
Tumor Mutation Burden, Expressed Neoantigens and the Immune Microenvironment in Diffuse Gliomas

 , , , , , , , and
, , , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Sample Characteristics
2.2. Pathogenic Germline Mutations
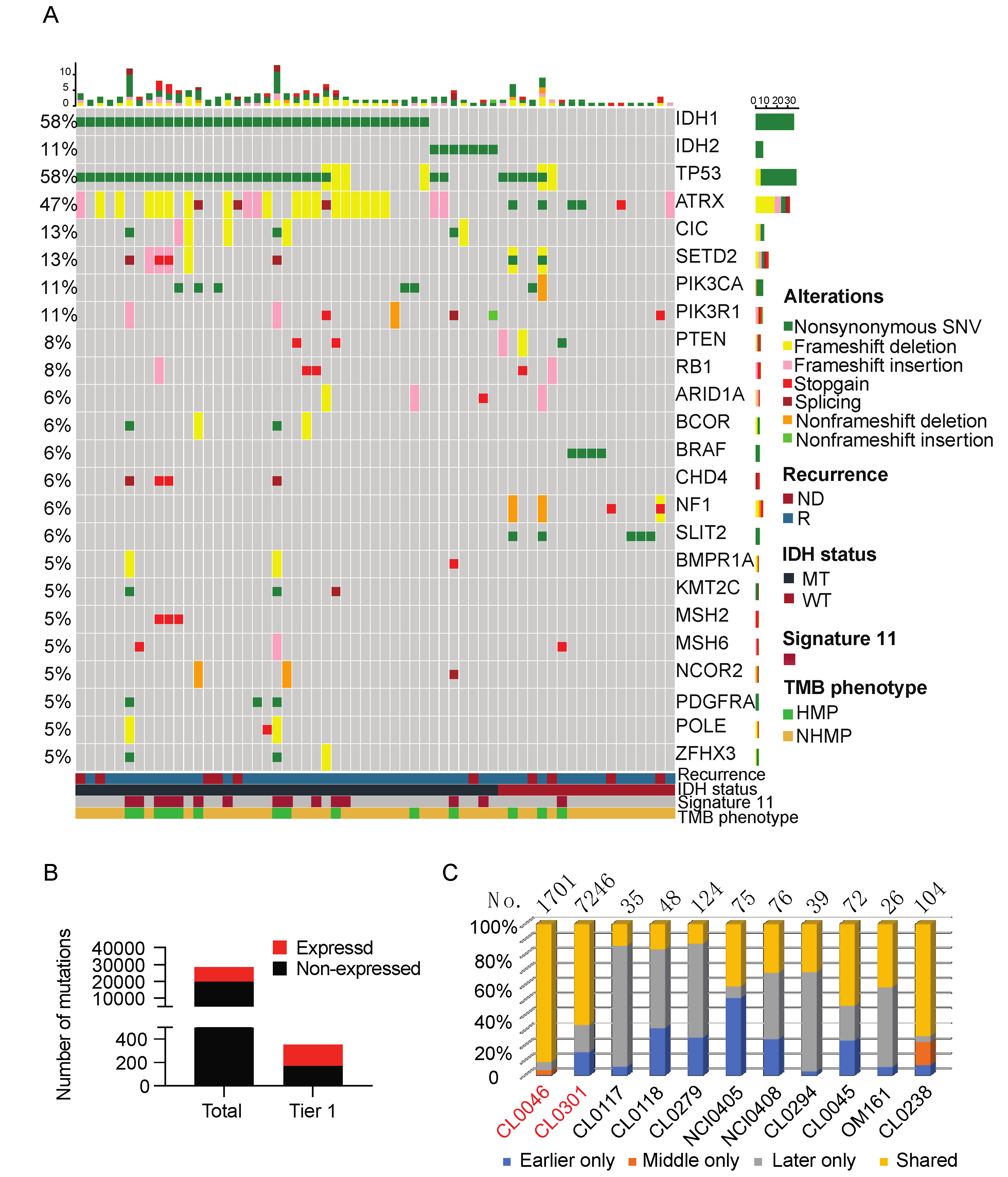
2.3. Mutational Landscape
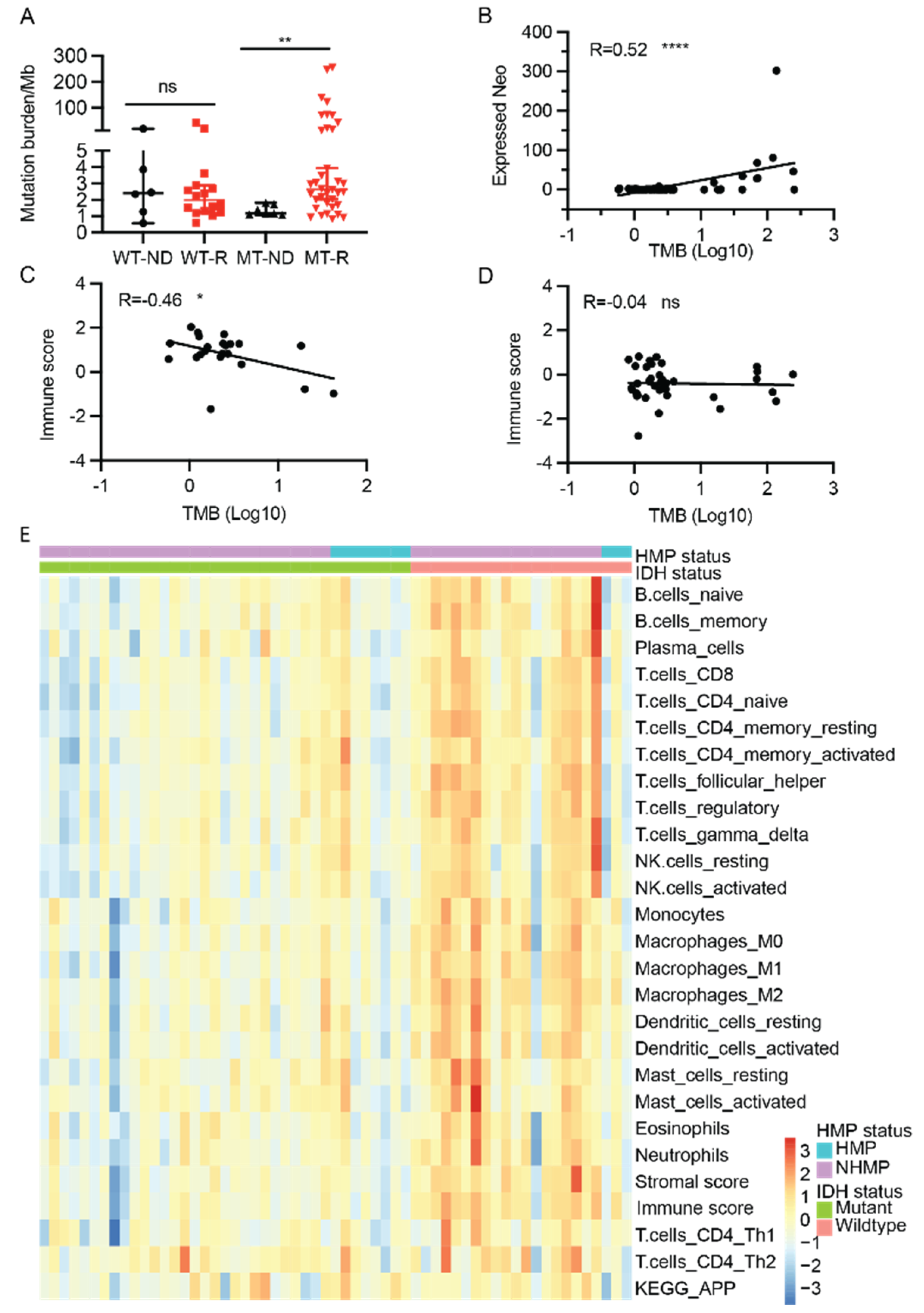
2.4. TMB, Neoantigens, and Immune Signatures
2.5. Antigen Processing and Presentation
2.6. Immunosuppressive Gene Expression in Gliomas
3. Discussion
3.1. Germline Variants of P/LP CPGs in Gliomas
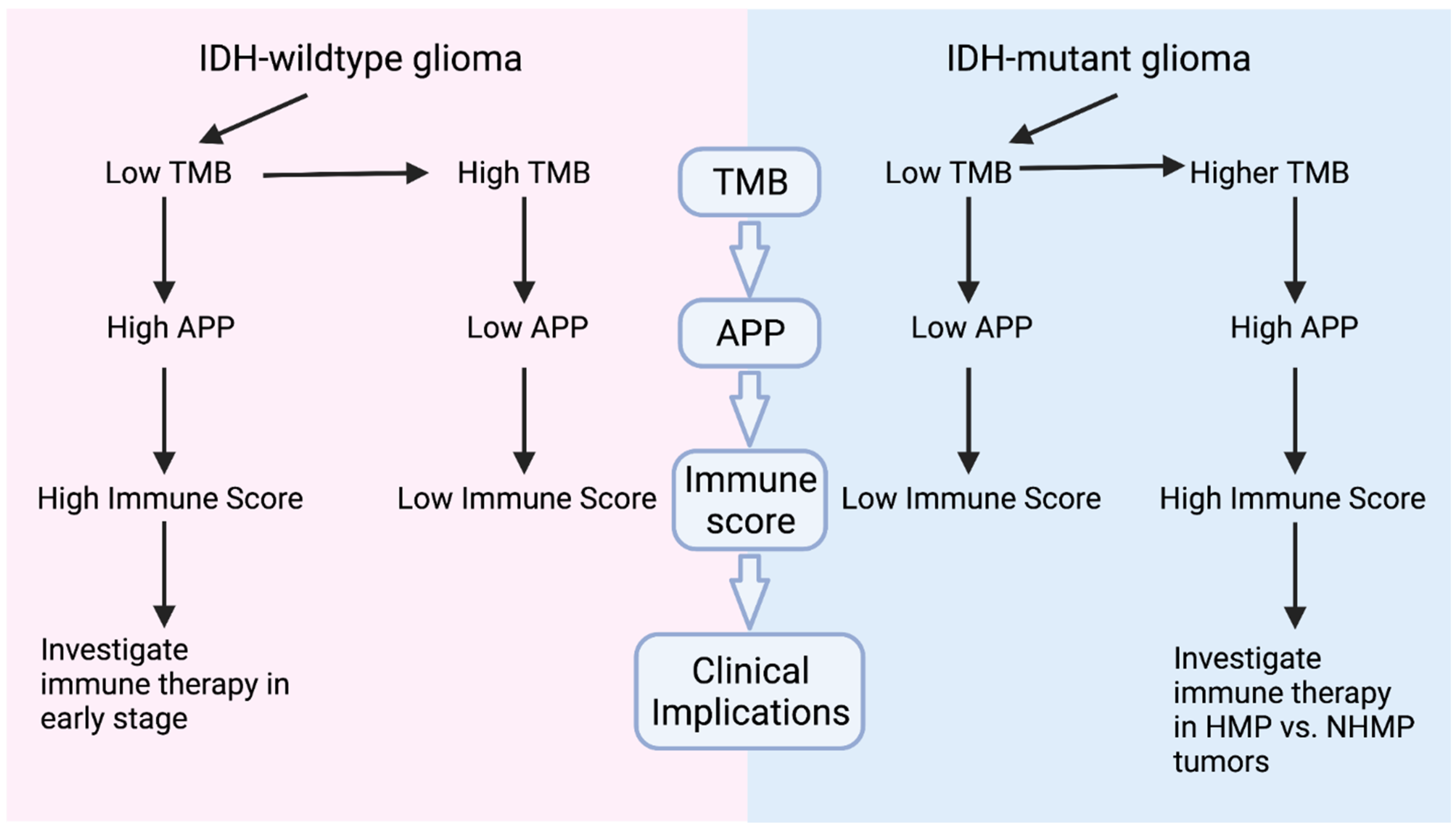
3.2. TMB, Immune Signatures, and IDH Mutation Status
3.3. Clinical Implications, Prospectives and Limitations
4. Conclusions
5. Materials and Methods
5.1. Patients and Samples
5.2. mRNA Sequencing (RNAseq)
5.3. Whole Exome Sequencing
5.4. Identification of Somatic Mutation
5.5. Neoantigen Prediction from Mutations and Fusions, and Expressed Neoantigen Computation
5.6. Statistical Analysis
5.7. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Prasad, V.; Addeo, A. The FDA approval of pembrolizumab for patients with TMB >10 mut/Mb: Was it a wise decision? No. Ann. Oncol. 2020, 31, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.J.N. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Ranjan, A.; Pang, Y.; Yu, G.; Kim, O.; Khan, J.; Wu, J. Tumor mutational burden and immunotherapy in gliomas. Trends Cancer 2021, 7, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Turkalp, Z.; Karamchandani, J.; Das, S. IDH mutation in glioma: New insights and promises for the future. JAMA Neurol. 