.; Yeung, M.Y.; Brooks, Y.S.; Chen, H.; Ichimura, T.; Henderson, J.M.; Bonventre, J.V. KIM-1-/TIM-1-mediated phagocytosis links ATG 5-/ ULK 1-dependent clearance of apoptotic cells to antigen presentation. EMBO J. 2015, 34, 2441–2464. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Li, S.; Jiang, N.; Jin, H.; Shao, X.; Zhu, X.; Wu, J.; Zhang, M.; Zhang, Z.; Shen, J.; et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 2020, 17, 2975–2990. [Google Scholar] [CrossRef]
- Kim, S.-M.; Kim, Y.G.; Kim, D.-J.; Park, S.H.; Jeong, K.-H.; Lee, Y.H.; Lim, S.J.; Lee, S.-H.; Moon, J.-Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, J.; Xu, W.; Ding, F.; Ding, W. Prohibitin 2-mediated mitophagy attenuates renal tubular epithelial cells injury by regulating mitochondrial dysfunction and NLRP3 inflammasome activation. Am. J. Physiol. Physiol. 2019, 316, F396–F407. [Google Scholar] [CrossRef]
- Peng, X.; Wang, Y.; Li, H.; Fan, J.; Shen, J.; Yu, X.; Zhou, Y.; Mao, H. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Andrianova, N.V.; Zorova, L.D.; Babenko, V.A.; Pevzner, I.B.; Popkov, V.A.; Silachev, D.N.; Plotnikov, E.Y.; Zorov, D.B. Rapamycin Is Not Protective against Ischemic and Cisplatin-Induced Kidney Injury. Biochemistry (Moscow) 2019, 84, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Nishihara, K.; Inui, K.-I.; Masuda, S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur. J. Pharmacol. 2012, 696, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucali, P.A.; De Pas, T.; Palmieri, G.; Favaretto, A.; Chella, A.; Tiseo, M.; Caruso, M.; Simonelli, M.; Perrino, M.; De Vincenzo, F.; et al. Phase II Study of Everolimus in Patients With Thymoma and Thymic Carcinoma Previously Treated With Cisplatin-Based Chemotherapy. J. Clin. Oncol. 2018, 36, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Mosley, J.; Poirier, J.; Seachrist, D.D.; Landis, M.D.; Keri, R.A. Rapamycin inhibits multiple stages of c-Neu/ErbB2–induced tumor progression in a transgenic mouse model of HER2-positive breast cancer. Mol. Cancer Ther. 2007, 6, 2188–2197. [Google Scholar] [CrossRef] [Green Version]
- Mabuchi, S.; Altomare, D.A.; Connolly, D.C.; Klein-Szanto, A.; Litwin, S.; Hoelzle, M.K.; Hensley, H.H.; Hamilton, T.C.; Testa, J.R. RAD001 (Everolimus) Delays Tumor Onset and Progression in a Transgenic Mouse Model of Ovarian Cancer. Cancer Res. 2007, 67, 2408–2413. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gui, Y.; Ren, J.; Liu, X.; Feng, Y.; Zeng, Z.; He, W.; Yang, J.; Dai, C. Metformin Protects Against Cisplatin-Induced Tubular Cell Apoptosis and Acute Kidney Injury via AMPKα-regulated Autophagy Induction. Sci. Rep. 2016, 6, 23975. [Google Scholar] [CrossRef] [Green Version]
- Ben Sahra, I.; Le Marchand-Brustel, Y.; Tanti, J.-F.; Bost, F. Metformin in Cancer Therapy: A New Perspective for an Old Antidiabetic Drug? Mol. Cancer Ther. 2010, 9, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Cui, J.; Wang, H.; Medina, R.; Zhang, S.; Zhang, X.; Zhuang, Z.; Lin, Y. Metformin enhances anti-cancer effects of cisplatin in meningioma through AMPK-mTOR signaling pathways. Mol. Ther. Oncol. 2021, 20, 119–131. [Google Scholar] [CrossRef]
