
Cell-of-Origin and Genetic, Epigenetic, and Microenvironmental Factors Contribute to the Intra-Tumoral Heterogeneity of Pediatric Intracranial Ependymoma

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Inter-Tumoral Heterogeneity and Clinicopathological Characteristics of Pediatric Intracranial EPN: A Brief Overview

{kind=link}
{kind=link}
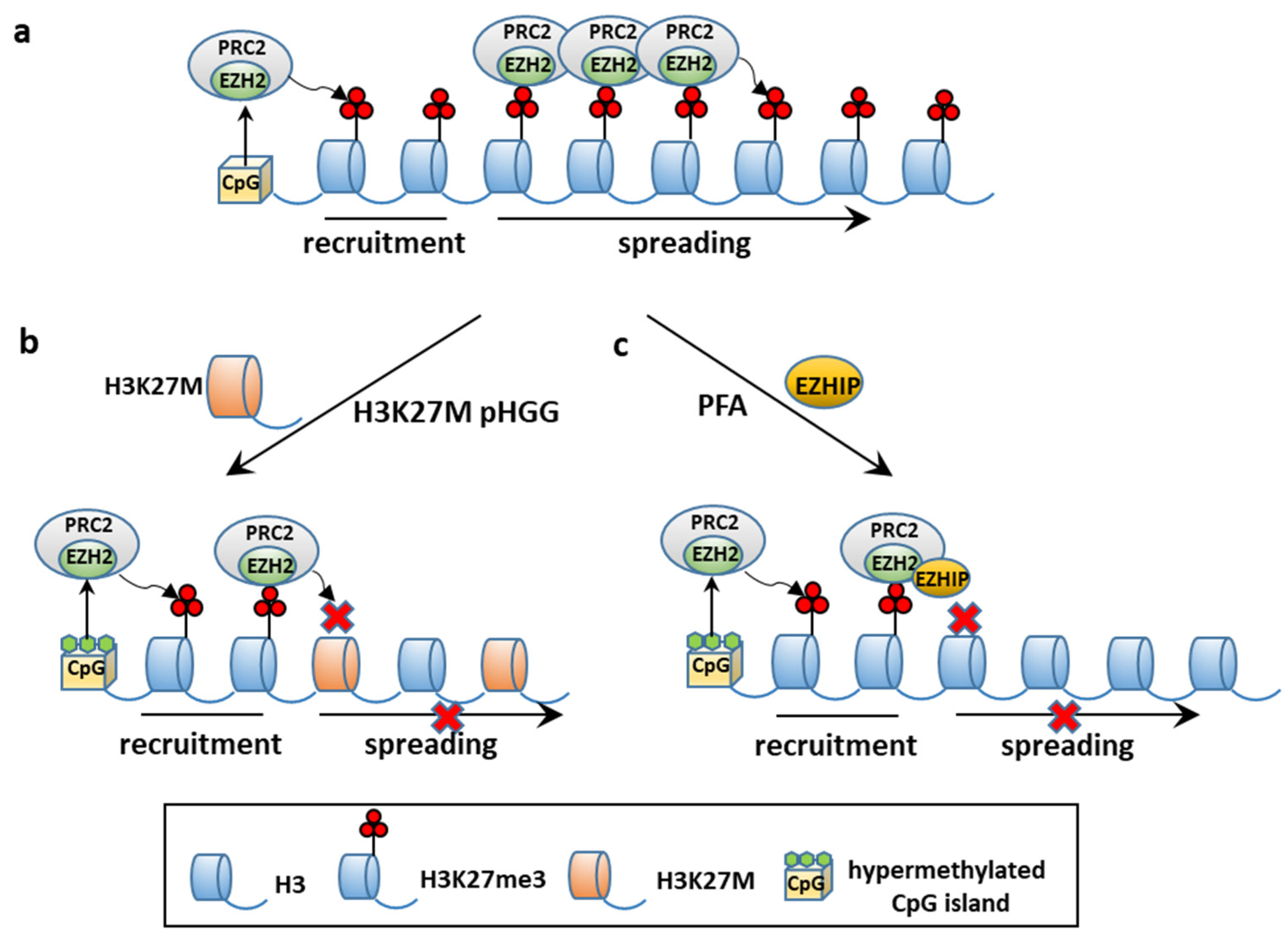{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Group | ST-RELA | ST-YAP1 | PFA | PFB | References |
|---|---|---|---|---|---|
| Location | ST, cerebral | ST, intra-periventricular | PF | PF | [25,40] |
| Age | children/adolescents median age 8 years | young children median age 1.4 years | young children median age 3 years | all age groups median age 30 years | [23] |
| Gender | [23] | ||||
| Male | 65% | 25% | 65% | 41% | |
| Female | 35% | 75% | 35% | 59% | |
| Molecular events | |||||
| Genetic | chromothripsis ZFTA-RELA fusions CDKN2a deletion loss of chromosome 9 | YAP1-fusions | balanced genome 1q gain 6q loss infrequent H3K27M substitution infrequent EZHIP mutations | chromosomal instability | [23] [23,24] [23,30] [35] [36] |
| Epigenetic | CIMP positive DNA hypomethylation H3K27me3 loss EZHIP overexpression | CIMP negative H3K27me3 retention | [31] [32] [32,34] [36] | ||
| Pathogenic impact | NF-κB pathway cell cycle cell migration MAPK pathway | Hippo pathway | angiogenesis RTK pathways cell cycle cell migration derepression of PRC2 target genes | ciliogenesis oxidative metabolism | [24,39] [23,25] [31] |
| Outcome | poor | favorable | poor | favorable | [23] |
3. CSCs as a Source of ITH
3.1. The CSC Model
3.2. CSC-Driven Preclinical Models of EPN
4. Determinants of ITH
4.1. Genetic ITH
4.1.1. CSCs and Genomic Instability: ST-Ependymomagenesis
4.2. Epigenetic ITH
4.2.1. CSCs and Epigenetic Alterations: PFA Ependymomagenesis
4.3. TME, CSCs, and EPN
4.3.1. The Perivascular TME
4.3.2. The Hypoxic TME
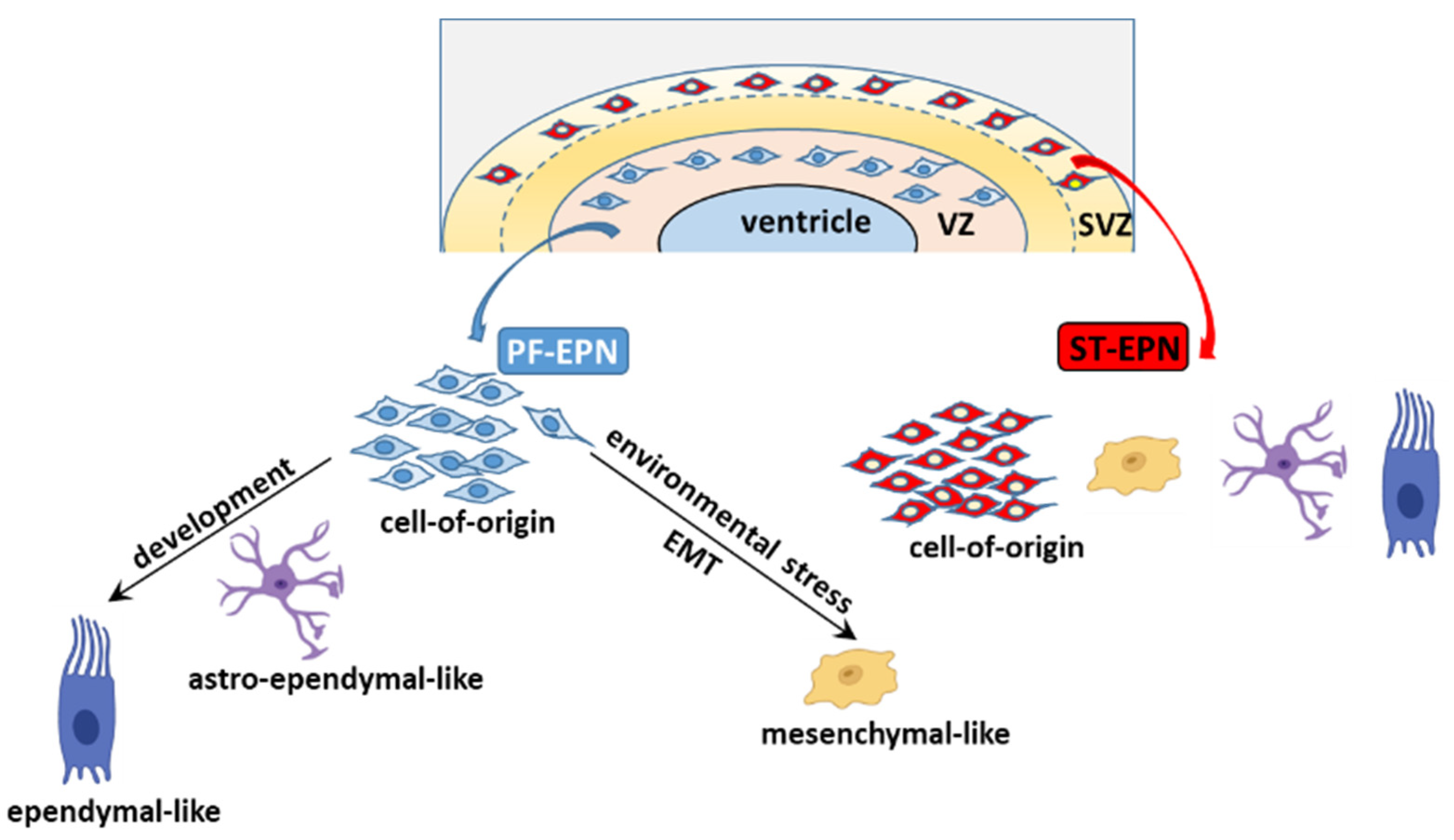
4.3.3. EMT
5. ITH of EPN: A Single-Cell Perspective
5.1. EPN Is Composed of Multiple Discrete Neoplastic Subpopulations
5.1.1. PF-EPN and scRNA-seq
5.1.2. ST-EPN and scRNA-seq
5.2. The Cell of Origin and Developmental Trajectories of EPN from an scRNA-seq Perspective
6. Therapeutic Applications
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BTSCs | brain tumor stem cells |
| CEC | ciliated ependymoma cells |
| CIMP | CpG island methylator phenotype |
| CSCs | cancer stem cells |
| DIPG | diffuse intrinsic pontine glioma |
| ecDNA | extrachromosomal DNA |
| EMT | epithelial-to-mesenchymal transition |
| EPN | ependymoma |
| ESCs | embryonic stem cells |
| GBM | glioblastoma |
| GSCs | glioblastoma stem cells |
| ITH | intra-tumoral heterogeneity |
| MEC | mesenchymal ependymoma cells |
| MET | mesenchymal epithelial transition |
| NSCs | neural stem cells |
| NSs | neurospheres |
| OPCs | oligodendrocyte precursor cells |
| oRGCs | outer radial glia cells |
| oSVZ | outer subventricular zone |
| PBTs | pediatric brain tumors |
| PF | posterior fossa |
| PF-NSC-like | posterior fossa-Neural-Stem-Cell-like |
| pHGG | pediatric high-grade gliomas |
| RGCs | radial glia cells |
| RTK | receptor tyrosine kinase |
| RT-PCR | reverse transcriptase polymerase chain reaction |
| scRNA-seq | single-cell RNA sequencing |
| ST | supratentorial |
| ST_EPN | supratentorial ependymoma |
| TCA | tricarboxylic acid cycle |
| TECs | transportive ependymoma cells |
| TF | transcription factor |
| TI | trajectory inference |
| TME | tumor microenvironment |
| UEC-1 | undifferentiated ependymoma cells-1 |
| vRGCs | ventricular radial glia cells |
| VZ | ventricular zone |
References
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, M.R.; de Sauvage, F.J. Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R. Tumor Heterogeneity—A “Contemporary Concept” Founded on Historical Insights and Predictions. Cancer Res. 2016, 76, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppner, G.H. Tumor Heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar]
- Merlo, L.M.F.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an Evolutionary and Ecological Process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-Tumour Heterogeneity: A Looking Glass for Cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.C.; Cunningham, J.J.; Bui, M.M.; Gillies, R.J.; Brown, J.S.; Gatenby, R.A. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer Res. 2016, 76, 3136–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A. Epigenetic Plasticity, Selection, and Tumorigenesis. Biochem. Soc. Trans. 2020, 48, 1609–1621. [Google Scholar] [CrossRef]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-like States, and Drug Resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Clevers, H. Cancer Stem Cells Revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Inda, M.-M.; Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.Y.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; et al. Tumor Heterogeneity Is an Active Process Maintained by a Mutant EGFR-Induced Cytokine Circuit in Glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Marusyk, A. Cancer: Clonal Cooperation. Nature 2014, 508, 52–53. [Google Scholar] [CrossRef]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-Oncogenic Signals That Maintain Intratumoral Heterogeneity. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional Communication in the Microenvirons of Glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Tabassum, D.P.; Castaño, Z.; Cristea, S.; Yamamoto, K.N.; Kingston, N.L.; Murphy, K.C.; Shu, S.; Harper, N.W.; Del Alcazar, C.G.; et al. Subclonal Cooperation Drives Metastasis by Modulating Local and Systemic Immune Microenvironments. Nat. Cell Biol. 2019, 21, 879–888. [Google Scholar] [CrossRef]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Naffar-Abu Amara, S.; Kuiken, H.J.; Selfors, L.M.; Butler, T.; Leung, M.L.; Leung, C.T.; Kuhn, E.P.; Kolarova, T.; Hage, C.; Ganesh, K.; et al. Transient Commensal Clonal Interactions Can Drive Tumor Metastasis. Nat. Commun. 