2014, 71, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Roper, N.; Brown, A.L.; Wei, J.S.; Pack, S.; Trindade, C.; Kim, C.; Restifo, O.; Gao, S.; Sindiri, S.; Mehrabadi, F.; et al. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep. Med. 2020, 1, 100007. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Harmanci, A.S.; Erson-Omay, E.Z.; Li, J.; Coskun, S.; Simon, M.; Krischek, B.; Ozduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.; Yu, G.; Butler, M.; Sindiri, S.; Song, Y.K.; Wei, J.S.; Wen, X.; Chou, H.C.; Quezado, M.; Pack, S.; et al. Report of Canonical BCR-ABL1 Fusion in Glioblastoma. JCO Precis. Oncol. 2021, 5, PO.20.00519. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Long, W.; Ma, Q.; Xiao, K.; Li, Y.; Xiao, Q.; Peng, G.; Yuan, J.; Liu, Q. Identification of a Tumor Microenvironment-Related Eight-Gene Signature for Predicting Prognosis in Lower-Grade Gliomas. Front. Genet. 2019, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehling, M.; Simon, P.; Mittelbronn, M.; Meyermann, R.; Ferrone, S.; Weller, M.; Wiendl, H. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: Does it reflect a potential immune escape mechanism? Acta Neuropathol. 2007, 114, 111–119. [Google Scholar] [CrossRef]
- Facoetti, A.; Nano, R.; Zelini, P.; Morbini, P.; Benericetti, E.; Ceroni, M.; Campoli, M.; Ferrone, S. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin. Cancer Res. 2005, 11, 8304–8311. [Google Scholar] [CrossRef] [Green Version]
- Yeung, J.T.; Hamilton, R.L.; Ohnishi, K.; Ikeura, M.; Potter, D.M.; Nikiforova, M.N.; Ferrone, S.; Jakacki, R.I.; Pollack, I.F.; Okada, H. LOH in the HLA class I region at 6p21 is associated with shorter survival in newly diagnosed adult glioblastoma. Clin. Cancer Res. 2013, 19, 1816–1826. [Google Scholar] [CrossRef] [Green Version]
