

FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. FGFs and Their Receptors in Cancer Progression
3. The Role of Cell Signaling Pathways in the Development of Anti-Cancer Drug Resistance
3.1. MAPK Cascade
3.2. PI3K/AKT Cascade
3.3. STAT Cascade
3.4. PLCγ/PKC Cascade
4. Signals from the Tumor Microenvironment
5. Cross-Talks between FGF/FGFR Signaling Pathways in Cancer
6. Dysregulation of Apoptosis in Cancer by FGFs/FGFRs System
7. Role of FGFs/FGFRs Axis during Cancer-Associated Angiogenesis
8. Contribution of FGFs/FGFRs to EMT
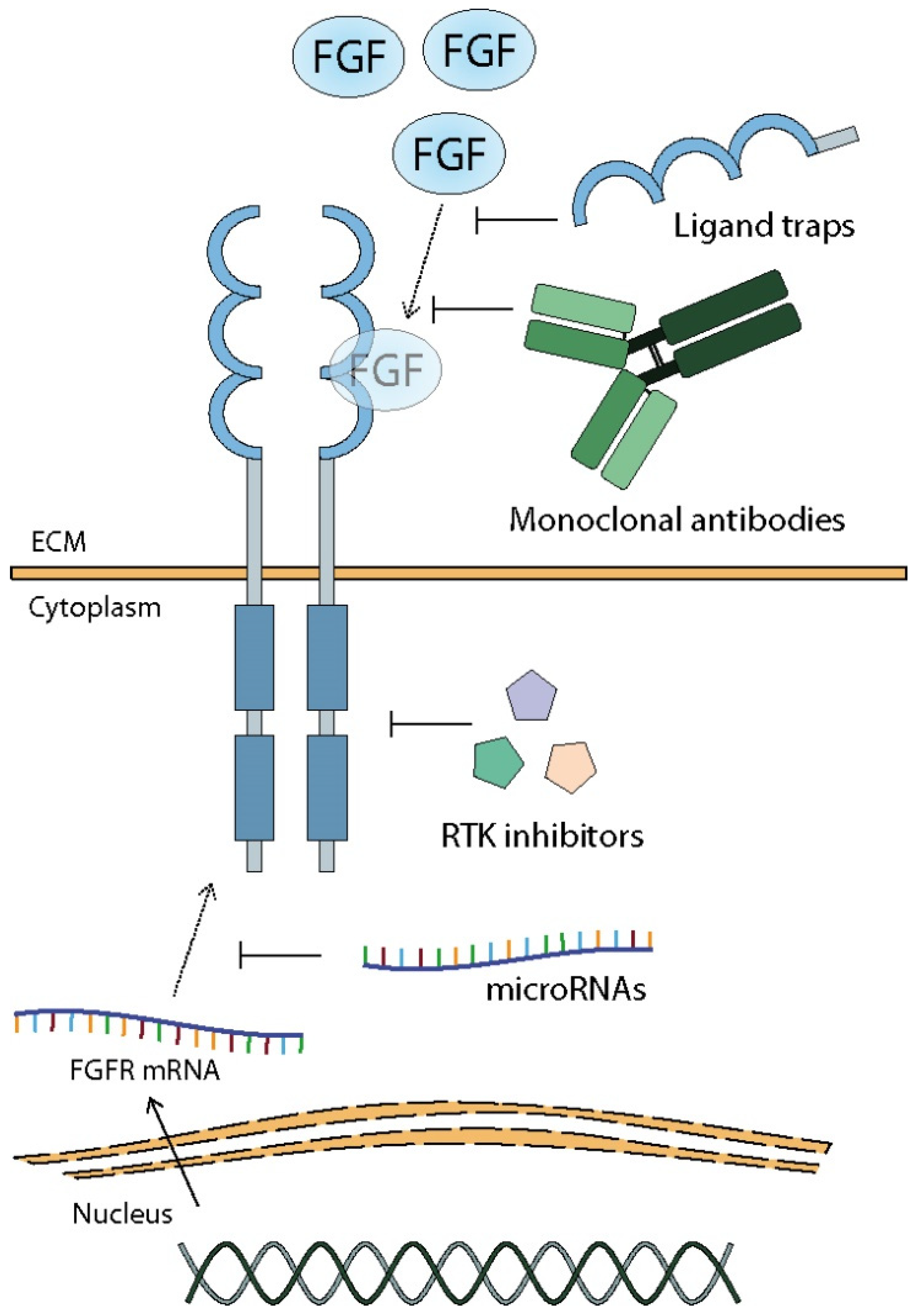
9. Sensitization of Tumor Cells to Chemotherapy by Inhibition of FGF/FGFR Complex Activity
9.1. TK Inhibitors
9.2. Monoclonal Antibodies and Ligand Traps
9.3. MicroRNAs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class of Drug | Name | Eliminating Resistance to | Cancer Type | References |
|---|---|---|---|---|
| Chemical inhibitors | AZD4547 | Gefitinib | NSCLC | [82] |
| PLX51107 | Melanoma | [199] | ||
| Tamoxifen | Breast cancer | [203] | ||
| PD173074 | ||||
| Gefitinib | NSCLC | [83] | ||
| Lapatinib | ESCC | [200] | ||
| Cisplatin | SCLC | [201] | ||
| Bevacizumab | HNSCC | [53] | ||
| Pemetrexed | Lung cancer | [176] | ||
| Doxorubicin | Endometrial cancer | [202] | ||
| Paclitaxel | ||||
| NSCLC | [205] | |||
| Epidermoid carcinoma | [204] | |||
| Vincristine | ||||
| Erdafitinib (JNJ-42756493) | Colchicine | [206] | ||
| ASP5878 | Gemcitabine | Urothelial cancer | [153] | |
| Doxorubicin | ||||
| BGJ398 (Infigratinib) | Paclitaxel/carboplatin | Ovarian cancer | [208] | |
| 5-fluorouracil | Colorectal cancer | [52] | ||
| Oxaliplatin | ||||
| Imatinib | GIST | [46] | ||
| Doxorubicin | [212] | |||
| Gefitinib | NSCLC | [213] | ||
| Alofanib | Paclitaxel/carboplatin | Ovarian cancer | [211] | |
| LY2874455 | Vemurafenib | Melanoma | [47] | |
| Ki23057 | Irinotecan | Gastric cancer | [214] | |
| Paclitaxel | ||||
| Etoposide | ||||
| Ligand trap | Suramin | Doxorubicin | Prostate cancer | [218] |
| miRNAs | miR-205 | Paclitaxel/doxorubicin/cyclophosphamide | Breast cancer | [223] |
| miR-3116 | Temozolomide | Glioma | [57] |
10. Limitations of FGFR Inhibition Therapy—FGFR Mutations and Molecular Cross-Talks with Other Protein
11. Enhancement of Chemotherapy by FGFs/FGFRs Action
12. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADC | antibody–drug conjugate |
| BAD | BCL-2 antagonist of cell death |
| BAX | apoptosis regulator BAX |
| BCL-2 | B-cell CLL/lymphoma 2 |
| BCL-XL | B-cell lymphoma-extra large |
| BIM | BCL-2-like protein 11 |
| BRAF | serine/threonine-protein kinase B-raf |
| CAFs | cancer-associated fibroblasts |
| CAM | cell adhesion molecule |
| CC | colon cancer |
| CK2 | casein kinase 2 |
| CMF | cyclophosphamide/methotrexate/5-fluorouracil chemotherapy |
| CRC | colorectal cancer |
| DAG | diacyloglycerol |
| E12/E47 | immunoglobulin enhancer-binding factor lub transcription factor E2-alpha |
| ECM | extracellular matrix |
| EF1/ZEB1 | elongation factor 1/zinc finger E-box-binding homeobox 1 |
| EGF | epithelial growth factor |
| EGFR | epithelial growth factor receptor |
| EMT | epithelial-to-mesenchymal transition |
| ENO1 | alpha-enolase |
| ER | estrogen receptor |
| ERCC1 | excision repair-cross complementing gene 1 |
| ERK | extracellular signal-regulated kinase |
| ESCC | esophageal squamous cell carcinoma |
| FGF | fibroblast growth factor |
| FGFR | fibroblast growth factor receptor |
| FGFRL1 | fibroblast growth factor receptor-like 1 |
| ESCC | esophageal squamous cell carcinoma |
| FLIP | FLICE-like inhibitory protein |
| FOXO1 | forkhead box protein O1 |
| FRA1 | Fos-related antigen 1 |
| FRS2α | fibroblast growth factor receptor substrate 2 |
| GAB1 | GRB2-associated binding protein 1 |
| GC | gastric cancer |
| GIST | gastrointestinal stromal tumor |
| GPCR | G-protein-coupled receptor |
| GRB2 | growth factor receptor-bound 2 |
| GSK3β | glycogen synthase kinase 3β |
| HA | hyaluronan |
| HER2/3 | receptor tyrosine-protein kinase erbB-2/3 |
| HES1 | hairy and enhancer of split1 |
| HCC | hepatocellular carcinoma |
| HGF | hepatocyte growth factor |
| HNC | head and neck cancer |
| HNSCC | head and neck squamous cell carcinoma |
| HO-1 | hemeoxygenase 1 |
| HSC | hepatic stellate cells |
| HSP90 | heat shock protein 90 |
| IP3 | inositol-1,4,5-triphosphate |
| ICC | intrahepatic cholangiocarcinoma |
| IGF | insulin-like growth factor |
| IGFR | insulin-like growth factor receptor |
| IKK-β | inhibitors of NFκB kinase-β |
| JAK | Janus kinases |
| JNK | c-Jun N-terminal kinase |
| KLB | βKlotho |
| LUAC | lung adenocarcinoma |
| mAb | monoclonal antibody |
| MACOM | MDS1 and EVI1 complex locus protein EVI1 |
| MAPK | mitogen-activated protein kinases |
| MCL1 | myeloid cell leukemia sequence 1 |
| MDM2 | mouse double minute 2 homolog |
| MEK | mitogen-activated protein kinase kinase |
| MMTV | mouse mammary tumor virus |
| miRNA | microRNA |
| MRP7 | multidrug resistance protein 7 |
| MT1-MMP | membrane type 1 matrix metalloproteinase |
| mTOR | mammalian target of rapamycin |
| mTORC1/2 | mammalian target of rapamycin complex 1/2 |
| NCT | neoadjuvant chemotherapy |
| NFκB | nuclear factor-κB |
| Notch | neurogenic locus notch homolog protein |
| NRP1 | neuropilin-1 |
| Nrf2 | nuclear factor E2-related factor 2 |
| NSCLC | non-small-cell lung cancer |
| P53 | cellular tumor antigen p53 |
| PARP | poly (ADP-ribose) polymerase |
| PD-ECGF | platelet-derived endothelial cell growth factor |
| PDGFRα | platelet-derived growth factor receptor α |
| P-gp | P-glycoprotein 1 |
| PI3K | phosphoinositide 3-kinase |
| PIP2 | phosphatidylinositol-4,5-bisphosphate |
| PKB | protein kinase B |
| PKC | protein kinase C |
| PLA2G4C | phospholipase A2γ |
| PLCγ | phospholipase Cγ |
| PPHLN1 | periphilin-1 |
| PRKACB | cAMP-dependent protein kinase catalytic subunit beta |
| PUMA | p53 upregulated modulator of apoptosis |
| RAP1 | RAS-proximate-1 or Ras-related protein 1 |
| RAS | rat sarcoma virus protein |
| ROS | reactive oxygen species |
| RTK | receptor tyrosine kinase |
| S6K2 | ribosomal p70 S6 kinase 2 (jest teżskrót S6K1) |
| SCLC | small cell lung cancer |
| SIP1/ZEB2 | smad interacting protein 1/zinc finger E-box-binding homeobox 2 |
| SNT-1 | suc1-associated neurotrophic factor-induced tyrosine-phosphorylated target |
| SOS1 | son of sevenless 1 |
| SOX2 | Sry-related HMG box 2 |
| STAT | signal transducers and activators of transcription |
| TAB | tumor-associated B cells |
| TAC | taxol, doxorubicin, cyclophosphamide chemotherapy |
| TGFβ | transforming growth factor β |
| Tin-PP | Tin-Protoporphyrin |
| TKI | tyrosine kinase inhibitor |
| TSC1/2 | tuberous sclerosis complex |
| TYK2 | tyrosine kinase 2 |
| UPP1 | uridinephosphorylase 1 |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
References
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhang, H.; Jia, Z.; Cui, M.; Tian, J. Chemoresistance and targeting of growth factors/cytokines signalling pathways: Towards the development of effective therapeutic strategy for endometrial cancer. Am. J. Cancer Res. 2018, 8, 1317–1331. [Google Scholar] [PubMed]
- Savant, S.S.; Sriramkumar, S.; O’hagan, H.M. The role of inflammation and inflammatory mediators in the development, progression, metastasis, and chemoresistance of epithelial ovarian cancer. Cancers 2018, 10, 251. [Google Scholar] [CrossRef] [Green Version]
- Caetano-Pinto, P.; Jamalpoor, A.; Ham, J.; Goumenou, A.; Mommersteeg, M.; Pijnenburg, D.; Ruijtenbeek, R.; Sanchez-Romero, N.; Van Zelst, B.; Heil, S.G.; et al. Cetuximab Prevents Methotrexate-Induced Cytotoxicity in Vitro through Epidermal Growth Factor Dependent Regulation of Renal Drug Transporters. Mol. Pharm. 2017, 14, 2147–2157. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.; Adachi, Y.; Yamamoto, H.; Imsumran, A.; Arimura, Y.; Endo, T.; Hinoda, Y.; Lee, C.T.; Nadaf, S.; Carbone, D.P.; et al. Insulin-like growth factor I receptor blockade enhances chemotherapy and radiation responses and inhibits tumour growth in human gastric cancer xenografts. Gut 2005, 54, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Zeng, L.; Wang, J.; Zhang, X.; Ruan, Q.; Wang, J.; Cui, S.; Yang, D. Reversal of 5-fluorouracil resistance by EGCG is mediate by inactivation of TFAP2A/VEGF signaling pathway and downregulation of MDR-1 and P-gp expression in gastric cancer. Oncotarget 2017, 8, 82842–82853. [Google Scholar] [CrossRef] [Green Version]
- Massarweh, S.; Osborne, C.K.; Creighton, C.J.; Qin, L.; Tsimelzon, A.; Huang, S.; Weiss, H.; Rimawi, M.; Schiff, R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol. Cancer Res. 2010, 8, 1439–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gospodarowicz, D. Humoral control of cell proliferation: The role of fibroblast growth factor in regeneration, angiogenesis, wound healing, and neoplastic growth. Prog. Clin. Biol. Res. 1976, 9, 1–19. [Google Scholar] [PubMed]