2020, 11, 5799. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajtler, K.W.; Mack, S.C.; Ramaswamy, V.; Smith, C.A.; Witt, H.; Smith, A.; Hansford, J.R.; von Hoff, K.; Wright, K.D.; Hwang, E.; et al. The Current Consensus on the Clinical Management of Intracranial Ependymoma and Its Distinct Molecular Variants. Acta Neuropathol. 2017, 133, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.W.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95-RELA Fusions Drive Oncogenic NF-ΚB Signalling in Ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, H.; Mack, S.C.; Ryzhova, M.; Bender, S.; Sill, M.; Isserlin, R.; Benner, A.; Hielscher, T.; Milde, T.; Remke, M.; et al. Delineation of Two Clinically and Molecularly Distinct Subgroups of Posterior Fossa Ependymoma. Cancer Cell 2011, 20, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef]
- Stuart, T.; Satija, R. Integrative Single-Cell Analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.; Perry, A.; Wesseling, P.; Brat, D.; Cree, I.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Baroni, L.V.; Sundaresan, L.; Heled, A.; Coltin, H.; Pajtler, K.W.; Lin, T.; Merchant, T.E.; McLendon, R.; Faria, C.; Buntine, M.; et al. Ultra High-Risk PFA Ependymoma Is Characterized by Loss of Chromosome 6q. Neuro Oncol. 2021, 23, 1360–1370. [Google Scholar] [CrossRef]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stütz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic Alterations Define Lethal CIMP-Positive Ependymomas of Infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, J.; Mukherjee, P.; Lu, C.; Jain, S.U.; Chung, C.; Martinez, D.; Sabari, B.; Margol, A.S.; Panwalkar, P.; Parolia, A.; et al. Lowered H3K27me3 and DNA Hypomethylation Define Poorly Prognostic Pediatric Posterior Fossa Ependymomas. Sci. Transl. Med. 2016, 8, 366ra161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panwalkar, P.; Clark, J.; Ramaswamy, V.; Hawes, D.; Yang, F.; Dunham, C.; Yip, S.; Hukin, J.; Sun, Y.; Schipper, M.J.; et al. Immunohistochemical Analysis of H3K27me3 Demonstrates Global Reduction in Group-A Childhood Posterior Fossa Ependymoma and Is a Powerful Predictor of Outcome. Acta Neuropathol. 2017, 134, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Gessi, M.; Capper, D.; Sahm, F.; Huang, K.; von Deimling, A.; Tippelt, S.; Fleischhack, G.; Scherbaum, D.; Alfer, J.; Juhnke, B.-O.; et al. Evidence of H3 K27M Mutations in Posterior Fossa Ependymomas. Acta Neuropathol. 2016, 132, 635–637. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Wen, J.; Sill, M.; Lin, T.; Orisme, W.; Tang, B.; Hübner, J.-M.; Ramaswamy, V.; Jia, S.; Dalton, J.D.; et al. Molecular Heterogeneity and CXorf67 Alterations in Posterior Fossa Group A (PFA) Ependymomas. Acta Neuropathol. 2018, 136, 211–226. [Google Scholar] [CrossRef]
- Cavalli, F.M.G.; Hübner, J.-M.; Sharma, T.; Luu, B.; Sill, M.; Zapotocky, M.; Mack, S.C.; Witt, H.; Lin, T.; Shih, D.J.H.; et al. Heterogeneity within the PF-EPN-B Ependymoma Subgroup. Acta Neuropathol. 2018, 136, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Wei, Y.; Okonechnikov, K.; Silva, P.B.G.; Vouri, M.; Zhang, L.; Brabetz, S.; Sieber, L.; Gulley, M.; Mauermann, M.; et al. YAP1 Subgroup Supratentorial Ependymoma Requires TEAD and Nuclear Factor I-Mediated Transcriptional Programmes for Tumorigenesis. Nat. Commun. 2019, 10, 3914. [Google Scholar] [CrossRef] [Green Version]
- Andreiuolo, F.; Varlet, P.; Tauziède-Espariat, A.; Jünger, S.T.; Dörner, E.; Dreschmann, V.; Kuchelmeister, K.; Waha, A.; Haberler, C.; Slavc, I.; et al. Childhood Supratentorial Ependymomas with YAP1-MAMLD1 Fusion: An Entity with Characteristic Clinical, Radiological, Cytogenetic and Histopathological Features. Brain Pathol. Zurich Switz. 2019, 29, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuoka, K.; Kanemura, Y.; Shofuda, T.; Fukushima, S.; Yamashita, S.; Narushima, D.; Kato, M.; Honda-Kitahara, M.; Ichikawa, H.; Kohno, T.; et al. Significance of Molecular Classification of Ependymomas: C11orf95-RELA Fusion-Negative Supratentorial Ependymomas Are a Heterogeneous Group of Tumors. Acta Neuropathol. Commun. 2018, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Tamai, S.; Nakano, Y.; Kinoshita, M.; Sabit, H.; Nobusawa, S.; Arai, Y.; Hama, N.; Totoki, Y.; Shibata, T.; Ichimura, K.; et al. Ependymoma with C11orf95-MAML2 Fusion: Presenting with Granular Cell and Ganglion Cell Features. Brain Tumor Pathol. 2021, 38, 64–70. [Google Scholar] [CrossRef]
- Zheng, T.; Ghasemi, D.R.; Okonechnikov, K.; Korshunov, A.; Sill, M.; Maass, K.K.; Benites Goncalves da Silva, P.; Ryzhova, M.; Gojo, J.; Stichel, D.; et al. Cross-Species Genomics Reveals Oncogenic Dependencies in ZFTA/C11orf95 Fusion-Positive Supratentorial Ependymomas. Cancer Discov. 2021, 11, 2230–2247. [Google Scholar] [CrossRef]
- Tauziède-Espariat, A.; Siegfried, A.; Nicaise, Y.; Kergrohen, T.; Sievers, P.; Vasiljevic, A.; Roux, A.; Dezamis, E.; Benevello, C.; Machet, M.-C.; et al. Supratentorial Non-RELA, ZFTA-Fused Ependymomas: A Comprehensive Phenotype Genotype Correlation Highlighting the Number of Zinc Fingers in ZFTA-NCOA1/2 Fusions. Acta Neuropathol. Commun. 2021, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Zschernack, V.; Jünger, S.T.; Mynarek, M.; Rutkowski, S.; Garre, M.L.; Ebinger, M.; Neu, M.; Faber, J.; Erdlenbruch, B.; Claviez, A.; et al. Supratentorial Ependymoma in Childhood: More than Just RELA or YAP. Acta Neuropathol. 2021, 141, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Sievers, P.; Henneken, S.C.; Blume, C.; Sill, M.; Schrimpf, D.; Stichel, D.; Okonechnikov, K.; Reuss, D.E.; Benzel, J.; Maaß, K.K.; et al. Recurrent Fusions in PLAGL1 Define a Distinct Subset of Pediatric-Type Supratentorial Neuroepithelial Tumors. Acta Neuropathol. 2021, 142, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Aldape, K.D.; Capper, D.; Fouladi, M.; Gilbert, M.R.; Gilbertson, R.J.; Hawkins, C.; Merchant, T.E.; Pajtler, K.; Venneti, S.; et al. CIMPACT-NOW Update 7: Advancing the Molecular Classification of Ependymal Tumors. Brain Pathol. Zurich Switz. 