
Vaccine-Based Immunotherapy for Head and Neck Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Therapeutic Vaccines
1.2. Main Therapeutic Vaccine Strategies
1.2.1. Peptide-Based Vaccines
1.2.2. DC-Based Vaccines
1.2.3. DNA Vaccines
1.2.4. RNA Vaccines
1.2.5. Live Vector-Based Vaccines
1.2.6. Personalized Vaccination
2. Development of Vaccines for Viral-Induced Head and Neck Cancers
2.1. Human Papilloma Virus in HNSCC
2.2. Development of Therapeutic Vaccines against HPV-Related Antigens for HNSCC
2.3. Vaccination in Epstein–Barr Virus-Induced Undifferentiated Nasopharyngeal Carcinoma
3. Therapeutic Vaccines Targeting Non-Viral Antigens in HNSCC
4. Combining Immune Therapies
5. Challenges and Future Perspectives of Vaccines in HNSCC
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIM | Absent in melanoma |
| APC | Antigen-presenting cells |
| ASCO | American Society of Clinical Oncology |
| cGAS | cGAMP synthase |
| CDCA1 | Cell division cycle associated gene 1 |
| CIN | Cervical intraepithelial neoplasia |
| COPV | Canine papillomavirus-associated oral cancer |
| CR | Complete response |
| CTL | Cytotoxic T lymphocytes |
| CTLA | Cytotoxic T lymphocyte-associated protein |
| DCs | Dendritic cells |
| FDA | Food and Drug Administration |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HLA | Human leukocyte antigen |
| HPV | Human papillomavirus |
| ICI | Immune checkpoint inhibitors |
| IFI | Interferon-γ-inducible protein |
| IL | Interleukin |
| IMP3 | Insulin-like growth factor-II mRNA-binding protein 3 |
| INF | Interferon |
| LP | Long peptide |
| LY6K | Lymphocyte antigen 6 complex locus K (LY6K) |
| MAGE-A3 | Melanoma antigen E-A3 |
| MHC | Major histocompatibility complex |
| NSCLC | Non-small cell lung cancer |
| OPC | Oropharyngeal cancers |
| ORR | Objective response rate |
| OS | Overall survival |
| PD | Progressive disease |
| PD-(L) | Programmed death–(ligand) |
| PFS | Progression-free survival |
| PR | Partial response |
| RB | Retinoblastoma |
| R/M | Recurrent/metastatic |
| SAE | Severe adverse event |
| HNSCC | Squamous cell carcinoma of the head and neck |
| SD | Stable disease |
| SP | Short peptide |
| TAA | Tumor-associated antigen |
| TCR | T cell receptor |
| TIL | Tumor-invasive T lymphocyte |
| TIM | T cell immunoglobulin- and mucin-domain-containing molecule |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| T-reg | Regulatory T cells |
References
- Burtness, B.; Harrington, K.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.-T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, F.; Leidner, R.; Bell, R.B. Immunotherapy for head and neck cancer. Oral Maxillofac. Surg. Clin. N. Am. 2019, 31, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilakopoulou, M.; Avgeris, M.; Velcheti, V.; Kotoula, V.; Rampias, T.; Chatzopoulos, K.; Perisanidis, C.; Kontos, C.K.; Giotakis, A.I.; Scorilas, A.; et al. Evaluation of PD-L1 expression and associated tumor-infiltrating lymphocytes in laryngeal squamous cell carcinoma. Clin. Cancer Res. 2015, 22, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troiano, G.; Rubini, C.; Togni, L.; Caponio, V.C.A.; Zhurakivska, K.; Santarelli, A.; Cirillo, N.; Muzio, L.L.; Mascitti, M. The immune phenotype of tongue squamous cell carcinoma predicts early relapse and poor prognosis. Cancer Med. 2020, 9, 8333–8344. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zhang, S.; Zeng, X.; Ouyang, Y.; Wang, Y.; Hu, Z.; Ye, Y.; Wu, W.; Jin, F.; Zhou, S.; et al. Development of an immunogenomic landscape-based prognostic index of head and neck squamous cell carcinoma. Front. Mol. Biosci. 2020, 7, 586344. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immuno-therapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulie, P.G.; Somville, M.; Lehmann, F.; Hainaut, P.; Brasseur, F.; Devos, R.; Boon, T. Precursor frequency analysis of human cytolytic T lymphocytes directed against autologous melanoma cells. Int. J. Cancer 1992, 50, 289–297. [Google Scholar] [CrossRef]
- Hanagiri, T.; Van Baren, N.; Neyns, B.; Boon, T.; Coulie, P.G. Analysis of a rare melanoma patient with a spontaneous CTL response to a MAGE-A3 peptide presented by HLA-A1. Cancer Immunol. Immunother. 2006, 55, 178–184. [Google Scholar] [CrossRef]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed] [Green Version]