- Lambert, I.H.; Nielsen, D.; Stürup, S. Impact of the histone deacetylase inhibitor trichostatin A on active uptake, volume-sensitive release of taurine, and cell fate in human ovarian cancer cells. Am. J. Physiol. Physiol. 2020, 318, C581–C597. [Google Scholar] [CrossRef]
- Tang, J.; Shi, Y.; Liu, N.; Xu, L.; Zang, X.; Li, P.; Zhang, J.; Zheng, X.; Qiu, A.; Zhuang, S. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiang, S.; Williams, K.A.; Dong, H.; Bai, W.; Nicosia, S.V.; Khochbin, S.; Bepler, G.; Zhang, X. Depletion of HDAC6 Enhances Cisplatin-Induced DNA Damage and Apoptosis in Non-Small Cell Lung Cancer Cells. PLoS ONE 2012, 7, e44265. [Google Scholar] [CrossRef] [Green Version]
- Selvi, S.K.; Vinoth, A.; Varadharajan, T.; Weng, C.F.; Padma, V.V. Neferine augments therapeutic efficacy of cisplatin through ROS- mediated non-canonical autophagy in human lung adenocarcinoma (A549 cells). Food Chem. Toxicol. 2017, 103, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Wang, J.; Chen, Y.; Yin, X.; Shi, G.; Li, H.; Hu, Z.; Liang, X. Emodin enhances cisplatin-induced cytotoxicity in human bladder cancer cells through ROS elevation and MRP1 downregulation. BMC Cancer 2016, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.-C.; Su, Y.-J.; Lin, S.-T.; Jhan, J.-Y.; Ciou, S.-C.; Cheng, C.-M.; Chiu, Y.-F.; Kuo, Y.-H.; Tsai, M.-S.; Lin, Y.-W. Emodin enhances cisplatin-induced cytotoxicity via down-regulation of ERCC1 and inactivation of ERK1/2. Lung Cancer 2010, 69, 155–164. [Google Scholar] [CrossRef]
- Lee, D.; Bin Kang, K.; Kim, H.W.; Park, J.S.; Hwang, G.S.; Kang, K.S.; Choi, S.; Yamabe, N.; Kim, K.H. Unique Triterpenoid of Jujube Root Protects Cisplatin-induced Damage in Kidney Epithelial LLC-PK1 Cells via Autophagy Regulation. Nutrients 2020, 12, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-Y.; Nie, J.; Zheng, Z.-L.; Zhao, J.; Wu, L.-M.; Zhu, Y.; Su, Z.-Q.; Zheng, G.-J.; Feng, B. Renoprotective effect of scutellarin on cisplatin-induced renal injury in mice: Impact on inflammation, apoptosis, and autophagy. Biomed. Pharmacother. 2019, 112, 108647. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Zhu, Y.; Li, X.-F.; Wang, X.-Q.; Tang, L.-P.; Su, Z.-Q.; Li, C.-Y.; Zheng, G.-J.; Feng, B. Scutellarin Increases Cisplatin-Induced Apoptosis and Autophagy to Overcome Cisplatin Resistance in Non-small Cell Lung Cancer via ERK/p53 and c-met/AKT Signaling Pathways. Front. Pharmacol. 2018, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Del Bello, B.; Toscano, M.; Moretti, D.; Maellaro, E. Cisplatin-Induced Apoptosis Inhibits Autophagy, Which Acts as a Pro-Survival Mechanism in Human Melanoma Cells. PLoS ONE 2013, 8, e57236. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Potočnjak, I.; Šimić, L.; Vukelić, I.; Domitrović, R. Oleanolic acid attenuates cisplatin-induced nephrotoxicity in mice and chemosensitizes human cervical cancer cells to cisplatin cytotoxicity. Food Chem. Toxicol. 2019, 132, 110676. [Google Scholar] [CrossRef]
