
Mouse Models of Frequently Mutated Genes in Acute Myeloid Leukemia


Abstract
:Simple Summary
Abstract
1. Introduction
2. Mouse Models of Genes Involved in Cell Signaling Pathways in Myeloid Malignancies
2.1. FLT3
2.2. KIT
2.3. KRAS
2.4. NRAS
2.5. NF1
2.6. PTPN11
3. Mouse Models of Epigenetic Modifier Genes in Myeloid Malignancies
3.1. DNMT3A
3.2. TET2
3.3. IDH1 and IDH2
3.4. EZH2
3.5. ASXL1
3.6. ASXL2
4. Mouse Models of Nucleophosmin 1 (NPM1) in Myeloid Malignancies
5. Mouse Models of Transcription Factor Genes in Myeloid Malignancies
5.1. CEBPA
5.2. RUNX1
5.3. MYC
5.4. BCOR
5.5. CUX1
5.6. SETBP1
5.7. PHF6
6. Mouse Models of Tumor Suppressor Genes in Myeloid Malignancies
6.1. WT1
6.2. TP53
7. Mouse Models of Spliceosome Complex Genes in Myeloid Malignancies
7.1. SRSF2
7.2. U2AF1
7.3. SF3B1
8. Mouse Models of Cohesin Complex Genes in Myeloid Malignancies
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Saultz, J.N.; Garzon, R. Acute Myeloid Leukemia: A Concise Review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voso, M.T.; Ottone, T.; Lavorgna, S.; Venditti, A.; Maurillo, L.; Lo-Coco, F.; Buccisano, F. MRD in AML: The Role of New Techniques. Front. Oncol. 2019, 9, 655. [Google Scholar] [CrossRef] [Green Version]
- Carbonell, D.; Suárez-González, J.; Chicano, M.; Andrés-Zayas, C.; Triviño, J.C.; Rodríguez-Macías, G.; Bastos-Oreiro, M.; Font, P.; Ballesteros, M.; Muñiz, P.; et al. Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms. Cancers 2019, 11, 1364. [Google Scholar] [CrossRef] [Green Version]
- Almosailleakh, M.; Schwaller, J. Murine Models of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, D. FLT3 mutations: Biology and treatment. Hematol. Am. Soc. Hematol. Educ. Program. 2006, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Mackarehtschian, K.; Hardin, J.D.; Moore, K.A.; Boast, S.; Goff, S.P.; Lemischka, I.R. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity 1995, 3, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Kelly, L.M.; Liu, Q.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Gilliland, D.G. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood 2002, 99, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Miething, C.; Thiede, C.; Peschel, C.; Duyster, J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood 2005, 105, 4792–4799. [Google Scholar] [CrossRef]
- Lee, B.H.; Williams, I.R.; Anastasiadou, E.; Boulton, C.L.; Joseph, S.W.; Amaral, S.M.; Curley, D.P.; Duclos, N.; Huntly, B.J.; Fabbro, D.; et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene 2005, 24, 7882–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.H.; Tothova, Z.; Levine, R.L.; Anderson, K.; Buza-Vidas, N.; Cullen, D.E.; McDowell, E.P.; Adelsperger, J.; Fröhling, S.; Huntly, B.J.; et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell 2007, 12, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Piloto, O.; Nguyen, H.B.; Greenberg, K.; Takamiya, K.; Racke, F.; Huso, D.; Small, D. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood 2008, 111, 3849–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, E.; Li, L.; Duffield, A.S.; Ma, H.S.; Huso, D.L.; Small, D. FLT3/D835Y mutation knock-in mice display less aggressive disease compared with FLT3/internal tandem duplication (ITD) mice. Proc. Natl. Acad. Sci. USA 2013, 110, 21113–21118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viny, A.D.; Ott, C.J.; Spitzer, B.; Rivas, M.; Meydan, C.; Papalexi, E.; Yelin, D.; Shank, K.; Reyes, J.; Chiu, A.; et al. Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J. Exp. Med. 2015, 212, 1819–1832. [Google Scholar] [CrossRef]