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. 2021, 11, 1–20. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Pitroski, C.E.; Cossio, S.L.; Koehler-Santos, P.; Graudenz, M.; Prolla, J.C.; Ashton-Prolla, P. Frequency of the common germline MUTYH mutations p.G396D and p.Y179C in patients diagnosed with colorectal cancer in Southern Brazil. Int. J. Colorectal Dis. 2011, 26, 841–846. [Google Scholar] [CrossRef]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol. 2017, 8, 4–23. [Google Scholar] [CrossRef]
- Gromeier, M.; Brown, M.C.; Zhang, G.; Lin, X.; Chen, Y.; Wei, Z.; Beaubier, N.; Yan, H.; He, Y.; Desjardins, A.; et al. Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat. Commun. 2021, 12, 352. [Google Scholar] [CrossRef] [PubMed]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.H.; Mansour, S.; Pritchard-Jones, K.; Kumar, D.; MacSweeney, F.; Rahman, N. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat. Clin. Pract. Oncol. 2007, 4, 130–134. [Google Scholar] [CrossRef]
- Chang, W.; Brohl, A.S.; Patidar, R.; Sindiri, S.; Shern, J.F.; Wei, J.S.; Song, Y.K.; Yohe, M.E.; Gryder, B.; Zhang, S.; et al. Multidimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. Clin. Cancer Res. 2016, 22, 3810–3820. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Samples n = 66 | IDH Mutant n = 44 | IDH Wildtype n = 22 | |
|---|---|---|---|
| Tumor histological type | |||
| Astrocytoma | 54 | 32 | 22 |
| Oligodendroglioma | 12 | 12 | 0 |
| WHO grade | |||
| II | 4 | 4 | 0 |
| III | 28 | 23 | 5 |