- Checkley, L.A.; Rho, O.; Moore, T.; Hursting, S.; DiGiovanni, J. Rapamycin Is a Potent Inhibitor of Skin Tumor Promotion by 12-O-Tetradecanoylphorbol-13-Acetate. Cancer Prev. Res. 2011, 4, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Song, Q.; Hu, D.; Zhuang, X.; Yu, S.; Teng, D. Oleanolic acid induced autophagic cell death in hepatocellular carcinoma cells via PI3K/Akt/mTOR and ROS-dependent pathway. Korean J. Physiol. Pharmacol. 2016, 20, 237–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Jaramillo, M.C.; Zhang, Z.; Zheng, Y.; Yao, M.; Zhang, N.D.; Yi, X. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol. Med. Rep. 2014, 11, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Li, Z. Blockage of cisplatin-induced autophagy sensitizes cervical cancer cells to cisplatin. Genet. Mol. Res. 2015, 14, 16905–16912. [Google Scholar] [CrossRef]
- Zhang, M.; Hagan, C.T.; Min, Y.; Foley, H.; Tian, X.; Yang, F.; Mi, Y.; Au, K.M.; Medik, Y.; Roche, K.; et al. Nanoparticle co-delivery of wortmannin and cisplatin synergistically enhances chemoradiotherapy and reverses platinum resistance in ovarian cancer models. Biomaterials 2018, 169, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bar, J.; Lukaschuk, N.; Zalcenstein, A.; Wilder, S.; Seger, R.; Oren, M. The PI3K inhibitor LY294002 prevents p53 induction by DNA damage and attenuates chemotherapy-induced apoptosis. Cell Death Differ. 2005, 12, 1578–1587. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-F.; Lin, Y.-C.; Tsai, T.-F.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.-S. Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Dev. Ther. 2017, 11, 1517–1533. [Google Scholar] [CrossRef] [Green Version]
- Aga, T.; Endo, K.; Tsuji, A.; Aga, M.; Moriyama-Kita, M.; Ueno, T.; Nakanishi, Y.; Hatano, M.; Kondo, S.; Sugimoto, H.; et al. Inhibition of autophagy by chloroquine makes chemotherapy in nasopharyngeal carcinoma more efficient. Auris Nasus Larynx 2018, 46, 443–450. [Google Scholar] [CrossRef]
- Su, Z.; Li, G.; Liu, C.; Ren, S.; Deng, T.; Zhang, S.; Tian, Y.; Liu, Y.; Qiu, Y. Autophagy inhibition impairs the epithelial-mesenchymal transition and enhances cisplatin sensitivity in nasopharyngeal carcinoma. Oncol. Lett. 2017, 13, 4147–4154. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-Q.; Fang, N.; Liu, X.-M.; Xiong, S.-P.; Liao, Y.-Q.; Jin, W.-J.; Song, R.-F.; Wan, Y.-Y. Antitumor Activity of Chloroquine in Combination with Cisplatin in Human Gastric Cancer Xenografts. Asian Pac. J. Cancer Prev. 2015, 16, 3907–3912. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; Arimoto, T.; et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol. Oncol. 2015, 137, 538–545. [Google Scholar] [CrossRef]
- Shen, C.; Wang, W.; Tao, L.; Liu, B.; Yang, Z.; Tao, H. Chloroquine blocks the autophagic process in cisplatin-resistant osteosarcoma cells by regulating the expression of p62/SQSTM1. Int. J. Mol. Med. 2013, 32, 448–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in Cancer Therapy: A Double-Edged Sword of Autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-Mediated Tumor Promotion. Cell 2013, 155, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.P.; Cho, H.J.; Kim, J.T.; Baek, K.E.; Lee, H.G.; Kang, S.C. Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma. Cancers 2019, 11, 986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-Y.; White, E. Role of Autophagy in Cancer Prevention. Cancer Prev. Res. 2011, 4, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Jung, J.-Y.; Choi, S.; Lee, H.; Morales, L.D.; Koh, J.-T.; Kim, S.H.; Choi, Y.-D.; Choi, C.; Slaga, T.J.; et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy 2016, 13, 149–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Variation | Method | Target | Effect | Underlying Mechanisms | Reference |