2020, 30, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.; Graham, R.T.; Harreld, J.H.; Lucas, J.T.; Finkelstein, D.; Wheeler, D.; Li, X.; Dalton, J.; Upadhyaya, S.A.; Raimondi, S.C.; et al. Infratentorial C11orf95-Fused Gliomas Share Histologic, Immunophenotypic, and Molecular Characteristics of Supratentorial RELA-Fused Ependymoma. Acta Neuropathol. 2020, 140, 963–965. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem Cells, Cancer, and Cancer Stem Cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-Tumor Heterogeneity from a Cancer Stem Cell Perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct Isolation of Human Central Nervous System Stem Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the Principles of Stem-Cell Biology to Cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain Tumour Stem Cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Prestegarden, L.; Svendsen, A.; Wang, J.; Sleire, L.; Skaftnesmo, K.O.; Bjerkvig, R.; Yan, T.; Askland, L.; Persson, A.; Sakariassen, P.Ø.; et al. Glioma Cell Populations Grouped by Different Cell Type Markers Drive Brain Tumor Growth. Cancer Res. 2010, 70, 4274–4279. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem Cell Fate in Cancer Growth, Progression and Therapy Resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Kondo, T. Glioblastoma-Initiating Cell Heterogeneity Generated by the Cell-of-Origin, Genetic/Epigenetic Mutation and Microenvironment. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Muñoz, P.; Iliou, M.S.; Esteller, M. Epigenetic Alterations Involved in Cancer Stem Cell Reprogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Liu, W.; Engelmann, J.C.; Nuhn, P.; Gurel, M.; Haffner, M.C.; Esopi, D.; Irizarry, R.A.; Getzenberg, R.H.; Nelson, W.G.; et al. DNA Methylation Alterations Exhibit Intraindividual Stability and Interindividual Heterogeneity in Prostate Cancer Metastases. Sci. Transl. Med. 2013, 5, 169ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hide, T.; Takezaki, T.; Nakatani, Y.; Nakamura, H.; Kuratsu, J.; Kondo, T. Combination of a Ptgs2 Inhibitor and an Epidermal Growth Factor Receptor-Signaling Inhibitor Prevents Tumorigenesis of Oligodendrocyte Lineage-Derived Glioma-Initiating Cells. Stem Cells 2011, 29, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of Human Embryonic Stem Cells to Model Pediatric Gliomas with H3.3K27M Histone Mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, S.; Quezada, M.A.; Gillespie, S.M.; Arzt, M.; Lennon, J.J.; Woo, P.J.; Hovestadt, V.; Kambhampati, M.; Filbin, M.G.; Suva, M.L.; et al. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 2019, 76, 965–980.e12. [Google Scholar] [CrossRef] [PubMed]
- Haag, D.; Mack, N.; Benites Goncalves da Silva, P.; Statz, B.; Clark, J.; Tanabe, K.; Sharma, T.; Jäger, N.; Jones, D.T.W.; Kawauchi, D.; et al. H3.3-K27M Drives Neural Stem Cell-Specific Gliomagenesis in a Human IPSC-Derived Model. Cancer Cell 2021, 39, 407–422.e13. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Ellis, T.; Markant, S.L.; Read, T.-A.; Kessler, J.D.; Bourboulas, M.; Schüller, U.; Machold, R.; Fishell, G.; Rowitch, D.H.; et al. Medulloblastoma Can Be Initiated by Deletion of Patched in Lineage-Restricted Progenitors or Stem Cells. Cancer Cell 2008, 14, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous Stem Cells Can Arise from Pediatric Brain Tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Taylor, M.D.; Poppleton, H.; Fuller, C.; Su, X.; Liu, Y.; Jensen, P.; Magdaleno, S.; Dalton, J.; Calabrese, C.; Board, J.; et al. Radial Glia Cells Are Candidate Stem Cells of Ependymoma. Cancer Cell 2005, 8, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Götz, M.; Huttner, W.B. The Cell Biology of Neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Servidei, T.; Meco, D.; Trivieri, N.; Patriarca, V.; Vellone, V.G.; Zannoni, G.F.; Lamorte, G.; Pallini, R.; Riccardi, R. Effects of Epidermal Growth Factor Receptor Blockade on Ependymoma Stem Cells in Vitro and in Orthotopic Mouse Models. Int. J. Cancer 2012, 131, E791–E803. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Baxter, P.A.; Voicu, H.; Gurusiddappa, S.; Zhao, Y.; Adesina, A.; Man, T.-K.; Shu, Q.; Zhang, Y.-J.; Zhao, X.-M.; et al. A Clinically Relevant Orthotopic Xenograft Model of Ependymoma That Maintains the Genomic Signature of the Primary Tumor and Preserves Cancer Stem Cells in Vivo. Neuro Oncol. 2010, 12, 580–594. [Google Scholar] [CrossRef]
- Milde, T.; Kleber, S.; Korshunov, A.; Witt, H.; Hielscher, T.; Koch, P.; Kopp, H.-G.; Jugold, M.; Deubzer, H.E.; Oehme, I.; et al. A Novel Human High-Risk Ependymoma Stem Cell Model Reveals the Differentiation-Inducing Potential of the Histone Deacetylase Inhibitor Vorinostat. Acta Neuropathol. 2011, 122, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Amani, V.; Donson, A.M.; Lummus, S.C.; Prince, E.W.; Griesinger, A.M.; Witt, D.A.; Hankinson, T.C.; Handler, M.H.; Dorris, K.; Vibhakar, R.; et al. Characterization of 2 Novel Ependymoma Cell Lines with Chromosome 1q Gain Derived From Posterior Fossa Tumors of Childhood. J. Neuropathol. Exp. Neurol. 2017, 76, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically Engineered Cerebral Organoids Model Brain Tumor Formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Sabnis, D.H.; Liu, J.-F.; Simmonds, L.; Blackburn, S.; Grundy, R.G.; Kerr, I.D.; Coyle, B. BLBP Is Both a Marker for Poor Prognosis and a Potential Therapeutic Target in Paediatric Ependymoma. Cancers 2021, 13, 2100. [Google Scholar] [CrossRef]