- Behrens, K.; Maul, K.; Tekin, N.; Kriebitzsch, N.; Indenbirken, D.; Prassolov, V.; Muller, U.; Serve, H.; Cammenga, J.; Stocking, C. RUNX1 cooperates with FLT3-ITD to induce leukemia. J. Exp. Med. 2017, 214, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Rudorf, A.; Muller, T.A.; Klingeberg, C.; Kreutmair, S.; Poggio, T.; Gorantla, S.P.; Ruckert, T.; Schmitt-Graeff, A.; Gengenbacher, A.; Paschka, P.; et al. NPM1c alters FLT3-D835Y localization and signaling in acute myeloid leukemia. Blood 2019, 134, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.E.; Qin, T.; Muench, D.E.; Masuda, K.; Venkatasubramanian, M.; Orr, E.; Suarez, L.; Gore, S.D.; Delwel, R.; Paietta, E.; et al. DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer Discov. 2016, 6, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poitras, J.L.; Heiser, D.; Li, L.; Nguyen, B.; Nagai, K.; Duffield, A.S.; Gamper, C.; Small, D. Dnmt3a deletion cooperates with the Flt3/ITD mutation to drive leukemogenesis in a murine model. Oncotarget 2016, 7, 69124–69135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Liu, Y.; Lu, C.; Cross, J.R.; Morris, J.P.t.; Shroff, A.S.; Ward, P.S.; Bradner, J.E.; Thompson, C.; Lowe, S.W. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013, 27, 1974–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annesley, C.E.; Rabik, C.; Duffield, A.S.; Rau, R.E.; Magoon, D.; Li, L.; Huff, V.; Small, D.; Loeb, D.M.; Brown, P. Knock-in of the Wt1 R394W mutation causes MDS and cooperates with Flt3/ITD to drive aggressive myeloid neoplasms in mice. Oncotarget 2018, 9, 35313–35326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronier, E.; Bowman, R.L.; Ahn, J.; Glass, J.; Kandoth, C.; Merlinsky, T.R.; Whitfield, J.T.; Durham, B.H.; Gruet, A.; Hanasoge Somasundara, A.V.; et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood 2018, 132, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Jiang, Y.; Meydan, C.; Shank, K.; Pandey, S.; Barreyro, L.; Antony-Debre, I.; Viale, A.; Socci, N.; Sun, Y.; et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 2015, 27, 502–515. [Google Scholar] [CrossRef] [Green Version]
- Pacharne, S.; Dovey, O.M.; Cooper, J.L.; Gu, M.; Friedrich, M.J.; Rajan, S.S.; Barenboim, M.; Collord, G.; Vijayabaskar, M.S.; Ponstingl, H.; et al. SETBP1 overexpression acts in the place of class-defining mutations to drive FLT3-ITD-mutant AML. Blood Adv. 2021, 5, 2412–2425. [Google Scholar] [CrossRef]
- Supper, E.; Rudat, S.; Iyer, V.; Droop, A.; Wong, K.; Spinella, J.-F.; Thomas, P.; Sauvageau, G.; Adams, D.J.; Wong, C.C. Cut-like homeobox 1 (CUX1) tumor suppressor gene haploinsufficiency induces apoptosis evasion to sustain myeloid leukemia. Nat. Commun. 2021, 12, 2482. [Google Scholar] [CrossRef]
- Thanasopoulou, A.; Tzankov, A.; Schwaller, J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica 2014, 99, 1465–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, S.; Li, L.; Slape, C.; Nguyen, B.; Novak, R.; Duffield, A.; Huso, D.; Desiderio, S.; Borowitz, M.J.; Aplan, P.; et al. Knock-in of a FLT3/ITD mutation cooperates with a NUP98-HOXD13 fusion to generate acute myeloid leukemia in a mouse model. Blood 2012, 119, 2883–2894. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, M.C.; Kim, Y.M.; Krivtsov, A.V.; Wright, R.D.; Feng, Z.; Agarwal, J.; Kung, A.L.; Armstrong, S.A. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: Development of a model for rapid therapeutic assessment. Leukemia 2008, 22, 66–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schessl, C.; Rawat, V.P.; Cusan, M.; Deshpande, A.; Kohl, T.M.; Rosten, P.M.; Spiekermann, K.; Humphries, R.K.; Schnittger, S.; Kern, W.; et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J. Clin. Investig. 