- Pol, J.; Bloy, N.; Buqué, A.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; et al. Trial Watch: Peptide-based anticancer vaccines. OncoImmunology 2015, 4, e974411. [Google Scholar] [CrossRef] [Green Version]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Madan, R.A.; Pachynski, R.; Mulders, P.; Sheikh, N.A.; Trager, J.; Drake, C.G. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J. Natl. Cancer Inst. 2017, 109, djw261. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Schlom, J.; Hodge, J.W. Induction of an antigen cascade by diversified subcutaneous/intratumoral vaccination is associated with antitumor responses. Clin. Cancer Res. 2005, 11, 2416–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slingluff, C.L., Jr. The present and future of peptide vaccines for cancer: Single or multiple, long or short, alone or in combination? Cancer J. 2011, 17, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Kim, D.S. Peptide immunotherapy in vaccine development: From epitope to adjuvant. Adv. Protein Chem. Struct. Biol. 2015, 99, 1–14. [Google Scholar] [PubMed]
- Melief, C.J.; van der Burg, S.H. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat. Rev. Cancer 2008, 8, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.; Hayashi, R.J.; Lafond-Walker, A.; Lowenstein, C.J.; Pardoll, D.M.; I Levitsky, H. The central role of CD4+ T cells in the antitumor immune response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Dickie, J.; Sutavani, R.V.; Pointer, C.; Thomas, G.J.; Savelyeva, N. Targeting head and neck cancer by vaccination. Front. Immunol. 2018, 9, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, E.M.; Droin, N.; Lemmens, E.E.; Pinkoski, M.J.; Bensinger, S.J.; Ehst, B.D.; Griffith, T.S.; Green, D.; Schoenberger, S.P. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 2005, 434, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.W.; Tveita, A.; Fauskanger, M.; Schjesvold, F.; Lorvik, K.B.; Hofgaard, P.O.; Omholt, H.; Munthe, L.A.; Dembic, Z.; Corthay, A.; et al. How do CD4+ T cells detect and eliminate tumor cells that either lack or express MHC Class II molecules? Front. Immunol. 2014, 5, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayama, M.; Nishimura, Y. The present status and future prospects of peptide-based cancer vaccines. Int. Immunol. 2016, 28, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Gammoh, E.; Simon, R.M.; Khleif, S.N. Is the “3 + 3” dose-escalation phase I clinical trial design suitable for therapeutic cancer vaccine development? A recommendation for alternative design. Clin. Cancer Res. 2014, 20, 4758–4767. [Google Scholar] [CrossRef] [Green Version]
- Embgenbroich, M.; Burgdorf, S. Current concepts of antigen cross-presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic cell cancer therapy: Vaccinating the right patient at the right time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.; Lion, E.; Van Tendeloo, V.; Berneman, Z. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Draube, A.; Klein-González, N.; Mattheus, S.; Brillant, C.; Hellmich, M.; Engert, A.; Von Bergwelt-Baildon, M. Dendritic cell based tumor vaccination in prostate and renal cell cancer: A systematic review and meta-analysis. PLoS ONE 2011, 6, e18801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platten, M.; Wick, W.; Eynde, B.J.V.D. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [Green Version]
- Mastelic-Gavillet, B.; Balint, K.; Boudousquie, C.; Gannon, P.O.; Kandalaft, L.E. Personalized dendritic cell vaccines—Recent breakthroughs and encouraging clinical results. Front. Immunol. 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonerba, C.; Ferro, M.; Di Lorenzo, G. Sipuleucel-T for prostate cancer: The immunotherapy era has commenced. Expert Rev. Anticancer Ther. 2011, 11, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479–480, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Bonehill, A.; Tuyaerts, S.; Van Nuffel, A.; Heirman, C.; Bos, T.J.; Fostier, K.; Neyns, B.; Thielemans, K. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol. Ther. 2008, 16, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Farmer, E.; Lin, J.; Wu, T.-C.; Hung, C.-F. The current state of therapeutic and T cell-based vaccines against human papillomaviruses. Virus Res. 2016, 231, 148–165. [Google Scholar] [CrossRef] [Green Version]
- Grunwitz, C.; Kranz, L.M. mRNA Cancer Vaccines—Messages that Prevail. Cancer Vaccines 2017, 405, 145–164. [Google Scholar] [CrossRef]
- Dörrie, J.; Schaft, N.; Schuler, G.; Schuler-Thurner, B. Therapeutic Cancer Vaccination with Ex Vivo RNA-Transfected Dendritic Cells—An Update. Pharmaceutics 2020, 12, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, K.; Lazzaro, S.; Lutz, J.; Rauch, S.; Heidenreich, R. mRNA cancer vaccines. Recent Results Cancer Res. 2016, 209, 61–85. [Google Scholar] [PubMed]