- Holley, R.W.; Baldwin, J.H.; Kiernan, J.A.; Messmer, T.O. Control of growth of benzo[a]pyrene transformed 3T3 cells. Proc. Natl. Acad. Sci. USA 1976, 73, 3229–3232. [Google Scholar] [CrossRef] [Green Version]
- Baird, A.; Mormède, P.; Böhlen, P. Immunoreactive fibroblast growth factor (FGF) in a transplantable chondrosarcoma: Inhibition of tumor growth by antibodies to FGF. J. Cell. Biochem. 1986, 30, 79–85. [Google Scholar] [CrossRef]
- Peters, G.; Lee, A.E.; Dickson, C. Concerted activation of two potential proto-oncogenes in carcinomas induced by mouse mammary tumour virus. Nature 1986, 320, 628–631. [Google Scholar] [CrossRef]
- Ohashi, R.; Matsuda, Y.; Ishiwata, T.; Naito, Z. Downregulation of fibroblast growth factor receptor 2 and its isoforms correlates with a high proliferation rate and poor prognosis in high-grade glioma. Oncol. Rep. 2014, 32, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Colomer, R.; Aparicio, J.; Montero, S.; Guzmán, C.; Larrodera, L.; Cortés-Funes, H. Low levels of basic fibroblast growth factor (bFGF) are associated with a poor prognosis in human breast carcinoma. Br. J. Cancer 1997, 76, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Shin, K.H.; Park, J.G. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001, 61, 3541–3543. [Google Scholar] [PubMed]
- Chesi, M.; Nardini, E.; Brents, L.A.; Schrock, E.; Ried, T.; Kuehl, W.M.; Bergsagel, P.L. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 1997, 16, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Yamada, Y.; Okita, N.; Takashima, A.; Honma, Y.; Iwasa, S.; Kato, K.; Hamaguchi, T.; Shimada, Y. Amplification of FGFR2 gene in patients with advanced gastric cancer receiving chemotherapy: Prevalence and prognostic significance. Anticancer Res. 2015, 35, 5055–5062. [Google Scholar] [PubMed]
- Seo, A.N.; Jin, Y.; Lee, H.J.; Sun, P.L.; Kim, H.; Jheon, S.; Kim, K.; Lee, C.T.; Chung, J.H. FGFR1 amplification is associated with poor prognosis and smoking in non-small-cell lung cancer. Virchows Arch. 2014, 465, 547–558. [Google Scholar] [CrossRef]
- Pecqueux, C.; Arslan, A.; Heller, M.; Falkenstein, M.; Kaczorowski, A.; Tolstov, Y.; Sültmann, H.; Grüllich, C.; Herpel, E.; Duensing, A.; et al. FGF-2 is a driving force for chromosomal instability and a stromal factor associated with adverse clinico-pathological features in prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 365.e15–365.e26. [Google Scholar] [CrossRef]
- Menzel, T.; Rahman, Z.; Calleja, E.; White, K.; Wilson, E.L.; Wieder, R.; Gabrilove, J. Elevated intracellular level of basic fibroblast growth factor correlates with stage of chronic lymphocytic leukemia and is associated with resistance to fludarabine. Blood 1996, 87, 1056–1063. [Google Scholar] [CrossRef]
- Miyake, H.; Hara, I.; Gohji, K.; Yoshimura, K.; Arakawa, S.; Kamidono, S. Expression of basic fibroblast growth factor is associated with resistance to cisplatin in a human bladder cancer cell line. Cancer Lett. 1998, 123, 121–126. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Huang, C.; Lin, C.C.; Tang, C.H.; Lin, C.Y.; Lee, I.N.; Huang, H.C.; Chen, J.C. Basic fibroblast growth factor promotes doxorubicin resistance in chondrosarcoma cells by affecting XRCC5 expression. Mol. Carcinog. 2020, 59, 293–303. [Google Scholar] [CrossRef]
- Song, S.; Guillaume Wientjes, M.; Gan, Y.; Au, J.L.S. Fibroblast growth factors: An epigenetic mechanism of broad spectrum resistance to anticancer drugs. Proc. Natl. Acad. Sci. USA 2000, 97, 8658–8663. [Google Scholar] [CrossRef] [Green Version]
- Vandermoere, F.; Yazidi-Belkoura, I.E.; Adriaenssens, E.; Lemoine, J.; Hondermarck, H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-κB activation induced via interaction between Akt and IκB kinase-β in breast cancer cells. Oncogene 2005, 24, 5482–5491. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.; Wientjes, M.G.; Au, J.L.S. Expression of basic fibroblast growth factor correlates with resistance to paclitaxel in human patient tumors. Pharm. Res. 2006, 23, 1324–1331. [Google Scholar] [CrossRef]
- Smith, G.; Ng, M.T.H.; Shepherd, L.; Herrington, C.S.; Gourley, C.; Ferguson, M.J.; Wolf, C.R. Individuality in FGF1 expression significantly influences platinum resistance and progression-free survival in ovarian cancer. Br. J. Cancer 2012, 107, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Dorman, S.N.; Baranova, K.; Knoll, J.H.M.; Urquhart, B.L.; Mariani, G.; Carcangiu, M.L.; Rogan, P.K. Genomic signatures for paclitaxel and gemcitabine resistance in breast cancer derived by machine learning. Mol. Oncol. 2016, 10, 85–100. [Google Scholar] [CrossRef]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- McDermott, S.C.; Rodriguez-Ramirez, C.; McDermott, S.P.; Wicha, M.S.; Nör, J.E. FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget 2018, 9, 25148–25165. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Meng, Y.; Zhang, Z.; Liu, Y.; Wang, X. Downregulation of basic fibroblast growth factor increases cisplatin sensitivity in A549 non-small cell lung cancer cells. J. Cancer Res. Ther. 2018, 14, 1519–1524. [Google Scholar] [PubMed]
- Makondi, P.T.; Chu, C.-M.; Wei, P.-L.; Chang, Y.-J. Prediction of novel target genes and pathways involved in irinotecan-resistant colorectal cancer. PLoS ONE 2017, 12, e0180616. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Murata, K.; Hirose, R.; Matsuda, C.; Komatsu, T.; Ikekita, M.; Nakawatari, M.; Nakayama, F.; Wakatsuki, M.; Ohno, T.; et al. Upregulated expression of FGF13/FHF2 mediates resistance to platinum drugs in cervical cancer cells. Sci. Rep. 2013, 3, 2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Wang, X.; Tang, Y.; Huang, S.; Hu, C.A.A.; Teng, Y. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J. Exp. Clin. Cancer Res. 2017, 36, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatlen, M.A.; Schmidt-Kittler, O.; Sherwin, C.A.; Rozsahegyi, E.; Rubin, N.; Sheets, M.P.; Kim, J.L.; Miduturu, C.; Bifulco, N.; Brooijmans, N.; et al. Acquired on-target clinical resistance validates fgfr4 as a driver of hepatocellular carcinoma. Cancer Discov. 2019, 9, 1686–1695. [Google Scholar] [CrossRef] [Green Version]
- Seitz, T.; Freese, K.; Dietrich, P.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Fibroblast Growth Factor 9 is expressed by activated hepatic stellate cells and promotes progression of hepatocellular carcinoma. Sci. Rep. 2020, 10, 1–9. [Google Scholar]
- Hanker, A.B.; Garrett, J.T.; Estrada, M.V.; Moore, P.D.; Ericsson, P.G.; Koch, J.P.; Langley, E.; Singh, S.; Kim, P.S.; Frampton, G.M.; et al. HER2-Overexpressing Breast Cancers Amplify FGFR Signaling upon Acquisition of Resistance to Dual Therapeutic Blockade of HER2. Clin. Cancer Res. 2017, 23, 4323–4334. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wang, H.; Qie, A.; Wang, J.; Liu, Y.; Gu, G.; Yang, J.; Zhang, H.; Pan, W.; Tian, Z.; et al. FGF13 enhances resistance to platinum drugs by regulating hCTR1 and ATP7A via a microtubule-stabilizing effect. Cancer Sci. 2021, 112, 4655–4668. [Google Scholar] [CrossRef] [PubMed]
- Gammelgaard, K.R.; Vad-Nielsen, J.; Clement, M.S.; Weiss, S.; Daugaard, T.F.; Dagnæs-Hansen, F.; Meldgaard, P.; Sorensen, B.S.; Nielsen, A.L. Up-Regulated FGFR1 Expression as a Mediator of Intrinsic TKI Resistance in EGFR-Mutated NSCLC. Transl. Oncol. 2019, 12, 432–440. [Google Scholar] [CrossRef]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A novel receptor tyrosine kinase switch promotes gastrointestinal stromal tumor drug resistance. Molecules 2017, 22, 2152. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S. Bin Reactivation of Mitogen-activated Protein Kinase (MAPK) pathway by FGF Receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J. Biol. Chem. 2012, 287, 28087–28098. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Kharbanda, S.; Chen, D.; Bullocks, J.; Miller, D.L.; Ding, I.Y.F.; Hanfelt, J.; McLeskey, S.W.; Kern, F.G. MCF-7 breast carcinoma cells overexpressing FGF-1 form vascularized, metastatic tumors in ovariectomized or tamoxifen-treated nude mice. Oncogene 1997, 15, 2093–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, K.M.; Priedigkeit, N.; Basudan, A.; Tasdemir, N.; Sikora, M.J.; Sokol, E.S.; Hartmaier, R.J.; Ding, K.; Ahmad, N.Z.; Watters, R.J.; et al. FGFR4 overexpression and hotspot mutations in metastatic er+ breast cancer are enriched in the lobular subtype. npj Breast Cancer 2019, 5, 19. [Google Scholar] [CrossRef]
- Sahores, A.; Figueroa, V.; May, M.; Liguori, M.; Rubstein, A.; Fuentes, C.; Jacobsen, B.M.; Elía, A.; Rojas, P.; Sequeira, G.R.; et al. Increased High Molecular Weight FGF2 in Endocrine-Resistant Breast Cancer. Horm. Cancer 2018, 9, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Turczyk, L.; Kitowska, K.; Mieszkowska, M.; Mieczkowski, K.; Czaplinska, D.; Piasecka, D.; Kordek, R.; Skladanowski, A.C.; Potemski, P.; Romanska, H.M.; et al. FGFR2-Driven Signaling Counteracts Tamoxifen Effect on ERα-Positive Breast Cancer Cells. Neoplasia 2017, 19, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Turkington, R.C.; Longley, D.B.; Allen, W.L.; Stevenson, L.; McLaughlin, K.; Dunne, P.D.; Blayney, J.K.; Salto-Tellez, M.; Van Schaeybroeck, S.; Johnston, P.G. Fibroblast growth factor receptor 4 (FGFR4): A targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014, 5, e1046. [Google Scholar] [CrossRef] [Green Version]