- Meco, D.; Servidei, T.; Lamorte, G.; Binda, E.; Arena, V.; Riccardi, R. Ependymoma Stem Cells Are Highly Sensitive to Temozolomide in Vitro and in Orthotopic Models. Neuro Oncol. 2014, 16, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Servidei, T.; Meco, D.; Muto, V.; Bruselles, A.; Ciolfi, A.; Trivieri, N.; Lucchini, M.; Morosetti, R.; Mirabella, M.; Martini, M.; et al. Novel SEC61G-EGFR Fusion Gene in Pediatric Ependymomas Discovered by Clonal Expansion of Stem Cells in Absence of Exogenous Mitogens. Cancer Res. 2017, 77, 5860–5872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral Heterogeneity of Receptor Tyrosine Kinases EGFR and PDGFRA Amplification in Glioblastoma Defines Subpopulations with Distinct Growth Factor Response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Schulte, A.; Günther, H.S.; Martens, T.; Zapf, S.; Riethdorf, S.; Wülfing, C.; Stoupiec, M.; Westphal, M.; Lamszus, K. Glioblastoma Stem-like Cell Lines with Either Maintenance or Loss of High-Level EGFR Amplification, Generated via Modulation of Ligand Concentration. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1901–1913. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Cancer Genes and the Pathways They Control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging Patterns of Somatic Mutations in Cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability--an Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Siri, S.O.; Martino, J.; Gottifredi, V. Structural Chromosome Instability: Types, Origins, Consequences, and Therapeutic Opportunities. Cancers 2021, 13, 3056. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, T.; Kumar, P.; Koseoglu, M.M.; Dutta, A. Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet. 2018, 34, 270–278. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Bafna, V.; Mischel, P.S. Extrachromosomal Oncogene Amplification in Tumour Pathogenesis and Evolution. Nat. Rev. Cancer 2019, 19, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Turner, K.M.; Nguyen, N.; Raviram, R.; Erb, M.; Santini, J.; Luebeck, J.; Rajkumar, U.; Diao, Y.; Li, B.; et al. Circular EcDNA Promotes Accessible Chromatin and High Oncogene Expression. Nature 2019, 575, 699–703. [Google Scholar] [CrossRef]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal Oncogene Amplification Drives Tumour Evolution and Genetic Heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef] [PubMed]
- deCarvalho, A.C.; Kim, H.; Poisson, L.M.; Winn, M.E.; Mueller, C.; Cherba, D.; Koeman, J.; Seth, S.; Protopopov, A.; Felicella, M.; et al. Discordant Inheritance of Chromosomal and Extrachromosomal DNA Elements Contributes to Dynamic Disease Evolution in Glioblastoma. Nat. Genet. 2018, 50, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Nguyen, N.-P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA Is Associated with Oncogene Amplification and Poor Outcome across Multiple Cancers. Nat. Genet. 2020, 52, 891–897. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Ding, L.; Chang, T.-C.; Shao, Y.; Chiang, J.; Mulder, H.; Wang, S.; Shaw, T.I.; Wen, J.; Hover, L.; et al. Structure and Evolution of Double Minutes in Diagnosis and Relapse Brain Tumors. Acta Neuropathol. 2019, 137, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Ciriano, I.; Lee, J.J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Pellman, D.S.; et al. Comprehensive Analysis of Chromothripsis in 2,658 Human Cancers Using Whole-Genome Sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronina, N.; Wong, J.K.L.; Hübschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The Landscape of Chromothripsis across Adult Cancer Types. Nat. Commun. 2020, 11, 2320. [Google Scholar] [CrossRef]
- Johnson, R.A.; Wright, K.D.; Poppleton, H.; Mohankumar, K.M.; Finkelstein, D.; Pounds, S.B.; Rand, V.; Leary, S.E.S.; White, E.; Eden, C.; et al. Cross-Species Genomics Matches Driver Mutations and Cell Compartments to Model Ependymoma. Nature 2010, 466, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Mohankumar, K.M.; Currle, D.S.; White, E.; Boulos, N.; Dapper, J.; Eden, C.; Nimmervoll, B.; Thiruvenkatam, R.; Connelly, M.; Kranenburg, T.A.; et al. An in Vivo Screen Identifies Ependymoma Oncogenes and Tumor-Suppressor Genes. Nat. Genet. 2015, 47, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Arora, S.; Szulzewsky, F.; Juric-Sekhar, G.; Miyajima, Y.; Bolouri, H.; Yasui, Y.; Barber, J.; Kupp, R.; Dalton, J.; et al. A De Novo Mouse Model of C11orf95-RELA Fusion-Driven Ependymoma Identifies Driver Functions in Addition to NF-ΚB. Cell Rep. 2018, 23, 3787–3797. [Google Scholar] [CrossRef] [PubMed]
- Takadera, M.; Satomi, K.; Szulzewsky, F.; Cimino, P.J.; Holland, E.C.; Yamamoto, T.; Ichimura, K.; Ozawa, T. Phenotypic Characterization with Somatic Genome Editing and Gene Transfer Reveals the Diverse Oncogenicity of Ependymoma Fusion Genes. Acta Neuropathol. Commun. 2020, 8, 203. [Google Scholar] [CrossRef]
- de Almeida Magalhães, T.; Cruzeiro, G.A.V.; de Sousa, G.R.; da Silva, K.R.; Lira, R.C.P.; Scrideli, C.A.; Tone, L.G.; Valera, E.T.; Borges, K.S. Notch Pathway in Ependymoma RELA-Fused Subgroup: Upregulation and Association with Cancer Stem Cells Markers Expression. Cancer Gene Ther. 2020, 27, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.J.; Jillette, N.; Li, X.-N.; Cheng, A.W.; Lau, C.C. C11orf95-RELA Reprograms 3D Epigenome in Supratentorial Ependymoma. Acta Neuropathol. 2020, 140, 951–960. [Google Scholar] [CrossRef]
- Arabzade, A.; Zhao, Y.; Varadharajan, S.; Chen, H.-C.; Jessa, S.; Rivas, B.; Stuckert, A.J.; Solis, M.; Kardian, A.; Tlais, D.; et al. ZFTA-RELA Dictates Oncogenic Transcriptional Programs to Drive Aggressive Supratentorial Ependymoma. Cancer Discov. 2021, 11, 2200–2215. [Google Scholar] [CrossRef] [PubMed]
- Kupp, R.; Ruff, L.; Terranova, S.; Nathan, E.; Ballereau, S.; Stark, R.; Sekhar Reddy Chilamakuri, C.; Hoffmann, N.; Wickham-Rahrmann, K.; Widdess, M.; et al. ZFTA-Translocations Constitute Ependymoma Chromatin Remodeling and Transcription Factors. Cancer Discov. 2021, 11, 2216–2229. [Google Scholar] [CrossRef]