|---|---|---|---|---|---|
| Autophagy | Atg5 KO | Mouse proximal tubule | More sensitive | 1. More damaged mitochondria and abnormal protein aggregates accumulation 2. More DNA damage and p53 activation | [101] |
| Atg7 KO | Mouse proximal tubule | More sensitive | More activation of p53 and c-Jun N terminal kinase | [92] | |
| Beclin1 shRNA | NRK-52E cells | Prevent apoptosis | Suppressed apoptosis | [102] | |
| Beclin1 siRNA | RPTCs | Aggregated apoptosis and cell death | Sensitized cells to cisplatin-induced apoptosis | [69,70] | |
| Mitophagy | Pink1/parkin KO | Mouse global | More sensitive | Promoted Drp1-mediated mitochondrial fission | [94] |
| Pink1 KO | Rat global | Attenuated cisplatin-induced acute kidney injury | Promoted Drp1-mediated mitochondrial fission | [103] | |
| Pink1/parkin knockdown | HK2 cells | More cell injury | Accelerated cisplatin-induced mitochondrial dysfunction | [104] |
| Compound | Kidney | Cancer | |||||
|---|---|---|---|---|---|---|---|
| Model | Effects | Underlying Mechanism | Model | Effects | Underlying Mechanism | Reference | |
| Activate autophagy | |||||||
| Metformin | NRK-52E | Protective | Stimulate AMPKα phosphorylation | Meningioma cell | Lower cancer risk | AMPK-mTOR pathways | [157,158,159] |
| Rapamycin | C57BL/6 mice | Protective Harmful | Inhibition of mTOR | Breast cancer Skin tumors | Inhibit progression | 1. Inhibition of mTOR 2. Decreased proliferation | [152,155,172] |
| Everolimus | Wistar/ST rats HK-2 NRK-52E | harmful | Inhibition of mTOR to activates Ulk1 impairing tubular regeneration after acute injury | Ovarian cancer Patients with Thymoma and Thymic Carcinoma | 1. Delay development 2. Durable disease control | 1. Inhibition of mTOR 2. Reverse cell senescence | [153,154,156] |
| Trichostatin A (TSA) | C57BL/6mice Atg7-/- mice | Protective | AMPK activation and marginal inactivation of mTOR | A2780RES cells | More sensitive | Akt/mTOR Signaling | [114,160] |
| Tubastatin A (TA) | C57BL/6 HK2 cell | Protective | HDAC6 inhibition decreased renal oxidative stress and malondialdehyde levels | A549 and H292 cells | More sensitive | Increased DNA damage | [114,160] |
| Rottlerin | PT-Atg7-KO mice RPTCs | Protective | Suppress phosphorylation of AKT/ mTOR, p70S6 kinase, and ULK1 | A2780 human ovarian cancer cells | More sensitive | Inhibit proliferation, migration, and metastasis | [49,112] |
| Neferine | NRK-52E cell | Protective | AMPK-mTOR pathways | Human lung adenocarcinoma (A549 cells) | More sensitive | AMPK-mTOR pathways | [95,163] |
| Emodin | NRK-52E | Protective | Induce the phosphorylation and activation of AMPK, decrease mTOR activation | Human bladder cancer cells Non-small cell lung cancer (NSCLC) | More sensitive | ROS elevation and MRP1 Down-regulation of ERCC1 and inactivation of ERK1/2 | [164,165] |