- Gyanchandani, R.; Ortega Alves, M.V.; Myers, J.N.; Kim, S. A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol. Cancer Res. 2013, 11, 1585–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Han, L.L.; Du, F.; Liu, X.M.; Li, J.; Wang, H.H.; Song, M.H.; Li, Z.; Li, G.Y. FGFR1 induces acquired resistance against gefitinib by activating AKT/mTOR pathway in NSCLC. Onco Targets Ther. 2019, 12, 9809–9816. [Google Scholar] [CrossRef] [Green Version]
- Karajannis, M.A.; Vincent, L.; DiRenzo, R.; Shmelkov, S.V.; Zhang, F.; Feldman, E.J.; Bohlen, P.; Zhu, Z.; Sun, H.; Kussie, P.; et al. Activation of FGFR1β signaling pathway promotes survival, migration and resistance to chemotherapy in acute myeloid leukemia cells. Leukemia 2006, 20, 979–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manousakidi, S.; Guillaume, A.; Pirou, C.; Bouleau, S.; Mignotte, B.; Renaud, F.; Le Floch, N. FGF1 induces resistance to chemotherapy in ovarian granulosa tumor cells through regulation of p53 mitochondrial localization. Oncogenesis 2018, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.; Cao, Y.; Li, X.; Li, Z.; Xin, Y.; Meng, Y. MiR-3116 sensitizes glioma cells to temozolomide by targeting FGFR1 and regulating the FGFR1/PI3K/AKT pathway. J. Cell. Mol. Med. 2020, 24, 4677–4686. [Google Scholar] [CrossRef] [PubMed]
- Tomé, M.; Tchorz, J.; Gassmann, M.; Bettler, B. Constitutive activation of Notch2 signalling confers chemoresistance to neural stem cells via transactivation of fibroblast growth factor receptor-1. Stem Cell Res. 2019, 35, 101390. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Gibson, T.B.; Robinson, F.; Silvestro, L.; Pearson, G.; Xu, B.E.; Wright, A.; Vanderbilt, C.; Cobb, M.H. MAP kinases. Chem. Rev. 2001, 101, 2449–2476. [Google Scholar] [CrossRef]
- Manchado, E.; Weissmueller, S.; Morris, J.P.; Chen, C.C.; Wullenkord, R.; Lujambio, A.; De Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016, 534, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, V.E.; Xue, J.Y.; Frederick, D.T.; Cao, Y.; Lin, E.; Wilson, C.; Urisman, A.; Carbone, D.P.; Flaherty, K.T.; Bernards, R.; et al. Adaptive Resistance to Dual BRAF/MEK Inhibition in BRAF-Driven Tumors through Autocrine FGFR Pathway Activation. Clin. Cancer Res. 2019, 25, 7202–7217. [Google Scholar] [CrossRef] [Green Version]
- McLeskey, S.W.; Kurebayashi, J.; Honig, S.F.; Zwiebel, J.; Lippman, M.E.; Dickson, R.B.; Kern, F.G. Fibroblast growth factor 4 transfection of MCF-7 cells produces cell lines that are tumorigenic and metastatic in ovariectomized or tamoxifen-treated athymic nude mice. Cancer Res. 1993, 53, 2168–2177. [Google Scholar]
- Shee, K.; Yang, W.; Hinds, J.W.; Hampsch, R.A.; Varn, F.S.; Traphagen, N.A.; Patel, K.; Cheng, C.; Jenkins, N.P.; Kettenbach, A.N.; et al. Therapeutically targeting tumor microenvironment- mediated drug resistance in estrogen receptor-positive breast cancer. J. Exp. Med. 2018, 215, 895–910. [Google Scholar] [CrossRef] [Green Version]
- Thottassery, J.V.; Sun, Y.; Westbrook, L.; Rentz, S.S.; Manuvakhova, M.; Qu, Z.; Samuel, S.; Upshaw, R.; Cunningham, A.; Kern, F.G. Prolonged extracellular signal-regulated kinase 1/2 activation during fibroblast growth factor 1- of heregulin β1-induced antiestrogen-resistant growth of breast cancer cells is resistant to mitogen-activated protein/extracellular regulated kinase kinase. Cancer Res. 2004, 64, 4637–4647. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, D.C.; Knowles, M.A.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 2012, 130, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Cohen, O.; Kowalski, K.J.; Kusiel, J.G.; Buendia-Buendia, J.E.; Cuoco, M.S.; Exman, P.; Wander, S.A.; Waks, A.G.; Nayar, U.; et al. Acquired FGFR and FGF Alterations Confer Resistance to Estrogen Receptor (ER) Targeted Therapy in ER + Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 5974–5989. [Google Scholar] [CrossRef] [PubMed]
- Manuvakhova, M.; Thottassery, J.V.; Hays, S.; Qu, Z.; Rentz, S.S.; Westbrook, L.; Kern, F.G. Expression of the SNT-1/FRS2 phosphotyrosine binding domain inhibits activation of MAP kinase and PI3-kinase pathways and antiestrogen resistant growth induced by FGF-1 in human breast carcinoma cells. Oncogene 2006, 25, 6003–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/pbotein kinace B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.Z.; Zhou, X.D.; Qian, G.; Shi, X.; Fang, J.; Jiang, B.H. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70s6K1 pathway. Cancer Res. 2007, 67, 6325–6332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar]
- Fujiwara, M.; Izuishi, K.; Sano, T.; Hossain, M.A.; Kimura, S.; Masaki, T.; Suzuki, Y. Modulating effect of the PI3-kinase inhibitor LY294002 on cisplatin in human pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2008, 27, 76. [Google Scholar] [CrossRef] [Green Version]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lu, H.; Zhang, J.; Chen, J.; Chai, Z.; Zhang, J. Essential role of AKT in tumor cells addicted to FGFR. Anticancer Drugs 2014, 25, 183–188. [Google Scholar] [CrossRef]
- Sun, B.; Xu, H.; Zhang, G.; Zhu, Y.; Sun, H.; Hou, G. Basic fibroblast growth factor upregulates survivin expression in hepatocellular carcinoma cells via a protein kinase B-dependent pathway. Oncol. Rep. 2013, 30, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, J.; Master, Z.; Yu, J.L.; Rak, J.; Dumont, D.J.; Kerbel, R.S. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc. Natl. Acad. Sci. USA 2002, 99, 4349–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, S.A.; Marshall, M.E.; Ware, K.E.; Heasley, L.E. The fibroblast growth factor receptor signaling pathway as a mediator of intrinsic resistance to EGFR-specific tyrosine kinase inhibitors in non-small cell lung cancer. Drug Resist. Updates 2009, 12, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Kurimoto, R.; Iwasawa, S.; Ebata, T.; Ishiwata, T.; Sekine, I.; Tada, Y.; Tatsumi, K.; Koide, S.; Iwama, A.; Takiguchi, Y. Drug resistance originating from a TGF-β/FGF-2-driven epithelial-to-mesenchymal transition and its reversion in human lung adenocarcinoma cell lines harboring an EGFR mutation. Int. J. Oncol. 2016, 48, 1825–1836. [Google Scholar] [CrossRef] [Green Version]
- Ware, K.E.; Hinz, T.K.; Kleczko, E.; Singleton, K.R.; Marek, L.A.; Helfrich, B.A.; Cummings, C.T.; Graham, D.K.; Astling, D.; Tan, A.C.; et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2013, 2, e39. [Google Scholar] [CrossRef] [Green Version]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T.; et al. Activation of the FGF2-FGFR1 autocrine pathway: A novel mechanism of acquired resistance to gefitinib in NSCLC. Mol. Cancer Res. 2013, 11, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Heo, Y.M.; Jeong, K.I.; Kim, Y.M.; Jang, H.L.; Lee, K.Y.; Yeo, C.Y.; Kim, S.H.; Lee, H.K.; Kim, S.R.; et al. FGF-2 inhibits TNF-α mediated apoptosis through upregulation of Bcl2-A1 and Bcl-xL in ATDC5 cells. BMB Rep. 2012, 45, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Zhao, H.; Gao, L.; Zhang, W.; Shull, A.Y.; Shay, C. FGF19 protects hepatocellular carcinoma cells against endoplasmic reticulum stress via activation of FGFR4–GSK3β–Nrf2 signaling. Cancer Res. 2017, 77, 6215–6225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzner, T.; Bedeir, A.; Held, G.; Peter-Vörösmarty, B.; Ghassemi, S.; Heinzle, C.; Spiegl-Kreinecker, S.; Marian, B.; Holzmann, K.; Grasl-Kraupp, B.; et al. Fibroblast growth factor receptors as therapeutic targets in human melanoma: Synergism with BRAF inhibition. J. Investig. Dermatol. 2011, 131, 2087–2095. [Google Scholar] [CrossRef] [Green Version]
- Grimm, J.; Hufnagel, A.; Wobser, M.; Borst, A.; Haferkamp, S.; Houben, R.; Meierjohann, S. BRAF inhibition causes resilience of melanoma cell lines by inducing the secretion of FGF1. Oncogenesis 2018, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Jansen, V.M.; Koch, J.P.; Li, H.; Formisano, L.; Williams, J.A.; Grandis, J.R.; Arteaga, C.L. Treatment of triple-negative breast cancer with TORC1/2 inhibitors sustains a drug-resistant and notch-dependent cancer stem cell population. Cancer Res. 2016, 76, 440–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Mori, T.; Yamamoto, A.; Takagi, S.; Ueda, M. Proliferation of poorly differentiated endometrial cancer cells through autocrine activation of FGF receptor and HES1 expression. Hum. Cell 2019, 32, 367–378. [Google Scholar] [CrossRef]