- Ozawa, T.; Kaneko, S.; Szulzewsky, F.; Qiao, Z.; Takadera, M.; Narita, Y.; Kondo, T.; Holland, E.C.; Hamamoto, R.; Ichimura, K. C11orf95-RELA Fusion Drives Aberrant Gene Expression through the Unique Epigenetic Regulation for Ependymoma Formation. Acta Neuropathol. Commun. 2021, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Hong, W. The Emerging Role of the Hippo Pathway in Cell Contact Inhibition, Organ Size Control, and Cancer Development in Mammals. Cancer Cell 2008, 13, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Arora, S.; Hoellerbauer, P.; King, C.; Nathan, E.; Chan, M.; Cimino, P.J.; Ozawa, T.; Kawauchi, D.; Pajtler, K.W.; et al. Comparison of Tumor-Associated YAP1 Fusions Identifies a Recurrent Set of Functions Critical for Oncogenesis. Genes Dev. 2020, 34, 1051–1064. [Google Scholar] [CrossRef]
- Tabasaran, J.; Schuhmann, M.; Ebinger, M.; Honegger, J.; Renovanz, M.; Schittenhelm, J. PAX6 Is Frequently Expressed in Ependymal Tumours and Associated with Prognostic Relevant Subgroups. J. Clin. Pathol. 2021. [Google Scholar] [CrossRef]
- Brock, A.; Chang, H.; Huang, S. Non-Genetic Heterogeneity—A Mutation-Independent Driving Force for the Somatic Evolution of Tumours. Nat. Rev. Genet. 2009, 10, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Carja, O.; Plotkin, J.B. The Evolutionary Advantage of Heritable Phenotypic Heterogeneity. Sci. Rep. 2017, 7, 5090. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Laird, P.W. Interplay between the Cancer Genome and Epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA Methylation-Based Classification of Central Nervous System Tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The Landscape of Genomic Alterations across Childhood Cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The Landscape of Somatic Mutations in Epigenetic Regulators across 1,000 Paediatric Cancer Genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic Histone H3 Alterations in Pediatric Diffuse Intrinsic Pontine Gliomas and Non-Brainstem Glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.; Zhao, K. The Epigenetic Basis of Cellular Heterogeneity. Nat. Rev. Genet. 2021, 22, 235–250. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin Accessibility and the Regulatory Epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, S.C.; Hubert, C.G.; Miller, T.E.; Taylor, M.D.; Rich, J.N. An Epigenetic Gateway to Brain Tumor Cell Identity. Nat. Neurosci. 2016, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining Cell Identity: PRC2-Mediated Regulation of Transcription and Cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Kondo, Y.; Rosner, G.L.; Xiao, L.; Hernandez, N.S.; Vilaythong, J.; Houlihan, P.S.; Krouse, R.S.; Prasad, A.R.; Einspahr, J.G.; et al. MGMT Promoter Methylation and Field Defect in Sporadic Colorectal Cancer. J. Natl. Cancer Inst. 2005, 97, 1330–1338. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; van den Berg, P.R.; Markoulaki, S.; Soldner, F.; Dall’Agnese, A.; Henninger, J.E.; Drotar, J.; Rosenau, N.; Cohen, M.A.; Young, R.A.; et al. Dynamic Enhancer DNA Methylation as Basis for Transcriptional and Cellular Heterogeneity of ESCs. Mol. Cell 2019, 75, 905–920.e6. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.E.; Golan, T.; Sheinboim, D.; Malcov, H.; Amar, D.; Salamon, A.; Liron, T.; Gelfman, S.; Gabet, Y.; Shamir, R.; et al. Enhancer Methylation Dynamics Contribute to Cancer Plasticity and Patient Mortality. Genome Res. 2016, 26, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Holsten, T.; Börnigen, D.; Frank, S.; Mawrin, C.; Glatzel, M.; Schüller, U. Ependymoma Relapse Goes along with a Relatively Stable Epigenome, but a Severely Altered Tumor Morphology. Brain Pathol. 2021, 31, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic Heterogeneity in Cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.G.; Bell, R.J.A.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Magill, S.T.; Vasudevan, H.N.; Seo, K.; Villanueva-Meyer, J.E.; Choudhury, A.; John Liu, S.; Pekmezci, M.; Findakly, S.; Hilz, S.; Lastella, S.; et al. Multiplatform Genomic Profiling and Magnetic Resonance Imaging Identify Mechanisms Underlying Intratumor Heterogeneity in Meningioma. Nat. Commun. 2020, 11, 4803. [Google Scholar] [CrossRef]
- Liu, S.J.; Magill, S.T.; Vasudevan, H.N.; Hilz, S.; Villanueva-Meyer, J.E.; Lastella, S.; Daggubati, V.; Spatz, J.; Choudhury, A.; Orr, B.A.; et al. Multiplatform Molecular Profiling Reveals Epigenomic Intratumor Heterogeneity in Ependymoma. Cell Rep. 2020, 30, 1300–1309.e5. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [Green Version]
- Krug, B.; Harutyunyan, A.S.; Deshmukh, S.; Jabado, N. Polycomb Repressive Complex 2 in the Driver’s Seat of Childhood and Young Adult Brain Tumours. Trends Cell Biol. 2021, 31, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-Mediated Methylation on Lys27 of Histone H3 Pre-Marks Genes for de Novo Methylation in Cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.-L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.-C.; Baumer, K.; Le Bitoux, M.-A.; Marino, D.; Cironi, L.; et al. EZH2 Is Essential for Glioblastoma Cancer Stem Cell Maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.-H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessa, S.; Blanchet-Cohen, A.; Krug, B.; Vladoiu, M.; Coutelier, M.; Faury, D.; Poreau, B.; De Jay, N.; Hébert, S.; Monlong, J.; et al. Stalled Developmental Programs at the Root of Pediatric Brain Tumors. Nat. Genet. 2019, 51, 1702–1713. [Google Scholar] [CrossRef]
- Mills, A.A. Throwing the Cancer Switch: Reciprocal Roles of Polycomb and Trithorax Proteins. Nat. Rev. Cancer 2010, 10, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Hübner, J.-M.; Müller, T.; Papageorgiou, D.N.; Mauermann, M.; Krijgsveld, J.; Russell, R.B.; Ellison, D.W.; Pfister, S.M.; Pajtler, K.W.; Kool, M. EZHIP/CXorf67 Mimics K27M Mutated Oncohistones and Functions as an Intrinsic Inhibitor of PRC2 Function in Aggressive Posterior Fossa Ependymoma. Neuro Oncol. 2019, 21, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.U.; Do, T.J.; Lund, P.J.; Rashoff, A.Q.; Diehl, K.L.; Cieslik, M.; Bajic, A.; Juretic, N.; Deshmukh, S.; Venneti, S.; et al. PFA Ependymoma-Associated Protein EZHIP Inhibits PRC2 Activity through a H3 K27M-like Mechanism. Nat. Commun. 2019, 10, 2146. [Google Scholar] [CrossRef] [Green Version]