- Van Lint, S.; Renmans, D.; Broos, K.; Dewitte, H.; Lentacker, I.; Heirman, C.; Breckpot, K.; Thielemans, K. The ReNAissanCe of mRNA-based cancer therapy. Expert Rev. Vaccines 2015, 14, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Jeang, J.; Cheng, K.; Cheng, T.; Yang, B.; Wu, T.-C.; Hung, C.-F. Current state in the development of candidate therapeutic HPV vaccines. Expert Rev. Vaccines 2016, 15, 989–1007. [Google Scholar] [CrossRef] [Green Version]
- Schneider, K.; Grønhøj, C.; Hahn, C.H.; Von Buchwald, C. Therapeutic human papillomavirus vaccines in head and neck cancer: A systematic review of current clinical trials. Vaccine 2018, 36, 6594–6605. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Curry, J.M.; Sprandio, J.; Cognetti, D.; Luginbuhl, A.; Bar-Ad, V.; Pribitkin, E.; Tuluc, M. Tumor Microenvironment in Head and Neck Squamous Cell Carcinoma. Semin. Oncol. 2014, 41, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the tumor: A roadmap for T cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer [published correction appears in Nature. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrjänen, K.; Syrjänen, S.; Pyrhönen, S. Human Papilloma Virus (HPV) antigens in lesions of laryngeal squamous cell carcinomas. ORL 1982, 44, 323–334. [Google Scholar] [CrossRef]
- IARC. Working group on the evaluation of carcinogenic risks to humans. biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–441. [Google Scholar]
- Lacau St Guily, J.; Rousseau, A.; Baujat, B.; Périé, S.; Schultz, P.; Barry, B.; Dufour, X.; Malard, O.; Pretet, J.; Clavel, C.; et al. Oropharyngeal cancer prognosis by tumour HPV status in France: The multicentric Papillophar study. Oral Oncol. 2017, 67, 29–36. [Google Scholar] [CrossRef]
- Gooi, Z.; Chan, J.Y.K.; Fakhry, C. The epidemiology of the human papillomavirus related to oropharyngeal head and neck cancer. Laryngoscope 2016, 126, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.K.; Graubard, B.I.; Broutian, T.; Pickard, R.K.L.; Tong, Z.-Y.; Xiao, W.; Kahle, L.; Gillison, M.L. Effect of Prophylactic Human Papillomavirus (HPV) Vaccination on Oral HPV Infections Among Young Adults in the United States. J. Clin. Oncol. 2018, 36, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Zaravinos, A. An updated overview of HPV-associated head and neck carcinomas. Oncotarget 2014, 5, 3956–3969. [Google Scholar] [CrossRef] [Green Version]
- Drolet, M.; Bénard, É.; Pérez, N.; Brisson, M.; HPV Vaccination Impact Study Group. Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: Updated systematic review and meta-analysis. Lancet 2019, 394, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Arbyn, M.; Xu, L.; Simoens, C.; Martin-Hirsch, P.P. Prophylactic vaccination against human papillomaviruses to prevent cervical cancer and its precursors. Cochrane Database Syst. Rev. 2018, 5, CD009069. [Google Scholar] [CrossRef] [PubMed]
- Herrero, R.; Quint, W.; Hildesheim, A.; Gonzalez, P.; Struijk, L.; Katki, H.A.; Porras, C.; Schiffman, M.; Rodriguez, A.C.; Solomon, D.; et al. Reduced prevalence of oral human papillomavirus (HPV) 4 years after bivalent HPV vaccination in a randomized clinical trial in Costa Rica. PLoS ONE 2013, 8, e68329. [Google Scholar]
- Gillison, M. Chapter 8. HPV vaccines and potential prevention of HPV-positive head and neck cancer. In IARC HPV Working Group. Primary End-points for Prophylactic HPV Vaccine Trials; IARC Working Group Reports, No. 7; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of human papillomavirus–positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Sansanaphongpricha, K.; Prince, M.; Sun, D.; Wolf, G.; Lei, Y. Engineering vaccines to reprogram immunity against head and neck cancer. J. Dent. Res. 2018, 97, 627–634. [Google Scholar] [CrossRef]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.P.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Essahsah, F.; Fathers, L.M.; Offringa, J.W.; Drijfhout, Y.; et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandberg, D.P.; Rollins, S.; Goloubeva, O.; Morales, R.E.; Tan, M.; Taylor, R.; Wolf, J.S.; Schumaker, L.; Cullen, K.J.; Zimrin, A.; et al. A phase I dose escalation trial of MAGE-A3- and HPV16-specific peptide immunomodulatory vaccines in patients with recurrent/metastatic (RM) squamous cell carcinoma of the head and neck (SCCHN). Cancer Immunol. Immunother. 2014, 64, 367–379. [Google Scholar] [CrossRef]