- Liu, Z.H.; Dai, X.M.; Du, B. Hes1: A key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 2015, 16, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Su, Y.; Zhang, Y.; Han, B.; Liu, H.; Wang, X. Endothelial Cells Promote Docetaxel Resistance of Prostate Cancer Cells by Inducing ERG Expression and Activating Akt/mTOR Signaling Pathway. Front. Oncol. 2020, 10, 584505. [Google Scholar] [CrossRef]
- Huang, S.; Liang, S.; Chen, G.; Chen, J.; You, K.; Ye, H.; Li, Z.; He, S. Overexpression of glycosyltransferase 8 domain containing 2 confers ovarian cancer to CDDP resistance by activating FGFR/PI3K signalling axis. Oncogenesis 2021, 10, 55. [Google Scholar] [CrossRef]
- Pan, X.; Zhou, T.; Tai, Y.H.; Wang, C.; Zhao, J.; Cao, Y.; Chen, Y.; Zhang, P.J.; Yu, M.; Zhen, C.; et al. Elevated expression of CUEDC2 protein confers endocrine resistance in breast cancer. Nat. Med. 2011, 17, 708–714. [Google Scholar] [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. Jak-stat signaling: A double-edged sword of immune regulation and cancer progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohrer, L.R.; Chuntova, P.; Bade, L.K.; Beadnell, T.C.; Leon, R.P.; Brady, N.J.; Ryu, Y.; Goldberg, J.E.; Schmechel, S.C.; Koopmeiners, J.S.; et al. Activation of the FGFR-STAT3 pathway in breast cancer cells induces a hyaluronan-rich microenvironment that licenses tumor formation. Cancer Res. 2014, 74, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Plowright, E.E.; Li, Z.; Bergsagel, P.L.; Chesi, M.; Barber, D.L.; Branch, D.R.; Hawley, R.G.; Stewart, A.K. Ectopic expression of fibroblast growth factor receptor 3 promotes myeloma cell proliferation and prevents apoptosis. Blood 2000, 95, 992–998. [Google Scholar] [CrossRef]
- Li, P.; Huang, T.; Zou, Q.; Liu, D.; Wang, Y.; Tan, X.; Wei, Y.; Qiu, H. FGFR2 Promotes Expression of PD-L1 in Colorectal Cancer via the JAK/STAT3 Signaling Pathway. J. Immunol. 2019, 202, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Tang, W.; Peng, H.; Qi, X.; Li, J. FGFR leads to sustained activation of STAT3 to mediate resistance to EGFR-TKIs treatment. Investig. New Drugs 2021, 39, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.R.; Lyons-Lewis, J.; Seckl, M.J.; Costa-Pereira, A.P. A novel requirement for Janus Kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PLoS ONE 2011, 6, e19861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.J.; Suh, P.G.; Lee, Y.J.; Shin, K.J.; Cocco, L.; Chae, Y.C. PLCγ1: Potential arbitrator of cancer progression. Adv. Biol. Regul. 2018, 67, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Pardo, O.E.; Wellbrock, C.; Khanzada, U.K.; Aubert, M.; Arozarena, I.; Davidson, S.; Bowen, F.; Parker, P.J.; Filonenko, V.V.; Gout, I.T.; et al. FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCε, B-Raf and S6K2. EMBO J. 2006, 25, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Lamont, F.R.; Shnyder, S.D.; Knowles, M.A. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Res. 2009, 69, 4613–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef]
- Zhang, X.; Nie, D.; Chakrabarty, S. Growth factors in tumor microenvironment. Front. Biosci. 2010, 15, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Giulianelli, S.; Cerliani, J.P.; Lamb, C.A.; Fabris, V.T.; Bottino, M.C.; Gorostiaga, M.A.; Novaro, V.; Góngora, A.; Baldi, A.; Molinolo, A.; et al. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int. J. Cancer 2008, 123, 2518–2531. [Google Scholar] [CrossRef]
- Hegab, A.E.; Ozaki, M.; Kameyama, N.; Gao, J.; Kagawa, S.; Yasuda, H.; Soejima, K.; Yin, Y.; Guzy, R.D.; Nakamura, Y.; et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J. Pathol. 2019, 249, 193–205. [Google Scholar] [CrossRef]
- Kumar, D.; New, J.; Vishwakarma, V.; Joshi, R.; Enders, J.; Lin, F.; Dasari, S.; Gutierrez, W.R.; Leef, G.; Ponnurangam, S.; et al. Cancer-Associated Fibroblasts Drive Glycolysis in a Targetable Signaling Loop Implicated in Head and Neck Squamous Cell Carcinoma Progression. Cancer Res. 2018, 78, 3769–3782. [Google Scholar] [CrossRef] [Green Version]
- Awaji, M.; Futakuchi, M.; Heavican, T.; Iqbal, J.; Singh, R.K. Cancer-Associated Fibroblasts Enhance Survival and Progression of the Aggressive Pancreatic Tumor via FGF-2 and CXCL8. Cancer Microenviron. 2019, 12, 37–46. [Google Scholar] [CrossRef]
- Fernández-Nogueira, P.; Mancino, M.; Fuster, G.; López-Plana, A.; Jauregui, P.; Almendro, V.; Enreig, E.; Menéndez, S.; Rojo, F.; Noguera-Castells, A.; et al. Tumor-associated fibroblasts promote HER2-targeted therapy resistance through FGFR2 activation. Clin. Cancer Res. 2020, 26, 1432–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, J.; Kim, D.H.; Lee, Y.H.; Jang, J.H.; Surh, Y.J. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol. Carcinog. 2020, 59, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Kato, S.; Fox, J.C.; Shigemori, M.; Morimatsu, M. Autocrine signaling through ras regulates cell survival activity in human glioma cells: Potential cross-talk between ras and the phosphatidylinositol 3-kinase-akt pathway. J. Neuropathol. Exp. Neurol. 2002, 61, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.R.; Kim, J.Y.; Kang, Y.J.; Ahn, J.Y.; Kim, J.H.; Kim, B.W.; Choi, H.Y.; Jeong, M.Y.; Cho, S.G. Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 958–968. [Google Scholar] [CrossRef] [Green Version]
- Wee, S.; Jagani, Z.; Kay, X.X.; Loo, A.; Dorsch, M.; Yao, Y.M.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- Ban, M.J.; Byeon, H.K.; Yang, Y.J.; An, S.; Kim, J.W.; Kim, J.H.; Kim, D.H.; Yang, J.; Kee, H.; Koh, Y.W. Fibroblast growth factor receptor 3-mediated reactivation of ERK signaling promotes head and neck squamous cancer cell insensitivity to MEK inhibition. Cancer Sci. 2018, 109, 3816–3825. [Google Scholar] [CrossRef]
- Pardo, O.E.; Arcaro, A.; Salerno, G.; Tetley, T.D.; Valovka, T.; Gout, I.; Seckl, M.J. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene 2001, 20, 7658–7667. [Google Scholar] [CrossRef] [PubMed]
- Binju, M.; Amaya-Padilla, M.A.; Wan, G.; Gunosewoyo, H.; Rahmanto, Y.S.; Yu, Y. Therapeutic inducers of apoptosis in ovarian cancer. Cancers 2019, 11, 1786. [Google Scholar] [CrossRef] [Green Version]
- Pardo, O.E.; Arcaro, A.; Salerno, G.; Raguz, S.; Downward, J.; Seckl, M.J. Fibroblast growth factor-2 induces translational regulation of Bcl-XL and Bcl-2 via a MEK-dependent pathway: Correlation with resistance to etoposide-induced apoptosis. J. Biol. Chem. 2002, 277, 12040–12046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, O.E.; Lesay, A.; Arcaro, A.; Lopes, R.; Ng, B.L.; Warne, P.H.; McNeish, I.A.; Tetley, T.D.; Lemoine, N.R.; Mehmet, H.; et al. Fibroblast Growth Factor 2-Mediated Translational Control of IAPs Blocks Mitochondrial Release of Smac/DIABLO and Apoptosis in Small Cell Lung Cancer Cells. Mol. Cell. Biol. 2003, 24, 6887. [Google Scholar] [CrossRef] [Green Version]
- Shaulian, E.; Resnitzky, D.; Shifman, O.; Blandino, G.; Amsterdam, A.; Yayon, A.; Oren, M. Induction of Mdm2 and enhancement of cell survival by bFGF. Oncogene 1997, 15, 2717–2725. [Google Scholar] [CrossRef] [Green Version]
- Bouleau, S.; Grimal, H.; Rincheval, V.; Godefroy, N.; Mignotte, B.; Vayssière, J.L.; Renaud, F. FGF1 inhibits p53-dependent apoptosis and cell cycle arrest via an intracrine pathway. Oncogene 2005, 24, 7839–7849. [Google Scholar] [CrossRef]
- Jung, C.R.; Lim, J.H.; Choi, Y.; Kim, D.G.; Kang, K.J.; Noh, S.M.; Im, D.S. Enigma negatively regulates p53 through MDM2 and promotes tumor cell survival in mice. J. Clin. Investig. 2010, 120, 4493–4506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewska, M.; Sørensen, V.; Jin, Y.; Wiedlocha, A.; Olsnes, S. Translocation of exogenous FGF1 into cytosol and nucleus is a periodic event independent of receptor kinase activity. Exp. Cell Res. 2011, 317, 1005–1015. [Google Scholar] [CrossRef]
- Kostas, M.; Lampart, A.; Bober, J.; Wiedlocha, A.; Tomala, J.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. Translocation of Exogenous FGF1 and FGF2 Protects the Cell against Apoptosis Independently of Receptor Activation. J. Mol. Biol. 2018, 430, 4087–4101. [Google Scholar] [CrossRef]
- Sluzalska, K.D.; Slawski, J.; Sochacka, M.; Lampart, A.; Otlewski, J.; Zakrzewska, M. Intracellular partners of fibroblast growth factors 1 and 2—Implications for functions. Cytokine Growth Factor Rev. 2020, 57, 93–111. [Google Scholar] [CrossRef]
- Rodriguez-Enfedaque, A.; Bouleau, S.; Laurent, M.; Courtois, Y.; Mignotte, B.; Vayssière, J.L.; Renaud, F. FGF1 nuclear translocation is required for both its neurotrophic activity and its p53-dependent apoptosis protection. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1719–1727. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Payne, S.; Wang, F.; Claus, P.; Su, Z.; Groth, J.; Geradts, J.; de Ridder, G.; Alvarez, R.; Marcom, P.K.; et al. Nuclear basic fibroblast growth factor regulates triple-negative breast cancer chemo-resistance. Breast Cancer Res. 2015, 17, 91. [Google Scholar] [CrossRef] [Green Version]