- Piunti, A.; Smith, E.R.; Morgan, M.A.J.; Ugarenko, M.; Khaltyan, N.; Helmin, K.A.; Ryan, C.A.; Murray, D.C.; Rickels, R.A.; Yilmaz, B.D.; et al. CATACOMB: An Endogenous Inducible Gene That Antagonizes H3K27 Methylation Activity of Polycomb Repressive Complex 2 via an H3K27M-like Mechanism. Sci. Adv. 2019, 5, eaax2887. [Google Scholar] [CrossRef] [Green Version]
- Nambirajan, A.; Sharma, A.; Rajeshwari, M.; Boorgula, M.T.; Doddamani, R.; Garg, A.; Suri, V.; Sarkar, C.; Sharma, M.C. EZH2 Inhibitory Protein (EZHIP/Cxorf67) Expression Correlates Strongly with H3K27me3 Loss in Posterior Fossa Ependymomas and Is Mutually Exclusive with H3K27M Mutations. Brain Tumor Pathol. 2021, 38, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M Induces Defective Chromatin Spread of PRC2-Mediated Repressive H3K27me2/Me3 and Is Essential for Glioma Tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.U.; Rashoff, A.Q.; Krabbenhoft, S.D.; Hoelper, D.; Do, T.J.; Gibson, T.J.; Lundgren, S.M.; Bondra, E.R.; Deshmukh, S.; Harutyunyan, A.S.; et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 2020, 80, 726–735.e7. [Google Scholar] [CrossRef] [PubMed]
- Michealraj, K.A.; Kumar, S.A.; Kim, L.J.Y.; Cavalli, F.M.G.; Przelicki, D.; Wojcik, J.B.; Delaidelli, A.; Bajic, A.; Saulnier, O.; MacLeod, G.; et al. Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell 2020, 181, 1329–1345.e24. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb. Perspect. Med. 2016, 6, a026583. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Brown, J.S.; Anderson, A.R.A.; Gatenby, R.A. Eco-Evolutionary Causes and Consequences of Temporal Changes in Intratumoural Blood Flow. Nat. Rev. Cancer 2018, 18, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Hoefflin, R.; Lahrmann, B.; Warsow, G.; Hübschmann, D.; Spath, C.; Walter, B.; Chen, X.; Hofer, L.; Macher-Goeppinger, S.; Tolstov, Y.; et al. Spatial Niche Formation but Not Malignant Progression Is a Driving Force for Intratumoural Heterogeneity. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korenchan, D.E.; Flavell, R.R. Spatiotemporal PH Heterogeneity as a Promoter of Cancer Progression and Therapeutic Resistance. Cancers 2019, 11, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastola, S.; Pavlyukov, M.S.; Yamashita, D.; Ghosh, S.; Cho, H.; Kagaya, N.; Zhang, Z.; Minata, M.; Lee, Y.; Sadahiro, H.; et al. Glioma-Initiating Cells at Tumor Edge Gain Signals from Tumor Core Cells to Promote Their Malignancy. Nat. Commun. 2020, 11, 4660. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia Promotes Expansion of the CD133-Positive Glioma Stem Cells through Activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peleli, M.; Moustakas, A.; Papapetropoulos, A. Endothelial-Tumor Cell Interaction in Brain and CNS Malignancies. Int. J. Mol. Sci. 2020, 21, 7371. [Google Scholar] [CrossRef] [PubMed]
- Griesinger, A.M.; Josephson, R.J.; Donson, A.M.; Mulcahy Levy, J.M.; Amani, V.; Birks, D.K.; Hoffman, L.M.; Furtek, S.L.; Reigan, P.; Handler, M.H.; et al. Interleukin-6/STAT3 Pathway Signaling Drives an Inflammatory Phenotype in Group A Ependymoma. Cancer Immunol. Res. 2015, 3, 1165–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillen, A.E.; Riemondy, K.A.; Amani, V.; Griesinger, A.M.; Gilani, A.; Venkataraman, S.; Madhavan, K.; Prince, E.; Sanford, B.; Hankinson, T.C.; et al. Single-Cell RNA Sequencing of Childhood Ependymoma Reveals Neoplastic Cell Subpopulations That Impact Molecular Classification and Etiology. Cell Rep. 2020, 32, 108023. [Google Scholar] [CrossRef] [PubMed]
- Gojo, J.; Englinger, B.; Jiang, L.; Hübner, J.M.; Shaw, M.L.; Hack, O.A.; Madlener, S.; Kirchhofer, D.; Liu, I.; Pyrdol, J.; et al. Single-Cell RNA-Seq Reveals Cellular Hierarchies and Impaired Developmental Trajectories in Pediatric Ependymoma. Cancer Cell 2020, 38, 44–59.e9. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma Stem Cells and Their Roles within the Hypoxic Tumor Microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour Hypoxia Induces a Metabolic Shift Causing Acidosis: A Common Feature in Cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and Acidosis Independently Up-Regulate Vascular Endothelial Growth Factor Transcription in Brain Tumors in Vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- Mohyeldin, A.; Garzón-Muvdi, T.; Quiñones-Hinojosa, A. Oxygen in Stem Cell Biology: A Critical Component of the Stem Cell Niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Pistollato, F.; Chen, H.-L.; Schwartz, P.H.; Basso, G.; Panchision, D.M. Oxygen Tension Controls the Expansion of Human CNS Precursors and the Generation of Astrocytes and Oligodendrocytes. Mol. Cell. Neurosci. 2007, 35, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain Tumor Initiating Cells Adapt to Restricted Nutrition through Preferential Glucose Uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Englinger, B.; Hack, O.A.; Filbin, M.G. Into Thin Air: Hypoxia Drives Metabolic and Epigenomic Deregulation of Lethal Pediatric Ependymoma. Dev. Cell 2020, 54, 134–136. [Google Scholar] [CrossRef]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood Cerebellar Tumours Mirror Conserved Fetal Transcriptional Programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBrian, M.A.; Behbahan, I.S.; Ferrari, R.; Su, T.; Huang, T.-W.; Li, K.; Hong, C.S.; Christofk, H.R.; Vogelauer, M.; Seligson, D.B.; et al. Histone Acetylation Regulates Intracellular PH. Mol. Cell 2013, 49, 310–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande Voorde, J.; Blyth, K.; et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindrick, J.D.; Mole, D.R. Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect. Int. J. Mol. Sci. 2020, 21, 8320. [Google Scholar] [CrossRef]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour Hypoxia Causes DNA Hypermethylation by Reducing TET Activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Ramsawhook, A.; Lewis, L.; Coyle, B.; Ruzov, A. Medulloblastoma and Ependymoma Cells Display Increased Levels of 5-Carboxylcytosine and Elevated TET1 Expression. Clin. Epigenet. 2017, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A Multi-Tool for Tumor Progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic Roles of EMT-Inducing Transcription Factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Shu, C.; Wang, J. Bioinformatics Analysis of Microarray Data Reveals Epithelial-Mesenchymal-Transition in Pediatric Ependymoma. Anticancer. Drugs 2021, 32, 437–447. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Malgulwar, P.B.; Nambirajan, A.; Pathak, P.; Rajeshwari, M.; Suri, V.; Sarkar, C.; Singh, M.; Sharma, M.C. Epithelial-to-Mesenchymal Transition-Related Transcription Factors Are up-Regulated in Ependymomas and Correlate with a Poor Prognosis. Hum. Pathol. 2018, 82, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Wani, K.; Armstrong, T.S.; Vera-Bolanos, E.; Raghunathan, A.; Ellison, D.; Gilbertson, R.; Vaillant, B.; Goldman, S.; Packer, R.J.; Fouladi, M.; et al. A Prognostic Gene Expression Signature in Infratentorial Ependymoma. Acta Neuropathol. 2012, 123, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour Heterogeneity and Metastasis at Single-Cell Resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and Oncogenic Programs in H3K27M Gliomas Dissected by Single-Cell RNA-Seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, M.-I.; Peyrot, S.M.; LeBoeuf, S.; Park, T.J.; McGary, K.L.; Marcotte, E.M.; Wallingford, J.B. RFX2 Is Broadly Required for Ciliogenesis during Vertebrate Development. Dev. Biol. 2012, 363, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ng, C.P.; Habacher, H.; Roy, S. Foxj1 Transcription Factors Are Master Regulators of the Motile Ciliogenic Program. Nat. Genet. 2008, 40, 1445–1453. [Google Scholar] [CrossRef]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized Membrane Domains for Water Transport in Glial Cells: High-Resolution Immunogold Cytochemistry of Aquaporin-4 in Rat Brain. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Owler, B.K. Expression of AQP1 and AQP4 in Paediatric Brain Tumours. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2011, 18, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Noell, S.; Fallier-Becker, P.; Mack, A.F.; Hoffmeister, M.; Beschorner, R.; Ritz, R. Water Channels Aquaporin 4 and -1 Expression in Subependymoma Depends on the Localization of the Tumors. PLoS ONE 2015, 10, e0131367. [Google Scholar] [CrossRef]
- Han, S.; Dennis, D.J.; Balakrishnan, A.; Dixit, R.; Britz, O.; Zinyk, D.; Touahri, Y.; Olender, T.; Brand, M.; Guillemot, F.; et al. A Non-Canonical Role for the Proneural Gene Neurog1 as a Negative Regulator of Neocortical Neurogenesis. Dev. Camb. Engl. 2018, 145, dev157719. [Google Scholar] [CrossRef] [Green Version]
- Stauber, M.; Weidemann, M.; Dittrich-Breiholz, O.; Lobschat, K.; Alten, L.; Mai, M.; Beckers, A.; Kracht, M.; Gossler, A. Identification of FOXJ1 Effectors during Ciliogenesis in the Foetal Respiratory Epithelium and Embryonic Left-Right Organiser of the Mouse. Dev. Biol. 2017, 423, 170–188. [Google Scholar] [CrossRef]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic Targeting of Ependymoma as Informed by Oncogenic Enhancer Profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef]
- Hartfuss, E.; Galli, R.; Heins, N.; Götz, M. Characterization of CNS Precursor Subtypes and Radial Glia. Dev. Biol. 2001, 229, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Rowitch, D.H.; Kriegstein, A.R. Developmental Genetics of Vertebrate Glial-Cell Specification. Nature 2010, 468, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular Identity of Human Outer Radial Glia during Cortical Development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, E.R.; Mich, J.K.; Yao, Z.; Hodge, R.D.; Doyle, A.M.; Jang, S.; Shehata, S.I.; Nelson, A.M.; Shapovalova, N.V.; Levi, B.P.; et al. Fixed Single-Cell Transcriptomic Characterization of Human Radial Glial Diversity. Nat. Methods 2016, 13, 87–93. [Google Scholar] [CrossRef]
- Liu, Z.; Lou, H.; Xie, K.; Wang, H.; Chen, N.; Aparicio, O.M.; Zhang, M.Q.; Jiang, R.; Chen, T. Reconstructing Cell Cycle Pseudo Time-Series via Single-Cell Transcriptome Data. Nat. Commun. 2017, 8, 22. [Google Scholar] [CrossRef]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A Comparison of Single-Cell Trajectory Inference Methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Bertrand, K.C. Sub-Group, Sub-Type, and Cell-Type Heterogeneity of Ependymoma. Cancer Cell 2020, 38, 15–17. [Google Scholar] [CrossRef]
- Jenseit, A.; Camgöz, A.; Pfister, S.M.; Kool, M. EZHIP: A New Piece of the Puzzle towards Understanding Pediatric Posterior Fossa Ependymoma. Acta Neuropathol. 2021, 1–13. [Google Scholar] [CrossRef]
- Han, J.; Yu, M.; Bai, Y.; Yu, J.; Jin, F.; Li, C.; Zeng, R.; Peng, J.; Li, A.; Song, X.; et al. Elevated CXorf67 Expression in PFA Ependymomas Suppresses DNA Repair and Sensitizes to PARP Inhibitors. Cancer Cell 2020, 38, 844–856.e7. [Google Scholar] [CrossRef] [PubMed]
- Panwalkar, P.; Tamrazi, B.; Dang, D.; Chung, C.; Sweha, S.; Natarajan, S.K.; Pun, M.; Bayliss, J.; Ogrodzinski, M.P.; Pratt, D.; et al. Targeting Integrated Epigenetic and Metabolic Pathways in Lethal Childhood PFA Ependymomas. Sci. Transl. Med. 2021, 13, eabc0497. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A Novel Target for Cancer Treatment. J. Hematol. Oncol. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Medeiros, M.; Candido, M.; Valera, E.; Brassesco, M. The Multifaceted NF-KB: Are There Still Prospects of Its Inhibition for Clinical Intervention in Pediatric Central Nervous System Tumors? Cell Mol. Life Sci. 2021, 78, 6161–6200. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.S.; Gilbert, M.R. Clinical Trial Challenges, Design Considerations, and Outcome Measures in Rare CNS Tumors. Neuro Oncol. 2021, 23, S30–S38. [Google Scholar] [CrossRef]