- Bosch-Voskens, C.; Sewell, D.; Hertzano, R.; Ms, J.D.; Ms, S.R.; Lee, M.; Taylor, R.; Wolf, J.; Suntharalingam, M.; Gastman, B.; et al. inducTION of mage-A3 and HPV-16 immunity by Trojan vaccines in patients with head and neck carcinoma. Head Neck 2012, 34, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Cohen, R.B.; Morrow, M.P.; Kraynyak, K.A.; Sylvester, A.J.; Knoblock, D.M.; Bauml, J.M.; Weinstein, G.S.; Lin, A.; Boyer, J.; et al. Immunotherapy targeting HPV16/18 generates potent immune responses in HPV-associated head and neck cancer. Clin. Cancer Res. 2019, 25, 110–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, C.; Saba, N.; Algazi, A.; Sukari, A.; Seiwert, T.; Haigentz, M.; Porosnicu, M.; Bonomi, M.; Boyer, J.; Durham, N.; et al. 916MO Safety and efficacy of MEDI0457 plus durvalumab in patients (pts) with human papillomavirus-associated recurrent/metastatic head and neck squamous cell carcinoma (HPV+ R/M HNSCC). Ann. Oncol. 2020, 31, S661–S662. [Google Scholar] [CrossRef]
- Schmitz, S.; Duhoux, F.; Machiels, J.-P. Window of opportunity studies: Do they fulfil our expectations? Cancer Treat. Rev. 2016, 43, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ozbun, L.; Chong, N.; Wallecha, A.; Berzofsky, J.A.; Khleif, S.N. Episomal expression of truncated listeriolysin O in LmddA-LLO-E7 vaccine enhances antitumor efficacy by preferentially inducing expansions of CD4+FoxP3- and CD8+ T cells. Cancer Immunol. Res. 2014, 2, 911–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallecha, A.; French, C.; Petit, R.; Singh, R.; Amin, A.; Rothman, J. Lm-LLO-Based immunotherapies and HPV-associated disease. J. Oncol. 2012, 2012, 542851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupar, R.; Imai, N.; Miles, B.; Genden, E.; Misiukiewicz, K.; Saenger, Y.; Demicco, E.G.; Patel, J.; Herrera, P.C.; Parikh, F.; et al. Abstract LB-095: HPV E7 antigen-expressing Listeria-based immunotherapy (ADXS11-001) prior to robotic surgery for HPV-positive oropharyngeal cancer enhances HPV-specific T cell immunity. Immunology 2016, 76, LB-095. [Google Scholar] [CrossRef]
- Strauss, J.; Floudas, C.S.; Sater, H.A.; Manu, M.; Lamping, E.; Francis, D.C.; Cordes, L.M.; Marte, J.; Donahue, R.N.; Jochems, C.; et al. Phase II evaluation of the triple combination of PDS0101, M9241, and bintrafusp alfa in patients with HPV 16 positive malignancies. J. Clin. Oncol. 2021, 39, 2501. [Google Scholar] [CrossRef]
- Ho, A.L.; Posner, M.R.; Niu, J.; Fu, S.; Leidner, R.S.; Pearson, A.T.; Chung, K.Y.; Richardson, D.L.; Wang, D.; Pimentel, A.; et al. First report of the safety/tolerability and preliminary antitumor activity of HB-201 and HB-202, an arenavirus-based cancer immunotherapy, in patients with HPV16+ cancers. J. Clin. Oncol. 2021, 39, 2502. [Google Scholar] [CrossRef]
- Reuschenbach, M.; Pauligk, C.; Karbach, J.; Rafiyan, M.; Kloor, M.; Prigge, E.; Sauer, M.; Al-Batran, S.; Kaufmann, A.M.; Schneider, A.; et al. A phase 1/2a study to test the safety and immunogenicity of a p16(INK4a) peptide vaccine in patients with advanced human papillomavirus-associated cancers. Cancer 2016, 122, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: A phase 2 clinical trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Haugen, M.; Moger, T.A.; Tretli, S.; Aalen, O.O.; Grotmol, T. Age-Incidence curves of nasopharyngeal carcinoma worldwide: Bimodality in low-risk populations and aetiologic implications. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2356–2365. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Steven, N.M. Therapeutic vaccination strategies to treat nasopharyngeal carcinoma. Chin. Clin. Oncol. 2016, 5, 23. [Google Scholar] [CrossRef]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Léonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 vaccine for infectious mononucleosis: A phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Coghill, A.E.; Bu, W.; Nguyen, H.; Hsu, W.-L.; Yu, K.J.; Lou, P.-J.; Wang, C.-P.; Chen, C.-J.; Hildesheim, A.; Cohen, J.I. High levels of antibody that neutralize B-cell infection of epstein-barr virus and that bind EBV gp350 are associated with a lower risk of nasopharyngeal carcinoma. Clin. Cancer Res. 2016, 22, 3451–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zyl, D.G.; Mautner, J.; Delecluse, H.J. Progress in EBV vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, L.; Hu, G.-Q.; Zhang, N.; Zhu, X.-D.; Yang, K.-Y.; Jin, F.; Shi, M.; Chen, Y.P.; Hu, W.