2005, 115, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Kreisel, F.; Cain, J.; Colson, A.; Tomasson, M.H. Neoplasia driven by mutant c-KIT is mediated by intracellular, not plasma membrane, receptor signaling. Mol. Cell Biol. 2007, 27, 267–282. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Zhao, L.J.; Wu, C.F.; Liu, P.; Shi, L.; Liang, Y.; Xiong, S.M.; Mi, J.Q.; Chen, Z.; Ren, R.; et al. C-KIT mutation cooperates with full-length AML1-ETO to induce acute myeloid leukemia in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 2450–2455. [Google Scholar] [CrossRef] [Green Version]
- Di Siena, S.; Gimmelli, R.; Nori, S.L.; Barbagallo, F.; Campolo, F.; Dolci, S.; Rossi, P.; Venneri, M.A.; Giannetta, E.; Gianfrilli, D.; et al. Activated c-Kit receptor in the heart promotes cardiac repair and regeneration after injury. Cell Death Dis. 2016, 7, e2317. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Zhang, Y.; Sha, K.; Tang, Q.; Yang, X.; Yu, C.; Liu, Z.; Sun, W.; Cai, L.; Xu, C.; et al. KRAS (G12D) cooperates with AML1/ETO to initiate a mouse model mimicking human acute myeloid leukemia. Cell Physiol. Biochem. 2014, 33, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Braun, B.S.; Tuveson, D.A.; Kong, N.; Le, D.T.; Kogan, S.C.; Rozmus, J.; Le Beau, M.M.; Jacks, T.E.; Shannon, K.M. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc. Natl. Acad. Sci. USA 2004, 101, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, I.T.; Kutok, J.L.; Williams, I.R.; Cohen, S.; Kelly, L.; Shigematsu, H.; Johnson, L.; Akashi, K.; Tuveson, D.A.; Jacks, T.; et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J. Clin. Investig. 2004, 113, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koera, K.; Nakamura, K.; Nakao, K.; Miyoshi, J.; Toyoshima, K.; Hatta, T.; Otani, H.; Aiba, A.; Katsuki, M. K-Ras is essential for the development of the mouse embryo. Oncogene 1997, 15, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Greenbaum, D.; Cichowski, K.; Mercer, K.; Murphy, E.; Schmitt, E.; Bronson, R.T.; Umanoff, H.; Edelmann, W.; Kucherlapati, R.; et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997, 11, 2468–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, S.; Jyotsana, N.; Sharma, A.; Kloos, A.; Gabdoulline, R.; Othman, B.; Lai, C.K.; Schottmann, R.; Mandhania, M.; Schmoellerl, J.; et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia. Cancers 2020, 12, 2766. [Google Scholar] [CrossRef]
- Parikh, C.; Subrahmanyam, R.; Ren, R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood 2006, 108, 2349–2357. [Google Scholar] [CrossRef] [Green Version]
- Kogan, S.C.; Lagasse, E.; Atwater, S.; Bae, S.C.; Weissman, I.; Ito, Y.; Bishop, J.M. The PEBP2betaMYH11 fusion created by Inv(16)(p13;q22) in myeloid leukemia impairs neutrophil maturation and contributes to granulocytic dysplasia. Proc. Natl. Acad. Sci. USA 1998, 95, 11863–11868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, K.L.; Dolnikov, A.; Millington, M.; Shounan, Y.; Symonds, G. Mutant N-ras induces myeloproliferative disorders and apoptosis in bone marrow repopulated mice. Blood 1999, 93, 2043–2056. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Li, Z.; Wang, Z.; Tan, L.X.; Ryu, M.J.; Meline, B.; Du, J.; Young, K.H.; Ranheim, E.; et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood 2011, 118, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Umanoff, H.; Edelmann, W.; Pellicer, A.; Kucherlapati, R. The murine N-ras gene is not essential for growth and development. Proc. Natl. Acad. Sci. USA 1995, 92, 1709–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannan, C.I.; Perkins, A.S.; Vogel, K.S.; Ratner, N.; Nordlund, M.L.; Reid, S.W.; Buchberg, A.M.; Jenkins, N.A.; Parada, L.F.; Copeland, N.G. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994, 8, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacks, T.; Shih, T.S.; Schmitt, E.M.; Bronson, R.T.; Bernards, A.; Weinberg, R.A. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994, 7, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Mohi, M.G.; Williams, I.R.; Dearolf, C.R.; Chan, G.; Kutok, J.L.; Cohen, S.; Morgan, K.; Boulton, C.; Shigematsu, H.; Keilhack, H.; et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell 2005, 7, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.; Kalaitzidis, D.; Usenko, T.; Kutok, J.L.; Yang, W.; Mohi, M.G.; Neel, B.G. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood 2009, 113, 4414–4424. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Yu, W.M.; Zheng, H.; Loh, M.L.; Bunting, S.T.; Pauly, M.; Huang, G.; Zhou, M.; Broxmeyer, H.E.; Scadden, D.T.; et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 2016, 539, 304–308. [Google Scholar] [CrossRef] [Green Version]
- Saxton, T.M.; Henkemeyer, M.; Gasca, S.; Shen, R.; Rossi, D.J.; Shalaby, F.; Feng, G.S.; Pawson, T. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. 1997, 16, 2352–2364. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.Y.; Dai, Y.J.; Zhang, W.; Zhang, W.N.; Xiong, S.M.; Gu, Z.H.; Wang, K.K.; Zeng, R.; Chen, Z.; et al. DNMT3A Arg882 mutation drives chronic myelomonocytic leukemia through disturbing gene expression/DNA methylation in hematopoietic cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2620–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.J.; Wang, Y.Y.; Huang, J.Y.; Xia, L.; Shi, X.D.; Xu, J.; Lu, J.; Su, X.B.; Yang, Y.; Zhang, W.N.; et al. Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement. Proc. Natl. Acad. Sci. USA 2017, 114, 5237–5242. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Guryanova, O.A.; Lieu, Y.K.; Garrett-Bakelman, F.E.; Spitzer, B.; Glass, J.L.; Shank, K.; Martinez, A.B.; Rivera, S.A.; Durham, B.H.; Rapaport, F.; et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 2016, 30, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Miao, Z.; Jiang, Y.; Zou, P.; Li, W.; Tang, X.; Lv, Y.; Xing, D.; Chen, S.; Yang, F.; et al. Erratum: Characteristics of myeloid sarcoma in mice and patients with TET2 deficiency. Oncol. Lett. 2020, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Gorlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brustle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itsumi, M.; Inoue, S.; Elia, A.J.; Murakami, K.; Sasaki, M.; Lind, E.F.; Brenner, D.; Harris, I.S.; Chio, I.I.; Afzal, S.; et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP(+)/NADPH ratio. Cell Death Differ. 2015, 22, 1837–1845. [Google Scholar] [CrossRef] [Green Version]
- McKenney, A.S.; Lau, A.N.; Somasundara, A.V.H.; Spitzer, B.; Intlekofer, A.M.; Ahn, J.; Shank, K.; Rapaport, F.T.; Patel, M.A.; Papalexi, E.; et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J. Clin. Investig. 2018, 128, 789–804. [Google Scholar] [CrossRef]
- Shih, A.H.; Meydan, C.; Shank, K.; Garrett-Bakelman, F.E.; Ward, P.S.; Intlekofer, A.M.; Nazir, A.; Stein, E.M.; Knapp, K.; Glass, J.; et al. Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia. Cancer Discov. 2017, 7, 494–505. [Google Scholar] [CrossRef] [Green Version]
- White, K.; Kim, M.-J.; Han, C.; Park, H.-J.; Ding, D.; Boyd, K.; Walker, L.; Linser, P.; Meneses, Z.; Slade, C.; et al. Loss of IDH2 Accelerates Age-related Hearing Loss in Male Mice. Sci. Rep. 2018, 8, 5039. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Merchan, A.; Arranz, L.; Ligos, J.M.; de Molina, A.; Dominguez, O.; Gonzalez, S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat. Commun. 2012, 3, 623. [Google Scholar] [CrossRef]