| Ginsenoside Rb3 | HEK293 cell ICR mice | Protective | Regulation of AMPK/mTOR | Oral Cancer | More sensitive | Regulation of AMPK/mTOR | [97] |
| Oridonin | C57BL/6 mice inhibit A549 cell line activate | Protective | AMPK/Akt/mTOR-dependent autophagosome accumulation | Human lung carcinoma cell | More sensitive | AMPK/Akt/mTOR-dependent autophagosome accumulation | [98] |
| 3DC2ME | LLC-PK1 cells | Protective | AMPK/mTOR | - | - | - | [166] |
| Scutellarin | C57BL/6 mice | Protective | JNK, p38, ERK and Stat3. | Non-small Cell Lung Cancer | More sensitive | Activate ERK/p53 and c-met/AKT/mTOR signaling pathways | [167,168] |
| Trehalose | C57BL/6 mice HK2 cell | Protective | 1. Activate TFEB-mediated mitophagy 2. Attenuate mitochondrial dysfunction | Me21 cells | Protective | Activate mTOR-independent autophagy | [129,169] |
| Oleanolic acid | BALB/CN mice Hela cells | Protective | Suppress oxidative stress and inflammatory response | Hela cells hepatocellular carcinoma | More sensitive | Induce autophagic cell death via mTOR | [171,173] |
| Inhibit autophagy | |||||||
| 3-MA | LLC-PK1 cells RPTC cells | Harmful | Block autophagosome formation-PI3K inhibitors | Osteosarcoma cells Cervical cancer cells Osteosarcoma Human ovarian cancer | More sensitive | Enhanced apoptosis, reduced cell viability | [22,26,70,174,175] |
| Wortmannin | LLC-PK1 cells | Harmful | Block autophagosome formation-PI3K inhibitors | Platinum resistant ovarian cancer (PROC) | More sensitive | 1. Inhibit DNA repair 2. Enhance cellular CP uptake | [176] |
| LY294002 | LLC-PK1 cells | Harmful | Block autophagosome formation-PI3K inhibitors | HCT 166 human colon cancer cell | More sensitive | p53 induction by DNA damage | [177] |
| Bafilomycin (BAF) | RPTC cells | Harmful | Block fusion of the autophagosome with lysosome | Human bladder cancer | More sensitive | Enhanced apoptosis, reduced cell viability | [70,178] |
| Chloroquine (CQ) | C57BL/6 mice Adult Wistar rats | Harmful | Block autophagic flux | Human bladder cancer Endometrial cancer Osteosarcoma Nasopharyngeal carcinoma Human gastric cancer Cervical cancer cells | More sensitive | 1. Block autophagic flux 2. Autophagy- independent manner | [18,175,178,179,180,181,182,183,184,185] |
| Morin hydrate | HEK-293 Male ICR mice | Protective | Phosphorylation and activation of AMPK | Hepatocellular Carcinoma | More sensitive | 1. Impairing PARP1/HMGB1-dependent autophagy 2. Reverses Cisplatin Resistance | [99,186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Ma, Z.; Wen, L.; Li, S.; Dong, Z. Autophagy in Cisplatin Nephrotoxicity during Cancer Therapy. Cancers 2021, 13, 5618. https://doi.org/10.3390/cancers13225618
Hu X, Ma Z, Wen L, Li S, Dong Z. Autophagy in Cisplatin Nephrotoxicity during Cancer Therapy. Cancers. 2021; 13(22):5618. https://doi.org/10.3390/cancers13225618
Chicago/Turabian StyleHu, Xiaoru, Zhengwei Ma, Lu Wen, Siyao Li, and Zheng Dong. 2021. "Autophagy in Cisplatin Nephrotoxicity during Cancer Therapy" Cancers 13, no. 22: 5618. https://doi.org/10.3390/cancers13225618
APA StyleHu, X., Ma, Z., Wen, L., Li, S., & Dong, Z. (2021). Autophagy in Cisplatin Nephrotoxicity during Cancer Therapy. Cancers, 13(22), 5618. https://doi.org/10.3390/cancers13225618