-H.; et al. Gemcitabine and cisplatin induction chemotherapy in nasopharyngeal carcinoma. N. Engl. J. Med. 2019, 381, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Q.-Y.; He, J.; Li, Z.-L.; Tang, X.-F.; Chen, S.-P.; Xie, C.-M.; Li, Y.-Q.; Huang, L.-X.; Ye, S.-B.; et al. Phase I trial of adoptively transferred tumor-infiltrating lymphocyte immunotherapy following concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma. OncoImmunology 2015, 4, e976507. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Jia, H.; Harrington, K.; Lee, L.W.; Turner, J.; Ladell, K.; Price, D.; Tanday, M.; Matthews, J.; Roberts, C.; et al. A recombinant modified vaccinia ankara vaccine encoding Epstein–Barr Virus (EBV) target antigens: A phase I trial in UK patients with EBV-positive cancer. Clin. Cancer Res. 2014, 20, 5009–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-L.; Lo, W.-F.; Lee, T.-H.; Ren, Y.; Hwang, S.-L.; Cheng, Y.-F.; Chen, C.-L.; Chang, Y.-S.; Lee, S.P.; Rickinson, A.B.; et al. Immunization with Epstein-Barr Virus (EBV) peptide-pulsed dendritic cells induces functional CD8+ T-cell immunity and may lead to tumor regression in patients with EBV-positive nasopharyngeal carcinoma. Cancer Res. 2002, 62, 6952–6958. [Google Scholar]
- Li, F.; Song, D.; Lu, Y.; Zhu, H.; Chen, Z.; He, X. Delayed-Type Hypersensitivity (DTH) immune response related with EBV-DNA in nasopharyngeal carcinoma treated with autologous dendritic cell vaccination after radiotherapy. J. Immunother. 2013, 36, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.; Chan, S.; Ho, R.; Wong, W.-L.; Wilson, S.; Johnson, B.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding epstein–barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, W.K.; Wang, W.-W.; Teo, M.; Tai, W.M.; Lim, D.W.-T.; Tan, E.H.; Leong, S.S.; Sun, L.; Chen, J.J.; Gottschalk, S.; et al. A phase II study evaluating the safety and efficacy of an adenovirus-ΔLMP1-LMP2 transduced dendritic cell vaccine in patients with advanced metastatic nasopharyngeal carcinoma. Ann. Oncol. 2011, 23, 997–1005. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2014, 21, 632–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.K.; Bier, H.; Donnenberg, A.D.; Whiteside, T.L.; De Leo, A.B. p53 as an Immunotherapeutic Target in Head and Neck Cancer. Adv. Otorhinolaryngol. 2004, 62, 151–160. [Google Scholar] [CrossRef]
- DeLeo, A.B.; Whiteside, T.L. Development of multi-epitope vaccines targeting wild-type sequence p53 peptides. Expert Rev. Vaccines 2008, 7, 1031–1040. [Google Scholar] [CrossRef] [Green Version]
- Schuler, P.J.; Harasymczuk, M.; Visus, C.; DeLeo, A.; Trivedi, S.; Lei, Y.; Argiris, A.; Gooding, W.; Butterfield, L.; Whiteside, T.L.; et al. Phase I Dendritic cell p53 peptide vaccine for head and neck cancer. Clin. Cancer Res. 2014, 20, 2433–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, A.; Kobayashi, J.; Torigoe, T.; Hirohashi, Y.; Yamamoto, T.; Yamaguchi, A.; Asanuma, H.; Takahashi, A.; Michifuri, Y.; Nakamori, K.; et al. Phase I clinical trial of survivin-derived peptide vaccine therapy for patients with advanced or recurrent oral cancer. Cancer Sci. 2010, 102, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Gleich, L.L.; Gluckman, J.L.; Nemunaitis, J.; Suen, J.Y.; Hanna, E.; Wolf, G.T.; Coltrera, M.D.; Villaret, D.B.; Wagman, L.; Castro, D.; et al. Clinical experience with HLA-B7 plasmid DNA/lipid complex in advanced squamous cell carcinoma of the head and neck. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 775–779. [Google Scholar] [PubMed]
- Bann, D.; Deschler, D.G.; Goyal, N. Novel immunotherapeutic approaches for head and neck squamous cell carcinoma. Cancers 2016, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Weed, D.T.; Zilio, S.; Reis, I.M.; Sargi, Z.; Abouyared, M.; Gomez-Fernandez, C.R.; Civantos, F.J.; Rodriguez, C.P.; Serafini, P. The Reversal of immune exclusion mediated by tadalafil and an anti-tumor vaccine also induces PDL1 upregulation in recurrent head and neck squamous cell carcinoma: Interim analysis of a phase I clinical trial. Front. Immunol. 2019, 10, 1206. [Google Scholar] [CrossRef]