| IV | 34 | 17 | 17 |
| Disease status | |||
| Primary disease | 13 | 7 | 6 |
| Recurrent disease | 53 | 37 | 16 |
| No. recurrence | |||
| 0 | 13 | 7 | 6 |
| 1–2 | 33 | 19 | 14 |
| 3–5 | 18 | 16 | 2 |
| >5 | 2 | 2 | 0 |
| Tumor mutation burden * | |||
| NHMP | 51 | 32 | 19 |
| HMP | 14 | 11 | 3 |
| Prior brain tumor therapies ** | |||
| TMZ/TMZ+RT | 43 | 31 | 12 |
| XRT | 4 | 1 | 3 |
| Others *** | 19 | 12 | 7 |
| Patient | Diagnosis * | Gene | Mutation | Associated Mendelian Disease | Mendelian Inheritance | ACMG-Based Classification [10] | HMP |
|---|---|---|---|---|---|---|---|
| OM161 | Astrocytoma, IDH-mutant, WHO grade 4 | TP53 | p.R209Q | Li-Fraumeni syndrome | Autosomal Dominant | Likely pathogenic | No |
| CL0095 | Astrocytoma, IDH-mutant, WHO grade 4 | MUTYH | p.G396D | MUTYH associated polyposis | Autosomal Dominant | Likely pathogenic | No |
| CL0101 | Astrocytoma, IDH-mutant, grade 4 | BLM | p.Q548X | Bloom Syndrome | Autosomal Dominant | Pathogenic | No |
| CL0248 | Astrocytoma, IDH-mutant, WHO grade 3 | RET | p.K666N | Medullary thyroid carcinoma | Autosomal Dominant | Pathogenic/Likely pathogenic | No |
| CL0301 | Astrocytoma, IDH-mutant, WHO grade 4 | ERCC6 | p.R670W | Cockayne syndrome | Autosomal Dominant | Likely pathogenic | Yes |
| CL0326 | Astrocytoma, IDH-mutant, WHO grade 3 | MITF | p.E419K | Susceptibility to cutaneous melanomaWaardenburg syndrome | Risk factorAutosomal Dominant | Risk factor/Likely pathogenic for cutaneous melanoma | No |
| CL0332 | Astrocytoma, IDH-mutant, WHO grade 4 | MUTYH | p.G396D | MUTYH associated polyposis | Autosomal Dominant | Likely pathogenic | Yes |