- Grose, R.; Fantl, V.; Werner, S.; Chioni, A.M.; Jarosz, M.; Rudling, R.; Cross, B.; Hart, I.R.; Dickson, C. The role of fibroblast growth factor receptor 2b in skin homeostasis and cancer development. EMBO J. 2007, 26, 1268–1278. [Google Scholar] [CrossRef]
- Chioni, A.M.; Grose, R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J. Cell Biol. 2012, 197, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Iida, M.; Dunn, E.F.; Ghia, A.J.; Wheeler, D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009, 28, 3801–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formisano, L.; Stauffer, K.M.; Young, C.D.; Bhola, N.E.; Guerrero-Zotano, A.L.; Jansen, V.M.; Estrada, M.M.; Hutchinson, K.E.; Giltnane, J.M.; Schwarz, L.J.; et al. Association of FGFR1 with ERα maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER+ breast cancer. Clin. Cancer Res. 2017, 23, 6138–6151. [Google Scholar] [CrossRef] [Green Version]
- Roidl, A.; Berger, H.J.; Kumar, S.; Bange, J.; Knyazev, P.; Ullrich, A. Resistance to chemotherapy is associated with fibroblast growth factor receptor 4 up-regulation. Clin. Cancer Res. 2009, 15, 2058–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R.J.; Fernando, M.; Hughes, D.; Brown, N.J.; Woll, P.J. Angiogenic growth factor expression in benign and malignant vascular tumours. Exp. Mol. Pathol. 2014, 97, 148–153. [Google Scholar] [CrossRef]
- Conconi, M.T.; Nico, B.; Guidolin, D.; Baiguera, S.; Spinazzi, R.; Rebuffat, P.; Malendowicz, L.K.; Vacca, A.; Carraro, G.; Parnigotto, P.P.; et al. Ghrelin inhibits FGF-2-mediated angiogenesis in vitro and in vivo. Peptides 2004, 25, 2179–2185. [Google Scholar] [CrossRef]
- Herbert, S.P.; Stainier, D.Y.R. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Slaton, J.W.; Karashima, T.; Yoshikawa, C.; Shuin, T.; Sweeney, P.; Millikan, R.; Dinney, C.P.N. The prognostic value of angiogenesis factor expression for predicting recurrence and metastasis of bladder cancer after neoadjuvant chemotherapy and radical cystectomy. Clin. Cancer Res. 2000, 6, 4866–4873. [Google Scholar]
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular mediators of angiogenesis. J. Burn Care Res. 2010, 31, 158–175. [Google Scholar] [CrossRef]
- Huang, X.; Yu, C.; Jin, C.; Kobayashi, M.; Bowles, C.A.; Wang, F.; McKeehan, W.L. Ectopic activity of fibroblast growth factor receptor 1 in hepatocytes accelerates hepatocarcinogenesis by driving proliferation and vascular endothelial growth factor-induced angiogenesis. Cancer Res. 2006, 66, 1481–1490. [Google Scholar] [CrossRef] [Green Version]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Schönau, K.K.; Steger, G.G.; Mader, R.M. Angiogenic effect of naive and 5-fluorouracil resistant colon carcinoma on endothelial cells in vitro. Cancer Lett. 2007, 257, 73–78. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Imarisio, I.; Ganini, C.; Sacchi, L.; Quaglini, S.; Giunta, V.; De Amici, M. Changes in circulating pro-angiogenic cytokines, other than VEGF, before progression to sunitinib therapy in advanced renal cell carcinoma patients. Oncology 2012, 84, 115–122. [Google Scholar] [CrossRef]
- Guerrin, M.; Scotet, E.; Malecaze, F.; Houssaint, E.; Plouët, J. Overexpression of vascular endothelial growth factor induces cell transformation in cooperation with fibroblast growth factor 2. Oncogene 1997, 14, 463–471. [Google Scholar] [CrossRef]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Hicklin, D.J.; Huber, J.; Nakatani, T.; Tsujinoue, H.; Yanase, K.; et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology 2002, 35, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Watanabe Miyano, S.; Minoshima, Y.; Matsui, J.; Funahashi, Y. Activated FGF2 signaling pathway in tumor vasculature is essential for acquired resistance to anti-VEGF therapy. Sci. Rep. 2020, 10, 2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, D.L.; Spinelli, F.M.; Del Dago, D.; Icardi, A.; Demarchi, G.; Caon, I.; García, M.; Bolontrade, M.F.; Passi, A.; Cristina, C.; et al. Co-treatment of tumor cells with hyaluronan plus doxorubicin affects endothelial cell behavior independently of VEGF expression. Oncotarget 2018, 9, 36585–36602. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Kim, P.J.; Shin, S.J.; Lee, J.L.; Cho, Y.M.; Go, H. FGFR1 is associated with c-MYC and proangiogenic molecules in metastatic renal cell carcinoma under anti-angiogenic therapy. Histopathology 2020, 76, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, A.; Taranto, S.; Rezzola, S.; Matarazzo, S.; Grillo, E.; Bugatti, M.; Scotuzzi, A.; Guerra, J.; Trani, M.D.; Presta, M.; et al. Inhibition of the fgf/fgfr system induces apoptosis in lung cancer cells via c-myc downregulation and oxidative stress. Int. J. Mol. Sci. 2020, 21, 9376. [Google Scholar] [CrossRef]
- Kikuchi, A.; Suzuki, T.; Nakazawa, T.; Iizuka, M.; Nakayama, A.; Ozawa, T.; Kameda, M.; Shindoh, N.; Terasaka, T.; Hirano, M.; et al. ASP5878, a selective FGFR inhibitor, to treat FGFR3-dependent urothelial cancer with or without chemoresistance. Cancer Sci. 2017, 108, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malchers, F.; Dietlein, F.; Schöttle, J.; Lu, X.; Nogova, L.; Albus, K.; Fernandez-Cuesta, L.; Heuckmann, J.M.; Gautschi, O.; Diebold, J.; et al. Cell-autonomous and non-cell-autonomous mechanisms of transformation by amplified FGFR1 in lung cancer. Cancer Discov. 2014, 4, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef]
- Kurimoto, R.; Ebata, T.; Iwasawa, S.; Ishiwata, T.; Tada, Y.; Tatsumi, K.; Takiguchi, Y. Pirfenidone may revert the epithelial-to-mesenchymal transition in human lung adenocarcinoma. Oncol. Lett. 2017, 14, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Suyama, K.; Shapiro, I.; Guttman, M.; Hazan, R.B. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2002, 2, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.T.; Tsunematsu, T.; Yanagisawa, S.; Kudo, Y.; Miyauchi, M.; Kamata, N.; Takata, T. The FGFR1 inhibitor PD173074 induces mesenchymal-epithelial transition through the transcription factor AP-1. Br. J. Cancer 2013, 109, 2248–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Mège, R.M. N-Cadherin and Fibroblast Growth Factor Receptors crosstalk in the control of developmental and cancer cell migrations. Eur. J. Cell Biol. 2016, 95, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ji, W.; Yu, Y.; Li, Z.; Niu, X.; Xia, W.; Lu, S. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial–mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene 2018, 37, 5340–5354. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ge, X.; Zhang, W.; Ding, P.; Du, Y.; Wang, Q.; Li, L.; Fang, L.; Sun, Y.; Zhang, P.; et al. PI3K/AKT inhibition reverses R-CHOP resistance by destabilizing SOX2 in diffuse large B cell lymphoma. Theranostics 2020, 10, 3151–3163. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, M.; Ishiwata, T.; Matsuda, K.; Lopez, M.E.; Fukahi, K.; Asano, G.; Beger, H.G.; Korc, M. IIIc isoform of fibroblast growth factor receptor 1 is overexpressed in human pancreatic cancer and enhances tumorigenicity of hamster ductal cells. Gastroenterology 2002, 123, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brennan, J.P.; Slavin, J.L.; Blick, T.; Thompson, E.W.; Williams, E.D. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: Role of fibroblast growth factor receptor-2. Cancer Res. 2006, 66, 11271–11278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri, D.; Rosato, B.; Nanni, M.; Magenta, A.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget 2016, 7, 5440–5460. [Google Scholar] [CrossRef] [Green Version]
- Ishiwata, T. Role of fibroblast growth factor receptor-2 splicing in normal and cancer cells. Front. Biosci. Landmark 2018, 23, 626–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Ingram, L.; Kim, S.; Beharry, Z.; Cooper, J.A.; Cai, H. Paracrine Fibroblast Growth Factor Initiates Oncogenic Synergy with Epithelial FGFR/Src Transformation in Prostate Tumor Progression. Neoplasia 2018, 20, 233–243. [Google Scholar] [CrossRef]
- Hopkins, A.; Coatham, M.L.; Berry, F.B. FOXC1 regulates FGFR1 isoform switching to promote invasion following TGFβ-induced EMT. Mol. Cancer Res. 2017, 15, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Shoji, K.; Teishima, J.; Hayashi, T.; Ohara, S.; McKeehan, W.L.; Matsubara, A. Restoration of fibroblast growth factor receptor 2IIIb enhances the chemosensitivity of human prostate cancer cells. Oncol. Rep. 2014, 32, 65–70. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z. De Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsen, K.R.; Demuth, C.; Madsen, A.T.; Hussmann, D.; Vad-Nielsen, J.; Nielsen, A.L.; Sorensen, B.S. MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells. Oncogenesis 2017, 6, e307. [Google Scholar] [CrossRef]