- Weed, D.T.; Vella, J.L.; Reis, I.M.; De La Fuente, A.C.; Gomez, C.; Sargi, Z.; Nazarian, R.; Califano, J.; Borrello, I.; Serafini, P. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshitake, Y.; Fukuma, D.; Yuno, A.; Hirayama, M.; Nakayama, H.; Tanaka, T.; Nagata, M.; Takamune, Y.; Kawahara, K.; Nakagawa, Y.; et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin. Cancer Res. 2015, 21, 312–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, N.; Vernet, R.; Belkouch, M.-C.; Luy, P.; Ancrenaz, V.; Teta, P.; Blazek, N.; Grandjean, N.; Wasem, J.; Grogg, J.; et al. MVX-ONCO-1 phase 1 final results of the first personalized cell-based immunotherapy using cell encapsulation technology. Ann. Oncol. 2016, 27, vi362. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, E.; Vernet, R.; Charrier, E.; Migliorini, D.; Joerger, M.; Belkouch, M.-C.; Urwyler, M.; Von Rohr, O.; Saingier, V.; Ancrenaz, V.; et al. MVX-ONCO-1 in advanced refractory cancers: Safety, feasibility, and preliminary efficacy results from all HNSCC patients treated in two ongoing clinical trials. J. Clin. Oncol. 2021, 39, e18005. [Google Scholar] [CrossRef]
- Chindavijak, S.; Har-noy, M.; Lausoontornsiri, W. Effect of therapeutic vaccine on CTLA4 and tumor debulking response in recurrent and metastic HNSCC. J. Clin. Oncol. 2018, 36, 115. [Google Scholar] [CrossRef]
- Perri, F.; Ionna, F.; Longo, F.; Scarpati, G.D.V.; DE Angelis, C.; Ottaiano, A.; Botti, G.; Caponigro, F. Immune response against head and neck cancer: Biological mechanisms and implication on therapy. Transl. Oncol. 2020, 13, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Mehra, R.; Seiwert, T.Y.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; Kang, H.; et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: Pooled analyses after long-term follow-up in KEYNOTE-012. Br. J. Cancer 2018, 119, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Bauml, J.; Seiwert, T.Y.; Pfister, D.G.; Worden, F.; Liu, S.V.; Gilbert, J.; Saba, N.F.; Weiss, J.; Wirth, L.; Sukari, A.; et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: Results from a single-arm, phase II study. J. Clin. Oncol. 2017, 35, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; Kos, F.J.; Hardwick, N.; Yuan, Y.; Chao, J.; Li, D.; Waisman, J.; Li, M.; Zurcher, K.; Frankel, P.; et al. Evaluation of safety and efficacy of p53MVA vaccine combined with pembrolizumab in patients with advanced solid cancers. Clin. Transl. Oncol. 2019, 21, 363–372. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Popa, X.; Martínez, O.; Mendoza, S.; Santiesteban, E.; Crespo, T.; Amador, R.M.; Fleytas, R.; Acosta, S.C.; Otero, Y.; et al. A phase III clinical trial of the epidermal growth factor vaccine CIMAvax-EGF as switch maintenance therapy in advanced non–small cell lung cancer patients. Clin. Cancer Res. 2016, 22, 3782–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saavedra, D.; Crombet, T. CIMAvax-EGF: A new therapeutic vaccine for advanced non-small cell lung cancer patients. Front. Immunol. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArdel, S.L.; Dugast, A.-S.; Hoover, M.E.; Bollampalli, A.; Hong, E.; Castano, Z.; Leonard, S.C.; Pawar, S.; Mellen, J.; Muriuki, K.; et al. Anti-tumor effects of RTX-240: An engineered red blood cell expressing 4-1BB ligand and interleukin-15. Cancer Immunol. Immunother. 2021, 70, 2701–2719. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, M.; Dastagir, S.R.; Nixon, M.; Khamhoung, A.; Schmidt, A.; Lee, A.; Subbiah, N.; McLaughlin, D.C.; Moore, C.L.; et al. Engineered red blood cells as an off-the-shelf allogeneic anti-tumor therapeutic. Nat. Commun. 2021, 12, 2637. [Google Scholar] [CrossRef] [PubMed]
- Grinshtein, N.; Bridle, B.; Wan, Y.; Bramson, J.L. Neoadjuvant vaccination provides superior protection against tumor relapse following surgery compared with adjuvant vaccination. Cancer Res. 2009, 69, 3979–3985. [Google Scholar] [CrossRef] [Green Version]
- Fisher, S.A.; Cleaver, A.; Lakhiani, D.D.; Khong, A.; Connor, T.; Wylie, B.; Lesterhuis, W.J.; Robinson, B.W.S.; Lake, R.A. Neoadjuvant anti-tumor vaccination prior to surgery enhances survival. J. Transl. Med. 2014, 12, 245. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Carroll, P.; Weinberg, V.; Chan, S.; Lewis, J.; Corman, J.; Amling, C.L.; Stephenson, R.A.; Simko, J.; Sheikh, N.A.; et al. Activated lymphocyte recruitment into the tumor microenvironment following preoperative sipuleucel-T for localized prostate cancer. J. Natl. Cancer Inst. 2014, 106, dju372. [Google Scholar] [CrossRef] [PubMed]
- Haug, M.; Brede, G.; Håkerud, M.; Nedberg, A.G.; Gederaas, O.A.; Flo, T.H.; Edwards, V.T.; Selbo, P.K.; Høgset, A.; Halaas, Ø. Photochemical internalization of peptide antigens provides a novel strategy to realize therapeutic cancer vaccination. Front. Immunol. 2018, 9, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selbo, P.K.; Janetzki, S.; Welters, M.; Håkerud, M.; Nedberg, A.; Edwards, V.; Olivecrona, H.; Van Der Burg, S.; Otterhaug, T.; Hogset, A. Phase I clinical study for validation of fimaporfin-based photochemical internalisation: A novel technology for enhancing cellular immune responses important for therapeutic effect of peptide-and protein-based vaccines. Ann. Oncol. 2019, 30, xi40–xi41. [Google Scholar] [CrossRef]