- Mochizuki-Kashio, M.; Mishima, Y.; Miyagi, S.; Negishi, M.; Saraya, A.; Konuma, T.; Shinga, J.; Koseki, H.; Iwama, A. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 2011, 118, 6553–6561. [Google Scholar] [CrossRef]
- Mochizuki-Kashio, M.; Aoyama, K.; Sashida, G.; Oshima, M.; Tomioka, T.; Muto, T.; Wang, C.; Iwama, A. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood 2015, 126, 1172–1183. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Kitaura, J.; Togami, K.; Nishimura, K.; Enomoto, Y.; Uchida, T.; Kagiyama, Y.; Kawabata, K.C.; Nakahara, F.; Izawa, K.; et al. Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations. J. Clin. Investig. 2013, 123, 4627–4640. [Google Scholar] [CrossRef]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Chiu, Y.C.; Lin, C.C.; Kuo, Y.Y.; Hou, H.A.; Tzeng, Y.S.; Kao, C.J.; Chuang, P.H.; Tseng, M.H.; Hsiao, T.H.; et al. The distinct biological implications of Asxl1 mutation and its roles in leukemogenesis revealed by a knock-in mouse model. J. Hematol. Oncol. 2017, 10, 139. [Google Scholar] [CrossRef]
- Uni, M.; Masamoto, Y.; Sato, T.; Kamikubo, Y.; Arai, S.; Hara, E.; Kurokawa, M. Modeling ASXL1 mutation revealed impaired hematopoiesis caused by depression of p16Ink4a through aberrant PRC1-mediated histone modification. Leukemia 2019, 33, 191–204. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; He, Y.; Pan, F.; Chen, S.; Rhodes, S.; Nguyen, L.; Yuan, J.; Jiang, L.; Yang, X.; et al. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 2014, 123, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Farber, C.R.; Bennett, B.J.; Orozco, L.; Zou, W.; Lira, A.; Kostem, E.; Kang, H.M.; Furlotte, N.; Berberyan, A.; Ghazalpour, A.; et al. Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet. 2011, 7, e1002038. [Google Scholar] [CrossRef] [Green Version]
- Vikas, M.; Lin, H.; Norimichi, H.; Weoi Woon, T.; Anand, M.; Qiao-Yang, S.; Ling-Wen, D.; Hazimah Binte Mohd, N.; Su Lin, L.; Pavithra, S.; et al. ASXL2 regulates hematopoiesis in mice and its deficiency promotes myeloid expansion. Haematologica 2018, 103, 1980–1990. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; He, F.; Zhang, P.; Chen, S.; Shi, H.; Sun, Y.; Guo, Y.; Yang, H.; Man, N.; Greenblatt, S.; et al. Loss of Asxl2 leads to myeloid malignancies in mice. Nat. Commun. 2017, 8, 15456. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Sportoletti, P.; Ito, K.; Clohessy, J.G.; Teruya-Feldstein, J.; Kutok, J.L.; Pandolfi, P.P. The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood 2010, 115, 3341–3345. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.H.; Ko, B.S.; Chiou, J.S.; Hsu, Y.C.; Tsai, M.H.; Chiu, Y.C.; Yu, I.S.; Lin, S.W.; Hou, H.A.; Kuo, Y.Y.; et al. A knock-in Npm1 mutation in mice results in myeloproliferation and implies a perturbation in hematopoietic microenvironment. PLoS ONE 2012, 7, e49769. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, G.S.; Cooper, J.L.; Rad, R.; Li, J.; Rice, S.; Uren, A.; Rad, L.; Ellis, P.; Andrews, R.; Banerjee, R.; et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 2011, 43, 470–475. [Google Scholar] [CrossRef]
