
Rational Development of Liquid Biopsy Analysis in Renal Cell Carcinoma

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
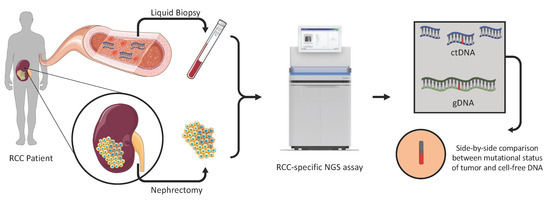
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animal Models of RCC
2.3. Collection of Blood Samples
2.4. Isolation of EV DNA from Blood Samples
2.5. Digital Droplet PCR (ddPCR)
2.6. Isolation of Genomic and Soluble Cell-Free DNA
2.7. Targeted Sequencing
2.8. Synthetic cfDNA Library Preparation
2.9. Bioinformatic Analysis
2.10. Statistical Analysis
3. Results
3.1. Characteristics of the ctDNA Repertoire in RCC Xenografts
3.2. Development of the RCC-Appropriate Targeted NGS Assay
3.3. Optimization of the NGS Assay for ctDNA Analysis
3.4. Assay Performance in RCC Liquid Biopsies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; Garcia-Carbonero, R.; Karthaus, M.; Smith, D.; Tabernero, J.; Van Cutsem, E.; Guan, X.; Boedigheimer, M.; Ang, A.; et al. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann. Oncol. 2018, 29, 119–126. [Google Scholar] [CrossRef]
- Pal, S.K.; Sonpavde, G.; Agarwal, N.; Vogelzang, N.J.; Srinivas, S.; Haas, N.B.; Signoretti, S.; McGregor, B.A.; Jones, J.; Lanman, R.B.; et al. Evolution of Circulating Tumor DNA Profile from First-line to Subsequent Therapy in Metastatic Renal Cell Carcinoma. Eur. Urol. 2017, 72, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.W.; Gill, D.M.; Maughan, B.; Agarwal, A.; Arjyal, L.; Gupta, S.; Streeter, J.; Bailey, E.; Pal, S.K.; Agarwal, N. Correlation of genomic alterations assessed by next-generation sequencing (NGS) of tumor tissue DNA and circulating tumor DNA (ctDNA) in metastatic renal cell carcinoma (mRCC): Potential clinical implications. Oncotarget 2017, 8, 33614–33620. [Google Scholar] [CrossRef] [Green Version]
- Riazalhosseini, Y.; Lathrop, M. Precision medicine from the renal cancer genome. Nat. Rev. Nephrol. 2016, 12, 655–666. [Google Scholar] [CrossRef]
- Rak, J. Extracellular vesicles—Biomarkers and effectors of the cellular interactome in cancer. Front Pharmacol. 2013, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Lázaro-Ibáñez, E.; Sanz-Garcia, A.; Visakorpi, T.; Escobedo-Lucea, C.; Siljander, P.; Ayuso-Sacido, A.; Yliperttula, M. Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: Apoptotic bodies, microvesicles, and exosomes. Prostate 2014, 74, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [Green Version]
- In ’t Veld, S.; Wurdinger, T. Tumor-educated platelets. Blood 2019, 133, 2359–2364. [Google Scholar] [CrossRef]
- Chennakrishnaiah, S.; Meehan, B.; D’Asti, E.; Montermini, L.; Lee, T.H.; Karatzas, N.; Buchanan, M.; Tawil, N.; Choi, D.; Divangahi, M.; et al. Leukocytes as a reservoir of circulating oncogenic DNA and regulatory targets of tumor-derived extracellular vesicles. J. Thromb. Haemost. 2018, 16, 1800–1813. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Uemura, M.; Fujita, M.; Maejima, K.; Koh, Y.; Matsushita, M.; Nakano, K.; Hayashi, Y.; Wang, C.; Ishizuya, Y.; et al. Clinical significance of the mutational landscape and fragmentation of circulating tumor DNA in renal cell carcinoma. Cancer Sci. 2019, 110, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.T.; Choi, Y.L.; Yun, J.W.; Kim, N.K.D.; Kim, S.Y.; Jeon, H.J.; Nam, J.Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat. Commun. 2017, 8, 1377. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Tyagi, M.; Vallania, F.; Bredemeyer, A.J.; Pfeifer, J.D.; Mitra, R.D.; Duncavage, E.J. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J. Mol. Diagn. 2014, 16, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Tracz, A.; Mastri, M.; Lee, C.R.; Pili, R.; Ebos, J.M. Modeling spontaneous metastatic renal cell carcinoma (mRCC) in mice following nephrectomy. J. Vis. Exp. 2014, 1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, L.R.; Pauley, R.J.; Santner, S.J.; Heppner, G.H.; Heng, H.H.; Rak, J.W.; Miller, F.R. Dynamic stromal-epithelial interactions during progression of MCF10DCIS.com xenografts. Int. J. Cancer 2007, 120, 2127–2134. [Google Scholar] [CrossRef]
- Jedeszko, C.; Paez-Ribes, M.; Di Desidero, T.; Man, S.; Lee, C.R.; Xu, P.; Bjarnason, G.A.; Bocci, G.; Kerbel, R.S. Postsurgical adjuvant or metastatic renal cell carcinoma therapy models reveal potent antitumor activity of metronomic oral topotecan with pazopanib. Sci. Transl. Med. 2015, 7, 282. [Google Scholar] [CrossRef]
- Bourgey, M.; Dali, R.; Eveleigh, R.; Chen, K.C.; Letourneau, L.; Fillon, J.; Michaud, M.; Caron, M.; Sandoval, J.; Lefebvre, F.; et al. GenPipes: An open-source framework for distributed and scalable genomic analyses. Gigascience 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FulcrumGenomics. Available online: https://github.com/fulcrumgenomics/fgbio (accessed on 1 September 2020).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome. Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broad Institute. Picard Toolkit. Available online: http://broadinstitute.github.io/picard/ (accessed on 1 September 2020).