- Azuma, K.; Kawahara, A.; Sonoda, K.; Nakashima, K.; Tashiro, K.; Watari, K.; Izumi, H.; Kage, M.; Kuwano, M.; Ono, M.; et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 2014, 5, 5908–5919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, A.; Akhand, S.S.; Paez, J.S.P.; Brown, W.; Pan, L.; Libring, S.; Badamy, M.; Dykuizen, E.; Solorio, L.; Andy Tao, W.; et al. Epigenetic targeting of neuropilin-1 prevents bypass signaling in drug-resistant breast cancer. Oncogene 2021, 40, 322–333. [Google Scholar] [CrossRef]
- Miura, K.; Oba, T.; Hamanaka, K.; Ito, K. ichi FGF2-FGFR1 pathway activation together with thymidylate synthase upregulation is induced in pemetrexed-resistant lung cancer cells. Oncotarget 2019, 10, 1171–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, W.S.; Akhand, S.S.; Wendt, M.K. FGFR signaling maintains a drug persistent cell population following epithelial-mesenchymal transition. Oncotarget 2016, 7, 83424–83436. [Google Scholar] [CrossRef] [Green Version]
- Koinis, F.; Corn, P.; Parikh, N.; Song, J.; Vardaki, I.; Mourkioti, I.; Lin, S.H.; Logothetis, C.; Panaretakis, T.; Gallick, G. Resistance to MET/VEGFR2 inhibition by cabozantinib is mediated by YAP/TBX5-dependent induction of FGFR1 in castration-resistant prostate cancer. Cancers 2020, 12, 244. [Google Scholar] [CrossRef] [Green Version]
- Spinola, M.; Leoni, V.P.; Tanuma, J.I.; Pettinicchio, A.; Frattini, M.; Signoroni, S.; Agresti, R.; Giovanazzi, R.; Pilotti, S.; Bertario, L.; et al. FGFR4 Gly388Arg polymorphism and prognosis of breast and colorectal cancer. Oncol. Rep. 2005, 14, 415–419. [Google Scholar] [CrossRef]
- Marmé, F.; Werft, W.; Benner, A.; Burwinkel, B.; Sinn, P.; Sohn, C.; Lichter, P.; Hahn, M.; Schneeweiss, A. FGFR4 Arg388 genotype is associated with pathological complete response to neoadjuvant chemotherapy for primary breast cancer. Ann. Oncol. 2010, 21, 1636–1642. [Google Scholar] [CrossRef]
- Thussbas, C.; Nahrig, J.; Streit, S.; Bange, J.; Kriner, M.; Kates, R.; Ulm, K.; Kiechle, M.; Hoefler, H.; Ullrich, A.; et al. FGFR4 Arg388 allele is associated with resistance to adjuvant therapy in primary breast cancer. J. Clin. Oncol. 2006, 24, 3747–3755. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Hong, C.S.; Kim, H.N.; Shin, M.H.; Kim, K.R.; Shim, H.J.; Hwang, J.E.; Bae, W.K.; Chung, I.J. FGFR4 Arg388 is correlated with poor survival in resected colon cancer promoting epithelial to mesenchymal transition. Cancer Res. Treat. 2017, 49, 766–777. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Li, J.; Jiang, D.; Li, J.; Xiao, C.; Li, Y.; Han, C.; Zhao, C. FGFR4 Gly388Arg polymorphism affects the progression of gastric cancer by activating STAT3 pathway to induce epithelial to mesenchymal transition. Cancer Res. Treat. 2020, 52, 1162–1177. [Google Scholar] [CrossRef] [PubMed]
- Ansell, A.; Farnebo, L.; Grénman, R.; Roberg, K.; Thunell, L.K. Polymorphism of FGFR4 in cancer development and sensitivity to cisplatin and radiation in head and neck cancer. Oral Oncol. 2009, 45, 23–29. [Google Scholar] [CrossRef]
- Marmé, F.; Hielscher, T.; Hug, S.; Bondong, S.; Zeillinger, R.; Castillo-Tong, D.C.; Sehouli, J.; Braicu, I.; Vergote, I.; Isabella, C.; et al. Fibroblast growth factor receptor 4 gene (FGFR4) 388Arg allele predicts prolonged survival and platinum sensitivity in advanced ovarian cancer. Int. J. Cancer 2012, 131, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Müller, S.; Gärtner, S.; Sures, I.; et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg388 allele. Cancer Res. 2002, 62, 840–847. [Google Scholar] [PubMed]
- Quintanal-Villalonga, Á.; Ojeda-Márquez, L.; Marrugal, Á.; Yagüe, P.; Ponce-Aix, S.; Salinas, A.; Carnero, A.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. The FGFR4-388arg Variant Promotes Lung Cancer Progression by N-Cadherin Induction. Sci. Rep. 2018, 8, 2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, S.B.; Reyes, S.; Du, M.; Gireud, M.; Zhang, L.; Woodfield, S.E.; Ittmann, M.; Scheurer, M.E.; Bean, A.J.; Zage, P.E. A polymorphism in the FGFR4 gene is associated with risk of neuroblastoma and altered receptor degradation. J. Pediatr. Hematol. Oncol. 2016, 38, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, N.; Varjosalo, M.; Meller, P.; Lohi, J.; Chan, K.M.; Zhou, Z.; Alitalo, K.; Taipale, J.; Keski-Oja, J.; Lehti, K. FGF receptor-4 (FGFR4) polymorphism acts as an activity switch of a membrane type 1 matrix metalloproteinase—FGFR4 complex. Proc. Natl. Acad. Sci. USA 2010, 107, 15786–15791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, V.; Zhang, K.; Savadelis, A.; Zmina, P.; Aguila, B.; Welford, S.M.; Abdul-Karim, F.; Bonk, K.W.; Keri, R.A.; Bedogni, B. The membrane tethered matrix metalloproteinase MT1-MMP triggers an outside-in DNA damage response that impacts chemo- and radiotherapy responses of breast cancer. Cancer Lett. 2019, 443, 115–124. [Google Scholar] [CrossRef]
- Udayakumar, T.S.; Nagle, R.B.; Bowden, G.T. Fibroblast Growth Factor-I Transcriptionally Induces Membrane Type-I Matrix Metalloproteinase Expression in Prostate Carcinoma Cell Line. Prostate 2004, 58, 66–75. [Google Scholar] [CrossRef]
- Nomura, S.; Yoshitomi, H.; Takano, S.; Shida, T.; Kobayashi, S.; Ohtsuka, M.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Kato, A.; et al. FGF10/FGFR2 signal induces cell migration and invasion in pancreatic cancer. Br. J. Cancer 2008, 99, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, E.; Valacca, C.; Mignatti, P. Membrane-type 1 matrix metalloproteinase downregulates fibroblast growth factor-2 binding to the cell surface and intracellular signaling. J. Cell. Physiol. 2015, 230, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Zhang, T.; Cheng, P.; Li, D.; Ogorodniitchouk, O.; Lahmamssi, C.; Wang, G.; Lan, M. Therapeutic implications of fibroblast growth factor receptor inhibitors in a combination regimen for solid tumors (Review). Oncol. Lett. 2020, 20, 2525–2536. [Google Scholar] [CrossRef] [PubMed]
- Repetto, M.; Crimini, E.; Giugliano, F.; Morganti, S.; Belli, C.; Curigliano, G. Selective FGFR/FGF pathway inhibitors: Inhibition strategies, clinical activities, resistance mutations, and future directions. Expert Rev. Clin. Pharmacol. 2021, 14, 1233–1252. [Google Scholar] [CrossRef]
- Yue, S.; Li, Y.; Chen, X.; Wang, J.; Li, M.; Chen, Y.; Wu, D. FGFR-TKI resistance in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [Green Version]
- Chua, V.; Orloff, M.; Teh, J.L.; Sugase, T.; Liao, C.; Purwin, T.J.; Lam, B.Q.; Terai, M.; Ambrosini, G.; Carvajal, R.D.; et al. Stromal fibroblast growth factor 2 reduces the efficacy of bromodomain inhibitors in uveal melanoma. EMBO Mol. Med. 2019, 11, e9081. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Morishima, K.; Ui, T.; Hoshino, H.; Matsubara, D.; Ishikawa, S.; Aburatani, H.; Fukayama, M.; Hosoya, Y.; Sata, N.; et al. The role of HGF/MET and FGF/FGFR in fibroblast-derived growth stimulation and lapatinib-resistance of esophageal squamous cell carcinoma. BMC Cancer 2015, 15, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, O.E.; Latigo, J.; Jeffery, R.E.; Nye, E.; Poulsom, R.; Spencer-Dene, B.; Lemoine, N.R.; Stamp, G.W.; Aboagye, E.O.; Seckl, M.J. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009, 69, 8645–8651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byron, S.A.; Loch, D.C.; Pollock, P.M. Fibroblast growth factor receptor inhibition synergizes with paclitaxel and doxorubicin in endometrial cancer cells. Int. J. Gynecol. Cancer 2012, 22, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Campbell, T.M.; Castro, M.A.A.; de Oliveira, K.G.; Ponder, B.A.J.; Meyer, K.B. Era binding by transcription factors NFIB and YBX1 enables FGFR2 signaling to modulate estrogen responsiveness in breast cancer. Cancer Res. 2018, 78, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; Tiwari, A.K.; Chufan, E.E.; Sodani, K.; Anreddy, N.; Singh, S.; Ambudkar, S.V.; Stephani, R.; Chen, Z.S. PD173074, a selective FGFR inhibitor, reverses ABCB1-mediated drug resistance in cancer cells. Cancer Chemother. Pharmacol. 2013, 72, 189–199. [Google Scholar] [CrossRef]
- Anreddy, N.; Patel, A.; Sodani, K.; Kathawala, R.J.; Chen, E.P.; Wurpel, J.N.D.; Chen, Z.-S. PD173074, a selective FGFR inhibitor, reverses MRP7 (ABCC10)-mediated MDR. Acta Pharm. Sin. B 2014, 4, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.P.; Hung, T.H.; Hsiao, S.H.; Huang, Y.H.; Hung, L.C.; Yu, Y.J.; Chang, Y.T.; Wang, S.P.; Wu, Y.S. Erdafitinib resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to cytotoxic anticancer drugs. Cancers 2020, 12, 1366. [Google Scholar] [CrossRef]