| NCI0391 | Gliosarcoma, IDH-wildtype, WHO grade 4 | BRIP1 | p.T997fs | Fanconi Anemia | Autosomal Dominant | Pathogenic | No |
| NCI0392 | Glioblastoma, IDH-wildtype, WHO grade 4 | MSH2 | c.1386+1G>A | Lynch syndrome | Autosomal Dominant | Pathogenic | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, G.; Pang, Y.; Merchant, M.; Kesserwan, C.; Gangalapudi, V.; Abdelmaksoud, A.; Ranjan, A.; Kim, O.; Wei, J.S.; Chou, H.-C.; et al. Tumor Mutation Burden, Expressed Neoantigens and the Immune Microenvironment in Diffuse Gliomas. Cancers 2021, 13, 6092. https://doi.org/10.3390/cancers13236092
Yu G, Pang Y, Merchant M, Kesserwan C, Gangalapudi V, Abdelmaksoud A, Ranjan A, Kim O, Wei JS, Chou H-C, et al. Tumor Mutation Burden, Expressed Neoantigens and the Immune Microenvironment in Diffuse Gliomas. Cancers. 2021; 13(23):6092. https://doi.org/10.3390/cancers13236092
Chicago/Turabian StyleYu, Guangyang, Ying Pang, Mythili Merchant, Chimene Kesserwan, Vineela Gangalapudi, Abdalla Abdelmaksoud, Alice Ranjan, Olga Kim, Jun S. Wei, Hsien-Chao Chou, and et al. 2021. "Tumor Mutation Burden, Expressed Neoantigens and the Immune Microenvironment in Diffuse Gliomas" Cancers 13, no. 23: 6092. https://doi.org/10.3390/cancers13236092
APA StyleYu, G., Pang, Y., Merchant, M., Kesserwan, C., Gangalapudi, V., Abdelmaksoud, A., Ranjan, A., Kim, O., Wei, J. S., Chou, H. -C., Wen, X., Sindiri, S., Song, Y. K., Xi, L., Kaplan, R. N., Armstrong, T. S., Gilbert, M. R., Aldape, K., Khan, J., & Wu, J. (2021). Tumor Mutation Burden, Expressed Neoantigens and the Immune Microenvironment in Diffuse Gliomas. Cancers, 13(23), 6092. https://doi.org/10.3390/cancers13236092