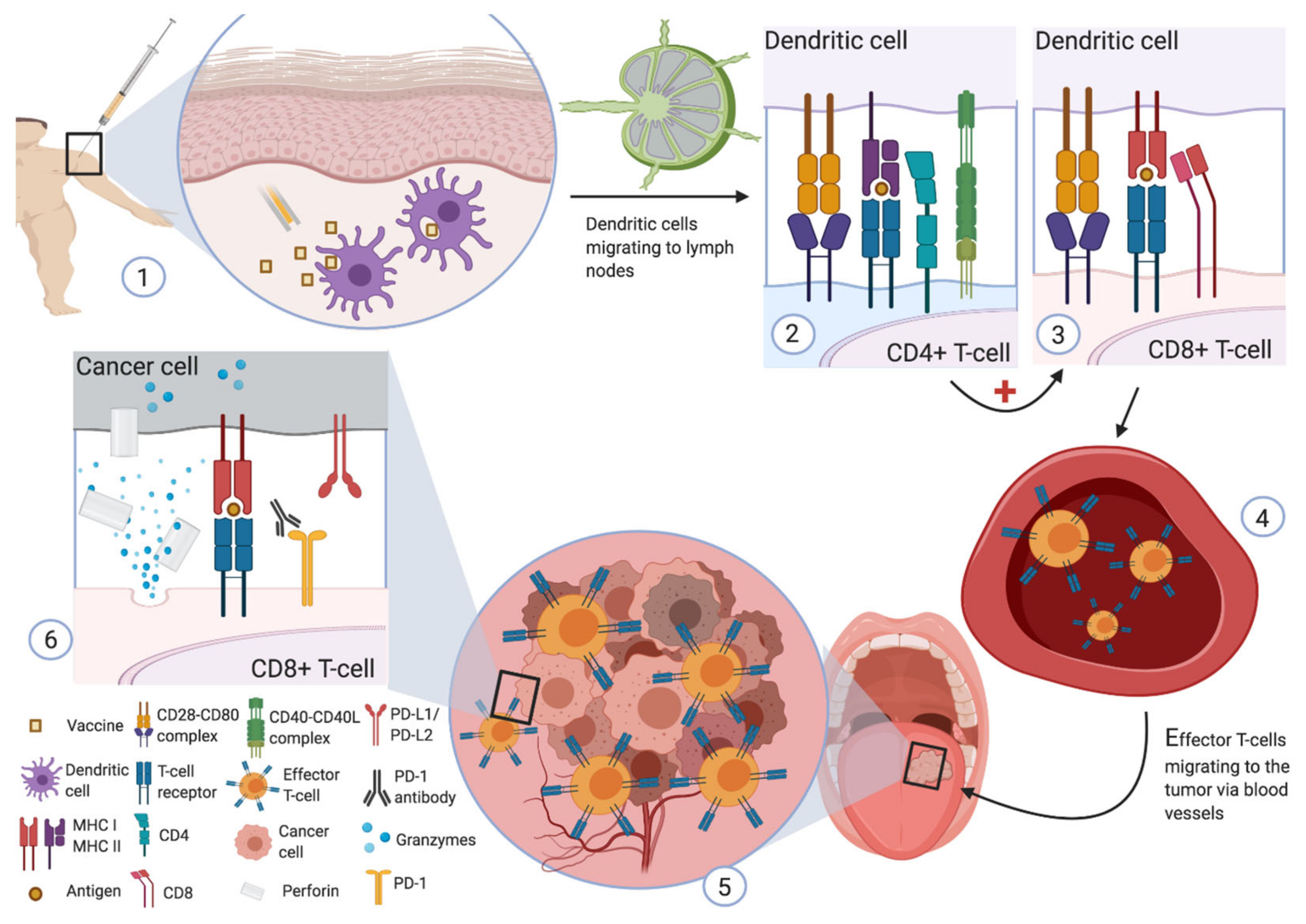{kind=link}
| Identifier and Reference | Vaccine ± Other Therapy | Phase | Type of Vaccine | N | Target Antigens | Population | Primary Endpoint | Status |
|---|---|---|---|---|---|---|---|---|
| NCT00257738 Ref. [65] | GL-0810/GL-0817 | I | Peptide | 16 | MAGE-A3 * (n = 7) /HPV16-E7 (n = 9) | HPV16-positive or MAGE-A3-positive R/M-HNSCC | Safety | Completed |
| NCT02163057 Ref. [66] | MEDI0457 | I/IIa | DNA | 22 | HPV16 E6/E7 HPV18 E6/E7 | Advanced HPV-related HNSCC | Safety | Completed |
| NCT03162224 NA | MEDI0457 + Durvalumab | I/IIa | DNA | ±35 | HPV16 E6/E7 HPV18 E6/E7 | R/M HNSCC HPV+ | Safety and efficacy | Completed |
| NCT02002182 NA | ADXS11 | II | Live (Listeria Monocytogenes) | ±15 | HPV16 E7 | Surgically elected HPV+ oropharyngeal SCC | Change in specific CD8+ CTL response and safety | Active, not recruiting |
| NCT02291055 NA | Durvalumab ± ADXS11 | I/II | Live (Listeria Monocytogenes) | ±66 | HPV16 E7 | R/M HPV16+ HNSCC or cervical cancer | I: Safety II: PFS and safety | Active, not recruiting |
| NCT03418480 NA | HARE-40 | I/II | RNA | ±44 | HPV16 E6/E7 | I: Advanced HPV16+ HNSCC II: Advanced HPV16+ cancer (HNSCC, anogenital, penile, cervical) | I: Safety II: Efficacy and significant increase in specific immune cells | Active, not recruiting |
| NCT02426892 Ref. [76] | ISA101 + Nivolumab | II | Peptide | 24 | HPV16 E6/E7 | Incurable HPV16+ cancers (22 oropharyngeal cancers, 1 anal cancer, and 1 cervical cancer) | Efficacy | Active, not recruiting |
| NCT03258008 NA | ISA101b + Utomilumab | II | Peptide | ±27 | HPV16 E6/E7 | HPV16+ incurable oropharyngeal cancer | Efficacy | Active, not recruiting |
| NCT02865135 NA | DPX-E7 | Ib/II | Peptide | ±11 | HPV16 E7 | Positive HLA-A*02 patients with HPV-related head and neck, cervical or anal cancer. | Safety | Active, not recruiting |
| NCT03260023 NA | TG4001 + Avelumab | Ib/II | Live (modified vaccinia Ankara virus) | ±52 | HPV16 E6/E7 | HPV-related carcinomas | I: Safety II: Efficacy | Recruiting |
| NCT02526316 NA | p16 vaccine + concurrent cisplatin-based chemotherapy | I | Peptide | ±11 | p16 | p16-positive cervical, vulvar, vaginal, penile, anal, or head and neck cancer | Immune response | Completed |
| NCT01462838 Ref. [75] | p16 vaccine | I/IIa | Peptide | 24 | p16 | HPV-associated cancers (including 6 HNSCC) | Immune response | Completed |
| NCT04260126 NA | PDS0101 + Pembrolizumab | II | Peptide | ±96 | HPV16 E6/E7 | HPV16+ R/M HNSCC and HPV-related esophageal SCC | Efficacy | Recruiting |
| NCT04369937 NA | ISA101b + Pembrolizumab + Cisplatin + radiotherapy | II | Peptide | ±50 | HPV16 E6/E7 | “Intermediate risk” HPV-16 associated HNSCC | Efficacy | Recruiting |
| NCT04534205 NA | BNT113 + Pembrolizumab vs. Pembrolizumab alone | II | RNA | 285 | HPV16 E6/E7 | HPV16 + and PD-L1+ R/M HNSCC | Part A: Safety Part B: Efficacy | Recruiting |
| NCT04287868 Ref. [73] | PDS0101 + M9241 + M7824 | I/II | Peptide | 21 | HPV16 E6/E7 | Advanced HPV16-positive cancers | Objective Response Rate | Suspended |
| NCT04180215 Ref. [74] | HB-201 ± HB-202 | I/II | Virus | ±200 | HPV16 E6/E7 | HPV16-positive cancers | I: dose-limiting toxicities II: ORR | Recruiting |
| NCT04672980 NA | RTX-231 | I | Allogenic aAPC | ±63 | HPV16 E7 | Advanced HPV16 positive cancers | I: safety | Recruiting |
| Identifier and Reference | Vaccine ± Other Therapy | Phase | Type of Vaccine | N | Target Antigens | Population | Primary Endpoint | Status |
|---|---|---|---|---|---|---|---|---|
| NCT01147991 Ref. [84] | MVA-EL | I | Live (MVA virus) | 16 | EBNA1 LMP2 | EBV-induced NPC in CR after first-line treatment | Safety and IR | Completed |
| NCT01256853 Ref. [87] | MVA-EL | I | Live (MVA virus) | 18 | EBNA1 LMP2 | EBV-induced NPC in CR or unconfirmed CR | Safety | Completed |
| NCT01800071 NA | MVA-EBNA1/LMP2 | Ib | Live (MVA virus) | 22 | EBNA1 LMP2 | EBV-induced NPC in remission or with current disease for whom no standard therapy is currently appropriate or required | IR and Safety | Completed |
| NCT01094405 NA | MVA EBNA1/LMP2 vaccine | II | Live (MVA virus) | 25 | EBNA1 LMP2 | Persistent, recurrent, or metastatic NPC that have residual EBV DNA following completion of conventional therapy | Efficacy | Completed |