- Feng, W.; Zhang, M.; Wu, Z.X.; Wang, J.Q.; Dong, X.D.; Yang, Y.; Teng, Q.X.; Chen, X.Y.; Cui, Q.; Yang, D.H. Erdafitinib Antagonizes ABCB1-Mediated Multidrug Resistance in Cancer Cells. Front. Oncol. 2020, 10, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.J.; Choi, J.H.; Park, I.C.; Kim, C.H.; An, S.K.; Kim, T.J.; Lee, J.H. Selective FGFR inhibitor BGJ398 inhibits phosphorylation of AKT and STAT3 and induces cytotoxicity in sphere-cultured ovarian cancer cells. Int. J. Oncol. 2017, 50, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberti, D.; Cristinziano, G.; Porru, M.; Leonetti, C.; Egan, J.B.; Shi, C.X.; Buglioni, S.; Amoreo, C.A.; Castellani, L.; Borad, M.J.; et al. HSP90 Inhibition Drives Degradation of FGFR2 Fusion Proteins: Implications for Treatment of Cholangiocarcinoma. Hepatology 2019, 69, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ding, X.; Wang, S.; Moser, C.D.; Shaleh, H.M.; Mohamed, E.A.; Chaiteerakij, R.; Allotey, L.K.; Chen, G.; Miyabe, K.; et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett. 2016, 380, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyulyandina, A.; Harrison, D.; Yin, W.; Stepanova, E.; Kochenkov, D.; Solomko, E.; Peretolchina, N.; Daeyaert, F.; Joos, J.B.; Van Aken, K.; et al. Alofanib, an allosteric FGFR2 inhibitor, has potent effects on ovarian cancer growth in preclinical studies. Investig. New Drugs 2017, 35, 127–133. [Google Scholar] [CrossRef]
- Sergei, B.; Pavel, D.; Aigul, G.; Firyuza, B.; Ilmira, N.; Ilshat, M.; Aida, A.; Refat, K.; Natalia, A.; Elena, S.; et al. Inhibition of FGFR2-signaling attenuates a homology-mediated dna repair in gist and sensitizes them to DNA-topoisomerase II inhibitors. Int. J. Mol. Sci. 2020, 21, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Qiu, H.; Yashiro, M.; Zhang, X.; Miwa, A.; Hirakawa, K. A FGFR2 inhibitor, Ki23057, enhances the chemosensitivity of drug-resistant gastric cancer cells. Cancer Lett. 2011, 307, 47–52. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Wilson, K.; Thayer, S.; Zanghi, J.; Gemo, A.T.; Kavanaugh, W.M.; Keer, H.N.; LoRusso, P.M. A phase I, first in human study of FP-1039 (GSK3052230), a novel FGF ligand trap, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 526–532. [Google Scholar] [CrossRef]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, S.; Yang, F.; Au, J.L.S.; Guillaume Wientjes, M. Nontoxic doses of suramin enhance activity of doxorubicin in prostate tumors. J. Pharmacol. Exp. Ther. 2001, 299, 426–433. [Google Scholar]
- Wu, Z.S.; Liu, C.F.; Fu, B.; Chou, R.H.; Yu, C. Suramin blocks interaction between human FGF1 and FGFR2 D2 domain and reduces downstream signaling activity. Biochem. Biophys. Res. Commun. 2016, 477, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Villalona-Calero, M.A.; Otterson, G.A.; Wientjes, M.G.; Weber, F.; Bekaii-Saab, T.; Young, D.; Murgo, A.J.; Jensen, R.; Yeh, T.K.; Wei, Y.; et al. Noncytotoxic suramin as a chemosensitizer in patients with advanced non-small-cell lung cancer: A phase II study. Ann. Oncol. 2008, 19, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Magee, P.; Shi, L.; Garofalo, M. Role of microRNAs in chemoresistance. Ann. Transl. Med. 2015, 3, 332. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Xie, W.; Yang, X.; Xia, N.; Yang, K. Inhibiting microRNA-449 Attenuates Cisplatin-Induced Injury in NRK-52E Cells Possibly via Regulating the SIRT1/P53/BAX Pathway. Med. Sci. Monit. 2016, 22, 818–823. [Google Scholar] [CrossRef]
- Hu, Y.; Qiu, Y.; Yagüe, E.; Ji, W.; Liu, J.; Zhang, J. MiRNA-205 targets VEGFA and FGF2 and regulates resistance to chemotherapeutics in breast cancer. Cell Death Dis. 2016, 7, e2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A phase Ib open-label multicenter study of AZD4547 in patients with advanced squamous cell lung cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogova, L.; Sequist, L.V.; Garcia, J.M.P.; Andre, F.; Delord, J.P.; Hidalgo, M.; Schellens, J.H.M.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast growth factor receptor 1-3 kinase inhibitor, in patientswith advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: Results of a global phase I, dose-escalation and dose-expansion stud. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Chen, H.; Wortmann, A.; Loch, D.; Gartside, M.G.; Dehkhoda, F.; Blais, S.P.; Neubert, T.A.; Mohammadi, M.; Pollock, P.M. The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia 2013, 15, 975–988. [Google Scholar] [CrossRef] [Green Version]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Chen, X.; Chen, D.; Xia, Q.; Liu, Z.; Li, F.; Yan, Y.; Cai, Y. Insight into ponatinib resistance mechanisms in rhabdomyosarcoma caused by the mutations in FGFR4 tyrosine kinase using molecular modeling strategies. Int. J. Biol. Macromol. 2019, 135, 294–302. [Google Scholar] [CrossRef]
- Ryan, M.R.; Sohl, C.D.; Luo, B.; Anderson, K.S. The FGFR1 V561M gatekeeper mutation drives AZD4547 resistance through STAT3 activation and EMT. Mol. Cancer Res. 2019, 17, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Tang, X.; Guo, J.; He, R.; Chan, S.; Song, X.; Tu, Z.; Wang, Y.; Ren, X.; Ding, K.; et al. GZD824 overcomes FGFR1-V561F/M mutant resistance in vitro and in vivo. Cancer Med. 2021, 10, 4874–4884. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krook, M.A.; Bonneville, R.; Chen, H.Z.; Reeser, J.W.; Wing, M.R.; Martin, D.M.; Smith, A.M.; Dao, T.; Samorodnitsky, E.; Paruchuri, A.; et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb. Mol. Case Stud. 2019, 5, a004002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, L.; Shi, L.; Liu, L.Y.; de la Cruz, F.F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 overcomes resistance to atp-competitive FGFR inhibitors in patients with FGFR2 fusion–positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Ahn, T.; Bang, H.; Ham, J.S.; Kim, J.; Kim, S.T.; Jang, J.; Shim, M.; Kang, S.Y.; Park, S.H.; et al. Acquired resistance to LY2874455 in FGFR2-amplified gastric cancer through an emergence of novel FGFR2-ACSL5 fusion. Oncotarget 2017, 8, 15014–15022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latko, M.; Czyrek, A.; Porębska, N.; Kucińska, M.; Otlewski, J.; Zakrzewska, M.; Opaliński, Ł. Cross-Talk between Fibroblast Growth Factor Receptors and Other Cell Surface Proteins. Cells 2019, 8, 455. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Watanabe, K.; Kita, K.; Kitai, H.; Kotani, H.; Sato, Y.; Inase, N.; Yano, S.; Ebi, H. Resistance mediated by alternative receptor tyrosine kinases in FGFR1-amplified lung cancer. Carcinogenesis 2017, 38, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Damodaran, S.; Parks, H.; Ocrainiciuc, C.; Miya, J.; Yu, L.; Gardner, E.P.; Samorodnitsky, E.; Wing, M.R.; Bhatt, D.; et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol. Cancer Ther. 2017, 16, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, S.; Zhang, Z.; Liu, X.; Wu, Z.; Geng, R.; Ge, X.; Dai, C.; Liu, R.; Zhang, Q.; et al. Multiple receptor tyrosine kinase activation attenuates therapeutic efficacy of the fibroblast growth factor receptor 2 inhibitor AZD4547 in FGFR2 amplified gastric cancer. Oncotarget 2015, 6, 2009–2022. [Google Scholar] [CrossRef] [Green Version]
- Prawira, A.; Le, T.B.U.; Ho, R.Z.W.; Huynh, H. Upregulation of the ErbB family by EZH2 in hepatocellular carcinoma confers resistance to FGFR inhibitor. J. Cancer Res. Clin. Oncol. 2021, 147, 2955–2968. [Google Scholar] [CrossRef]
- Wang, J.; Mikse, O.; Liao, R.G.; Li, Y.; Tan, L.; Janne, P.A.; Gray, N.S.; Wong, K.K.; Hammerman, P.S. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene 2014, 34, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Shen, J.; Xiang, J.; Li, H.; Li, B.; Zhang, T.; Zhang, L.; Mao, X.; Jian, H.; Shu, Y. Characterization of acquired receptor tyrosine–kinase fusions as mechanisms of resistance to EGFR tyrosine–kinase inhibitors. Cancer Manag. Res. 2019, 11, 6343–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Li, D.; Zheng, M.; Chen, B.; Wei, T.; Wang, Y.; Li, M.; Huang, W.; Tong, Q.; Wang, Q.; et al. FGFRL1 affects chemoresistance of small-cell lung cancer by modulating the PI3K/Akt pathway via ENO1. J. Cell. Mol. Med. 2020, 24, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.N.; Altamentova, S.M.; Kilkenny, D.M.; Rocheleau, J.V. Fibroblast Growth Factor Receptor Like-1 (FGFRL1) interacts with SHP-1 phosphatase at insulin secretory granules and induces beta-cell ERK1/2 protein activation. J. Biol. Chem. 2013, 288, 17859–17870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Hu, S.; Li, X.; Yan, S.; Xing, S.; Li, J.; Tan, H. MiRNA-495 inhibits cell proliferation and invasion abilities in gastric cancer cells by down-regulation of FGFRL1. Int. J. Clin. Exp. Pathol. 2016, 9, 7867–7877. [Google Scholar]
- Memarzadeh, S.; Cai, H.; Janzen, D.M.; Xin, L.; Lukacs, R.; Riedinger, M.; Zong, Y.; DeGendt, K.; Verhoeven, G.; Huang, J.; et al. Role of autonomous androgen receptor signaling in prostate cancer initiation is dichotomous and depends on the oncogenic signal. Proc. Natl. Acad. Sci. USA 2011, 108, 7962–7967. [Google Scholar] [CrossRef] [Green Version]
- Fenig, E.; Livnat, T.; Sharkon-Polak, S.; Wasserman, L.; Beery, E.; Lilling, G.; Yahalom, J.; Wieder, R.; Nordenberg, J. Basic fibroblast growth factor potentiates cisplatinum-induced cytotoxicity in MCF-7 human breast cancer cells. J. Cancer Res. Clin. Oncol. 1999, 125, 556–562. [Google Scholar] [CrossRef]