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome. Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Paila, U.; Chapman, B.A.; Kirchner, R.; Quinlan, A.R. GEMINI: Integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol. 2013, 9, e1003153. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Scelo, G.; Riazalhosseini, Y.; Greger, L.; Letourneau, L.; Gonzàlez-Porta, M.; Wozniak, M.; Lathrop, M. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat. Commun. 2014, 5, 5135. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.G.; Moser, T.; Mouliere, F.; Field-Rayner, J.; Eldridge, M.; Riediger, A.L.; Chandrananda, D.; Heider, K.; Wan, J.C.M.; Warren, A.Y.; et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome. Med. 2020, 12, 23. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef] [Green Version]
- Cimadamore, A.; Massari, F.; Santoni, M.; Mollica, V.; Di Nunno, V.; Cheng, L.; Lopez-Beltran, A.; Scarpelli, M.; Montironi, R.; Moch, H. Molecular characterization and diagnostic criteria of renal cell carcinoma with emphasis on liquid biopsies. Expert. Rev. Mol. Diagn. 2020, 20, 141–150. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Rowan, A.; Chambers, T.E.A. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell 2018, 173, 581–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med. 2017, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef] [Green Version]
- Chennakrishnaiah, S.; Tsering, T.; Aprikian, S.; Rak, J. Leukobiopsy—A Possible New Liquid Biopsy Platform for Detecting Oncogenic Mutations. Front Pharmacol. 2019, 10, 1608. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age | RCC Subtype | Pathological Tumor Stage | Pathological Tumor Grade | Mutated Genes |
|---|---|---|---|---|---|---|
| P1 | Male | 63 | ccRCC | T1b | 4/4 | VHL, PBRM1 |
| P2 | Male | 57 | ccRCC | T1a | 3/4 | VHL, PBRM1 |
| P3 | Female | 62 | ccRCC | T3a | 3/4 | VHL, SETD2, PBRM1, MET |
| P4 | Female | 78 | ccRCC | T1a | 3/4 | VHL, PBRM1 |
| P5 | Female | 58 | ccRCC | T1a | 2/4 | VHL, COL11A1, SETD2, PBRM1, TRRAP, ATM |
| P6 | Male | 66 | ccRCC | T1a | 3/4 | VHL, PBRM1, KDM5C |
| P7 | Female | 58 | ccRCC | T1a | 2/4 | VHL |
| P8 | Male | 77 | ccRCC | T3b | 4/4 | COL11A1, BAP1, PBRM1 |
| P9 | Female | 61 | ccRCC | T1a | 1/4 | VHL |
| P10 | Female | 57 | Unclassified RCC | T4 | 4/4 | KDM5C *, SETD2, NF2 * |
| P11 | Male | 43 | ccRCC | T4 | 4/4 | VHL, PBRM1 *, SETD2 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glennon, K.I.; Maralani, M.; Abdian, N.; Paccard, A.; Montermini, L.; Nam, A.J.; Arseneault, M.; Staffa, A.; Jandaghi, P.; Meehan, B.; et al. Rational Development of Liquid Biopsy Analysis in Renal Cell Carcinoma. Cancers 2021, 13, 5825. https://doi.org/10.3390/cancers13225825
Glennon KI, Maralani M, Abdian N, Paccard A, Montermini L, Nam AJ, Arseneault M, Staffa A, Jandaghi P, Meehan B, et al. Rational Development of Liquid Biopsy Analysis in Renal Cell Carcinoma. Cancers. 2021; 13(22):5825. https://doi.org/10.3390/cancers13225825
Chicago/Turabian StyleGlennon, Kate I., Mahafarin Maralani, Narges Abdian, Antoine Paccard, Laura Montermini, Alice Jisoo Nam, Madeleine Arseneault, Alfredo Staffa, Pouria Jandaghi, Brian Meehan, and et al. 2021. "Rational Development of Liquid Biopsy Analysis in Renal Cell Carcinoma" Cancers 13, no. 22: 5825. https://doi.org/10.3390/cancers13225825
APA StyleGlennon, K. I., Maralani, M., Abdian, N., Paccard, A., Montermini, L., Nam, A. J., Arseneault, M., Staffa, A., Jandaghi, P., Meehan, B., Brimo, F., Tanguay, S., Rak, J., & Riazalhosseini, Y. (2021). Rational Development of Liquid Biopsy Analysis in Renal Cell Carcinoma. Cancers, 13(22), 5825. https://doi.org/10.3390/cancers13225825