- Di, K.; Lloyd, G.K.; Abraham, V.; MacLaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D.A. Marizomib Activity as a Single Agent in Malignant Gliomas: Ability to Cross the Blood-Brain Barrier. Neuro Oncol. 2016, 18, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Donson, A.M.; Amani, V.; Warner, E.A.; Griesinger, A.M.; Witt, D.A.; Levy, J.M.M.; Hoffman, L.M.; Hankinson, T.C.; Handler, M.H.; Vibhakar, R.; et al. Identification of FDA-Approved Oncology Drugs with Selective Potency in High-Risk Childhood Ependymoma. Mol. Cancer Ther. 2018, 17, 1984–1994. [Google Scholar] [CrossRef] [Green Version]
- Antonelli, R.; Jiménez, C.; Riley, M.; Servidei, T.; Riccardi, R.; Soriano, A.; Roma, J.; Martínez-Saez, E.; Martini, M.; Ruggiero, A.; et al. CN133, a Novel Brain-Penetrating Histone Deacetylase Inhibitor, Hampers Tumor Growth in Patient-Derived Pediatric Posterior Fossa Ependymoma Models. Cancers 2020, 12, 1922. [Google Scholar] [CrossRef] [PubMed]
- Servidei, T.; Meco, D.; Martini, M.; Battaglia, A.; Granitto, A.; Buzzonetti, A.; Babini, G.; Massimi, L.; Tamburrini, G.; Scambia, G.; et al. The BET Inhibitor OTX015 Exhibits In Vitro and In Vivo Antitumor Activity in Pediatric Ependymoma Stem Cell Models. Int. J. Mol. Sci. 2021, 22, 1877. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, L.; Li, C.J.; Ezhilan, S.; Iyer, V.R.; Kuo, J.S. Emerging Epigenetic Therapies for Brain Tumors. Neuromol. Med. 2021, 1–9. [Google Scholar] [CrossRef]
- Lötsch, D.; Kirchhofer, D.; Englinger, B.; Jiang, L.; Okonechnikov, K.; Senfter, D.; Laemmerer, A.; Gabler, L.; Pirker, C.; Donson, A.M.; et al. Targeting Fibroblast Growth Factor Receptors to Combat Aggressive Ependymoma. Acta Neuropathol. 2021, 142, 339–360. [Google Scholar] [CrossRef] [PubMed]
- Vega-Benedetti, A.F.; Saucedo, C.; Zavattari, P.; Vanni, R.; Zugaza, J.L.; Parada, L.A. PLAGL1: An Important Player in Diverse Pathological Processes. J. Appl. Genet. 2017, 58, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Jünger, S.T.; Mynarek, M.; Wohlers, I.; Dörner, E.; Mühlen, A.Z.; Velez-Char, N.; von Hoff, K.; Rutkowski, S.; Warmuth-Metz, M.; Kortmann, R.-D.; et al. Improved Risk-Stratification for Posterior Fossa Ependymoma of Childhood Considering Clinical, Histological and Genetic Features—A Retrospective Analysis of the HIT Ependymoma Trial Cohort. Acta Neuropathol. Commun. 2019, 7, 181. [Google Scholar] [CrossRef]
- Rudà, R.; Reifenberger, G.; Frappaz, D.; Pfister, S.M.; Laprie, A.; Santarius, T.; Roth, P.; Tonn, J.C.; Soffietti, R.; Weller, M.; et al. EANO Guidelines for the Diagnosis and Treatment of Ependymal Tumors. Neuro Oncol. 2018, 20, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatua, S.; Mangum, R.; Bertrand, K.C.; Zaky, W.; McCall, D.; Mack, S.C. Pediatric Ependymoma: Current Treatment and Newer Therapeutic Insights. Future Oncol. 2018, 14, 3175–3186. [Google Scholar] [CrossRef] [PubMed]
- Adolph, J.E.; Fleischhack, G.; Gaab, C.; Mikasch, R.; Mynarek, M.; Rutkowski, S.; Schüller, U.; Pfister, S.M.; Pajtler, K.W.; Milde, T.; et al. Systemic Chemotherapy of Pediatric Recurrent Ependymomas: Results from the German HIT-REZ Studies. J. Neurooncol. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Home-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 17 November 2021).
- Jünger, S.T.; Timmermann, B.; Pietsch, T. Pediatric Ependymoma: An Overview of a Complex Disease. Childs Nerv. Syst. 2021, 37, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, A.K.S.; Szulzewsky, F.; Gilbert, M.R.; Gujral, T.S.; Holland, E.C. Utilizing Preclinical Models to Develop Targeted Therapies for Rare Central Nervous System Cancers. Neuro Oncol. 2021, 23, S4–S15. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.M.; Shelat, A.A.; Carcaboso, A.M.; Kranenburg, T.A.; Arnold, L.A.; Boulos, N.; Wright, K.; Johnson, R.A.; Poppleton, H.; Mohankumar, K.M.; et al. An Integrated in Vitro and in Vivo High-Throughput Screen Identifies Treatment Leads for Ependymoma. Cancer Cell 2011, 20, 384–399. [Google Scholar] [CrossRef] [Green Version]
- Rogers, H.A.; Chapman, R.; Kings, H.; Allard, J.; Barron-Hastings, J.; Pajtler, K.W.; Sill, M.; Pfister, S.; Grundy, R.G. Limitations of Current in Vitro Models for Testing the Clinical Potential of Epigenetic Inhibitors for Treatment of Pediatric Ependymoma. Oncotarget 2018, 9, 36530–36541. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Petralia, F.; Tignor, N.; Reva, B.; Koptyra, M.; Chowdhury, S.; Rykunov, D.; Krek, A.; Ma, W.; Zhu, Y.; Ji, J.; et al. Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell 2020, 183, 1962–1985.e31. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Servidei, T.; Lucchetti, D.; Navarra, P.; Sgambato, A.; Riccardi, R.; Ruggiero, A. Cell-of-Origin and Genetic, Epigenetic, and Microenvironmental Factors Contribute to the Intra-Tumoral Heterogeneity of Pediatric Intracranial Ependymoma. Cancers 2021, 13, 6100. https://doi.org/10.3390/cancers13236100
Servidei T, Lucchetti D, Navarra P, Sgambato A, Riccardi R, Ruggiero A. Cell-of-Origin and Genetic, Epigenetic, and Microenvironmental Factors Contribute to the Intra-Tumoral Heterogeneity of Pediatric Intracranial Ependymoma. Cancers. 2021; 13(23):6100. https://doi.org/10.3390/cancers13236100
Chicago/Turabian StyleServidei, Tiziana, Donatella Lucchetti, Pierluigi Navarra, Alessandro Sgambato, Riccardo Riccardi, and Antonio Ruggiero. 2021. "Cell-of-Origin and Genetic, Epigenetic, and Microenvironmental Factors Contribute to the Intra-Tumoral Heterogeneity of Pediatric Intracranial Ependymoma" Cancers 13, no. 23: 6100. https://doi.org/10.3390/cancers13236100
APA StyleServidei, T., Lucchetti, D., Navarra, P., Sgambato, A., Riccardi, R., & Ruggiero, A. (2021). Cell-of-Origin and Genetic, Epigenetic, and Microenvironmental Factors Contribute to the Intra-Tumoral Heterogeneity of Pediatric Intracranial Ependymoma. Cancers, 13(23), 6100. https://doi.org/10.3390/cancers13236100