| NA Ref. [85] | DC vaccine | I | Autologous DCs | 16 | LMP2 | Local recurrence or metastasis NPC | Safety | Completed |
| NA Ref. [86] | DC vaccine | ? | Autologous DCs | 16 | LMP2 | Stage II-III NPC | IR | Completed |
| NA Ref. [88] | Ad-ΔLMP1-LMP2 DC vaccine | II | Autologous DCs transducted with an adenovirus | 16 | LMP1 LMP2 | Refractory metastatic NPC | Efficacy | NA |
| NCT00078494 NA | LMP-2:340–349 or LMP-2:419–427 | I/II | Peptide | 99 | LMP2 | Locally controlled anaplastic NPC | IR | Completed |
| NCT00589186 NA | Ad5F35-LMP1/LMP2-transduced autologous DCs + Celecoxib | II | Autologous DCs transducted with an adenovirus | ±35 | LMP1 LMP2 | Metastatic NPC | Efficacy | Unknown |
| Identifier and Reference | Vaccine ± Other Therapy | Phase | Type of Vaccine | N | Target Antigens | Population | Primary Endpoint | Status |
|---|---|---|---|---|---|---|---|---|
| NCT00404339 Ref. [93] | Peptide pulsed DCs | I | DC | 16 | p53 | HLA-A2.1-positive patients with treated HNSCC | Safety | Completed |
| UMIN000000976 Ref. [94] | Survivin-2B vaccine | I | Peptide | 10 | Survivin-2B | 10 HLA-A24-positive patients with advanced or recurrent oral cancer | Safety | Completed |
| NCT00050388 Ref. [95] | Allovectin-7 | I/II | DNA | 69 | Restore HLA-B7/β2 | Persistent or recurrent HNSCC after (chemo)radiotherapy | Safety | Completed |
| NA Ref. [99] | Peptide vaccine | II | Peptide | 55 | LY6K, CDCA1 and IMP3 | HLA-A24-positive patients with advanced HNSCC | Overall survival | Completed |
| NCT02999646 [101] | MVX-ONCO-1 | II | Personalized | ±41 | Autologous tumor cells | Advanced HNSCC | Overall survival | Recruiting |
| NCT01998542 Ref. [102] | AlloVax | II | Personalized | 10 | Chaperone-enriched tumor cell lysate | Advanced chemo-resistant HNSCC | Efficacy | Completed |
| NCT03946358 NA | UCPVax | II | Peptide | ±47 | Telomerase | HPV+ cancers (head and neck, anal, and cervical cancers) | Efficacy | Recruiting |
| NCT03552718 NA | YE-NEO-001 | I | Personalized | ±16 | NA | Solid cancers in curative post-treatment surveillance period | Safety | Unknown |
| NCT03548467 NA | CB10.NEO + Bempegaldek-leukin | I/II | Personalized | ±65 | NA | Locally advanced or metastatic solid tumors | Safety | Recruiting |
| NCT00019331 NA | Ras vaccine | II | Peptide | NA | Ras | Metastatic solid tumors | IR, Efficacy, and Safety | Completed |
| NCT00021424 NA | TRICOM vaccine | I | Live (Fowlpox virus) | Max 20 | Expression of B7-1, ICAM-1, and LFA-3 | Advanced SCC of the oral cavity or oropharynx or nodal or dermal metastases | DLT | Completed |
| NCT02544880 Ref. [97] | MUC1 vaccine + Tadalafil | I/II | Peptide | 16 | MUC1 | Resectable and recurrent or second primary HNSCC | I: Safety II: IR | Completed |
| NCT04247282 NA | M7823 ± TriAd vaccine * ± N-803 | I/II | Live (Adenovirus) | ±40 | Brachyury, Mucin-1, and CEA | p16-negative resectable HNSCC | Efficacy | Suspended |
| NCT04266730 NA | PANDA-VAC | I | Personalized peptide | ±6 | NA | Advanced lung cancers or HNSCC under Pembrolizumab | Safety | Not yet recruiting |
| NCT03689192 NA | ARG1 vaccine | I | Peptide | ±10 | Arginase-1 | Metastatic solid tumors | Safety | Recruiting |
| NCT03311334 NA | DSP-7888 + ICI | Ib/II | Peptide | ±84 | WT1 | Advanced solid tumors | I: Safety II: Efficacy | Recruiting |
| NCT04445064 NA | IO102 | II | Peptide | 18 | IDO | Curable HNSCC | Biological activity | Recruiting |
| NCT04470024 NA | DPV-001 + delayed anti-PD1 ± anti-GITR | Ib | Autophagosome-enriched vaccine | 56 | NA | R/M HNSCC | Safety | Recruiting |
| NCT05075122 NA | UV1 vaccine + Pembrolizumab + Sargramostism | II | Peptide | 75 | Telomerase | R/M HNSCC with CPS ≥1 | Efficacy | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyaert, S.; Machiels, J.-P.; Schmitz, S. Vaccine-Based Immunotherapy for Head and Neck Cancers. Cancers 2021, 13, 6041. https://doi.org/10.3390/cancers13236041
Beyaert S, Machiels J-P, Schmitz S. Vaccine-Based Immunotherapy for Head and Neck Cancers. Cancers. 2021; 13(23):6041. https://doi.org/10.3390/cancers13236041
Chicago/Turabian StyleBeyaert, Simon, Jean-Pascal Machiels, and Sandra Schmitz. 2021. "Vaccine-Based Immunotherapy for Head and Neck Cancers" Cancers 13, no. 23: 6041. https://doi.org/10.3390/cancers13236041
APA StyleBeyaert, S., Machiels, J. -P., & Schmitz, S. (2021). Vaccine-Based Immunotherapy for Head and Neck Cancers. Cancers, 13(23), 6041. https://doi.org/10.3390/cancers13236041