- Wang, Q.; Maloof, P.; Wang, H.; Fenig, E.; Stein, D.; Nichols, G.; Denny, T.N.; Yahalom, J.; Wieder, R. Basic Fibroblast Growth Factor Downregulates Bcl-2 and Promotes Apoptosis in MCF-7 Human Breast Cancer Cells. Exp. Cell Res. 1998, 238, 177–187. [Google Scholar] [CrossRef]
- Coleman, A.B.; Metz, M.Z.; Donohue, C.A.; Schwarz, R.E.; Kane, S.E. Chemosensitization by fibroblast growth factor-2 is not dependent upon proliferation, S-phase accumulation, or p53 status. Biochem. Pharmacol. 2002, 64, 1111–1123. [Google Scholar] [CrossRef]
- Im, Y.S.; Shin, H.K.; Kim, H.R.; Jeong, S.H.; Kim, S.R.; Kim, Y.M.; Lee, D.H.; Jeon, S.H.; Lee, H.W.; Choi, J.K. Enhanced cytotoxicity of 5-FU by bFGF through up-regulation of uridine phosphorylase 1. Mol. Cells 2009, 28, 119–124. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, C.; Lu, W.; Xie, R.; Jin, C.; Huang, P.; Wang, F.; McKeehan, W.L. Metabolic Regulator βKlotho Interacts with Fibroblast Growth Factor Receptor 4 (FGFR4) to Induce Apoptosis and Inhibit Tumor Cell Proliferation. J. Biol. Chem. 2010, 285, 30069–30078. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, H.; Ding, S.; Qi, S.; Liu, S.; Sun, D.; Dong, W.; Yin, L.; Li, M.; Zhao, X.; et al. βklotho inhibits androgen/androgen receptor-associated epithelial-mesenchymal transition in prostate cancer through inactivation of ERK1/2 signaling. Oncol. Rep. 2018, 40, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Massabeau, C.; Sigal-Zafrani, B.; Belin, L.; Savignoni, A.; Richardson, M.; Kirova, Y.M.; Cohen-Jonathan-Moyal, E.; Mégnin-Chanet, F.; Hall, J.; Fourquet, A. The fibroblast growth factor receptor 1 (FGFR1), a marker of response to chemoradiotherapy in breast cancer? Breast Cancer Res. Treat. 2012, 134, 259–266. [Google Scholar] [CrossRef]
- Turkowski, K.; Herzberg, F.; Günther, S.; Brunn, D.; Weigert, A.; Meister, M.; Muley, T.; Kriegsmann, M.; Schneider, M.A.; Winter, H.; et al. Fibroblast Growth Factor-14 Acts as Tumor Suppressor in Lung Adenocarcinomas. Cells 2020, 9, 1755. [Google Scholar] [CrossRef]
- Sochacka, M.; Opalinski, L.; Szymczyk, J.; Zimoch, M.B.; Czyrek, A.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. FHF1 is a bona fide fibroblast growth factor that activates cellular signaling in FGFR-dependent manner. Cell Commun. Signal. 2020, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, P.; Martínez-Bosch, N.; Blidner, A.G.; Rabinovich, G.A. Impact of galectins in resistance to anticancer therapies. Clin. Cancer Res. 2020, 26, 6086–6101. [Google Scholar] [CrossRef]
- Markowska, A.I.; Liu, F.T.; Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucińska, M.; Porȩbska, N.; Lampart, A.; Latko, M.; Knapik, A.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Differential regulation of fibroblast growth factor receptor 1 trafficking and function by extracellular galectins. Cell Commun. Signal. 2019, 17, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porębska, N.; Poźniak, M.; Matynia, A.; Żukowska, D.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Galectins as modulators of receptor tyrosine kinases signaling in health and disease. Cytokine Growth Factor Rev. 2021, 60, 89–106. [Google Scholar] [CrossRef]
- Liang, Q.; Wang, J.; Zhao, L.; Hou, J.; Hu, Y.; Shi, J. Recent advances of dual FGFR inhibitors as a novel therapy for cancer. Eur. J. Med. Chem. 2021, 214, 113205. [Google Scholar] [CrossRef]
- Singleton, K.R.; Hinz, T.K.; Kleczko, E.K.; Marek, A.L.; Kwak, J.; Harp, T.; Kim, J.; Tan, A.C.; Heasley, L.E. Kinome RNAi screens reveal synergistic targeting of MTOR and FGFR1 Pathways for treatment of lung cancer and hnscc. Cancer Res. 2015, 75, 4398–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, L.M.; Geng, X.; Bonazzi, V.F.; Ju, R.J.; Mahon, C.E.; Cummings, M.C.; Stephenson, S.A.; Pollock, P.M. PI3K inhibitors synergize with FGFR inhibitors to enhance antitumor responses in FGFR2mutant endometrial cancers. Mol. Cancer Ther. 2017, 16, 637–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadone-Montaudié, B.; Laroche-Clary, A.; Mongis, A.; Chamorey, E.; Di Mauro, I.; Chaire, V.; Finetti, P.; Schiappa, R.; Le Loarer, F.; Birtwisle-Peyrottes, I.; et al. Novel Therapeutic Insights in Dedifferentiated Liposarcoma: A Role for FGFR and MDM2 Dual Targeting. Cancers 2020, 12, 3058. [Google Scholar] [CrossRef]
- Guo, T.; Gu, C.; Li, B.; Xu, C. Dual inhibition of FGFR4 and BCL-xL inhibits multi-resistant ovarian cancer with BCL2L1 gain. Aging 2021, 13, 19750–19759. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.; Bamodu, O.A.; Chen, J.H.; Wu, A.T.; Lee, W.H.; Chao, T.Y.; Yeh, C.T. Targeted PARP Inhibition Combined with FGFR1 Blockade is Synthetically Lethal to Malignant Cells in Patients with Pancreatic Cancer. Cells 2020, 9, 911. [Google Scholar] [CrossRef] [Green Version]
- Theocharopoulos, C.; Lialios, P.P.; Samarkos, M.; Gogas, H.; Ziogas, D.C. Antibody-drug conjugates: Functional principles and applications in oncology and beyond. Vaccines 2021, 9, 1111. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Puthenveetil, S.; Musto, S.; Loganzo, F.; Tumey, L.N.; O’Donnell, C.J.; Graziani, E. Development of Solid-Phase Site-Specific Conjugation and Its Application toward Generation of Dual Labeled Antibody and Fab Drug Conjugates. Bioconjug. Chem. 2016, 27, 1030–1039. [Google Scholar] [CrossRef]
- Świderska, K.W.; Szlachcic, A.; Opaliński, Ł.; Zakrzewska, M.; Otlewski, J. FGF2 dual warhead conjugate with monomethyl auristatin E and α-amanitin displays a cytotoxic effect towards cancer cells overproducing FGF receptor 1. Int. J. Mol. Sci. 2018, 19, 2098. [Google Scholar] [CrossRef] [Green Version]
- Schmid, S.L. Reciprocal regulation of signaling and endocytosis: Implications for the evolving cancer cell. J. Cell Biol. 2017, 216, 2623–2632. [Google Scholar] [CrossRef] [Green Version]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of endocytosis mechanisms for the receptors targeted by the currently approved adcs—A necessity for future adc research and development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef]
- Muley, H.; Fadó, R.; Rodríguez-Rodríguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Pharmacol. 2020, 177, 113959. [Google Scholar] [CrossRef] [PubMed]
- Pozniak, M.; Sokolowska-Wedzina, A.; Jastrzebski, K.; Szymczyk, J.; Porebska, N.; Krzyscik, M.A.; Zakrzewska, M.; Miaczynska, M.; Otlewski, J.; Opalinski, L. FGFR1 clustering with engineered tetravalent antibody improves the efficiency and modifies the mechanism of receptor internalization. Mol. Oncol. 2020, 14, 1998–2021. [Google Scholar] [CrossRef] [PubMed]





| Cancer Type | Drug | Involved Protein(s) | References |
|---|---|---|---|
| Breast cancer | Etoposide | FGF2 | [30] |
| 5-fluorouracil | |||
| Mifepristone, Telepristone | [50] | ||
| Paclitaxel | [31,33] | ||
| Tamoxifen | FGF1, FGFR2 | [48,51] | |
| Trastuzumab | FGFR4 | [49] | |
| FGF4 | [43] | ||
| Lapatinib | |||
| Colorectal cancer | 5-fluorouracil | FGFR4 | [52] |
| Irinotecan | FGF2, FGF9 | [37] | |
| Liver cancer | Sorafenib | FGF19, FGFR4 | [39] |
| FGF9 | [42] | ||
| Head and neck cancer | Paclitaxel | FGF2 | [31] |
| Cisplatin | FGF2, FGFR2 | [35] | |
| Bevacizumab | FGF2, FGFR3 | [53] | |
| Lung cancer | Gefitinib | FGFR1 | [54] |
| Cisplatin | FGF2 | [36] | |
| Erlotinib | FGFR1 | [45] | |
| Bladder cancer | Cisplatin | FGF2 | [27] |
| Paclitaxel | [31] | ||
| Prostate cancer | Paclitaxel | FGF2 | [31] |
| FGF1, FGF2 | [29] | ||
| Doxorubicin | |||
| 5-fluorouracil | |||
| Blood cancer | Cytarabine | FGF2, FGFR1 | [55] |
| Etoposide | |||
| Fludarabine | FGF2 | [26] | |
| Ovarian and cervical cancer | Paclitaxel | FGF2 | [31] |
| Etoposide | FGF1 | [56] | |
| Cisplatin | [32] | ||
| FGF13 | [38] | ||
| Brain cancer | Temozolomide | FGFR1 | [57] |
| FGF2 | [58] | ||
| Melanoma | Paclitaxel | FGF2 | [34] |
| Cisplatin | |||
| Vemurafenib | FGFR3 | [47] | |
| Bone cancer | Doxorubicin | FGF2 | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymczyk, J.; Sluzalska, K.D.; Materla, I.; Opalinski, L.; Otlewski, J.; Zakrzewska, M. FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers 2021, 13, 5796. https://doi.org/10.3390/cancers13225796
Szymczyk J, Sluzalska KD, Materla I, Opalinski L, Otlewski J, Zakrzewska M. FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers. 2021; 13(22):5796. https://doi.org/10.3390/cancers13225796
Chicago/Turabian StyleSzymczyk, Jakub, Katarzyna Dominika Sluzalska, Izabela Materla, Lukasz Opalinski, Jacek Otlewski, and Malgorzata Zakrzewska. 2021. "FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance" Cancers 13, no. 22: 5796. https://doi.org/10.3390/cancers13225796
APA StyleSzymczyk, J., Sluzalska, K. D., Materla, I., Opalinski, L., Otlewski, J., & Zakrzewska, M. (2021). FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers, 13(22), 5796. https://doi.org/10.3390/cancers13225796