- Mallardo, M.; Caronno, A.; Pruneri, G.; Raviele, P.R.; Viale, A.; Pelicci, P.G.; Colombo, E. NPMc+ and FLT3_ITD mutations cooperate in inducing acute leukaemia in a novel mouse model. Leukemia 2013, 27, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Grisendi, S.; Bernardi, R.; Rossi, M.; Cheng, K.; Khandker, L.; Manova, K.; Pandolfi, P.P. Role of nucleophosmin in embryonic development and tumorigenesis. Nature 2005, 437, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Sportoletti, P.; Grisendi, S.; Majid, S.M.; Cheng, K.; Clohessy, J.G.; Viale, A.; Teruya-Feldstein, J.; Pandolfi, P.P. Npm1 is a haploinsufficient suppressor of myeloid and lymphoid malignancies in the mouse. Blood 2008, 111, 3859–3862. [Google Scholar] [CrossRef]
- Bereshchenko, O.; Mancini, E.; Moore, S.; Bilbao, D.; Mansson, R.; Luc, S.; Grover, A.; Jacobsen, S.E.; Bryder, D.; Nerlov, C. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell 2009, 16, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.D.; Finegold, M.J.; Bradley, A.; Ou, C.N.; Abdelsayed, S.V.; Wilde, M.D.; Taylor, L.R.; Wilson, D.R.; Darlington, G.J. Impaired energy homeostasis in C/EBP alpha knockout mice. Science 1995, 269, 1108–1112. [Google Scholar] [CrossRef]
- Kirstetter, P.; Schuster, M.B.; Bereshchenko, O.; Moore, S.; Dvinge, H.; Kurz, E.; Theilgaard-Monch, K.; Mansson, R.; Pedersen, T.A.; Pabst, T.; et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell 2008, 13, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe-Okochi, N.; Kitaura, J.; Ono, R.; Harada, H.; Harada, Y.; Komeno, Y.; Nakajima, H.; Nosaka, T.; Inaba, T.; Kitamura, T. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 2008, 111, 4297–4308. [Google Scholar] [CrossRef] [Green Version]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Li, Q.; O’Neal, J.; Kreisel, F.; Le Beau, M.M.; Tomasson, M.H. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 2005, 106, 2452–2461. [Google Scholar] [CrossRef] [Green Version]
- Kawagoe, H.; Kandilci, A.; Kranenburg, T.A.; Grosveld, G.C. Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res. 2007, 67, 10677–10685. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Nakajima-Takagi, Y.; Aoyama, K.; Tara, S.; Oshima, M.; Saraya, A.; Koide, S.; Si, S.; Manabe, I.; Sanada, M.; et al. Internal deletion of BCOR reveals a tumor suppressor function for BCOR in T lymphocyte malignancies. J. Exp. Med. 2017, 214, 2901–2913. [Google Scholar] [CrossRef]
- Tara, S.; Isshiki, Y.; Nakajima-Takagi, Y.; Oshima, M.; Aoyama, K.; Tanaka, T.; Shinoda, D.; Koide, S.; Saraya, A.; Miyagi, S.; et al. Bcor insufficiency promotes initiation and progression of myelodysplastic syndrome. Blood 2018, 132, 2470–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wamstad, J.A.; Corcoran, C.M.; Keating, A.M.; Bardwell, V.J. Role of the transcriptional corepressor Bcor in embryonic stem cell differentiation and early embryonic development. PLoS ONE 2008, 3, e2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadieux, C.; Fournier, S.; Peterson, A.C.; Bédard, C.; Bedell, B.J.; Nepveu, A. Transgenic mice expressing the p75 CCAAT-displacement protein/Cut homeobox isoform develop a myeloproliferative disease-like myeloid leukemia. Cancer Res. 2006, 66, 9492–9501. [Google Scholar] [CrossRef] [Green Version]
- Luong, M.X.; van der Meijden, C.M.; Xing, D.; Hesselton, R.; Monuki, E.S.; Jones, S.N.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Neufeld, E.J.; et al. Genetic ablation of the CDP/Cux protein C terminus results in hair cycle defects and reduced male fertility. Mol. Cell Biol. 2002, 22, 1424–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwakarma, B.A.; Nguyen, N.; Makishima, H.; Hosono, N.; Gudmundsson, K.O.; Negi, V.; Oakley, K.; Han, Y.; Przychodzen, B.; Maciejewski, J.P.; et al. Runx1 repression by histone deacetylation is critical for Setbp1-induced mouse myeloid leukemia development. Leukemia 2016, 30, 200–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, D.; Kitaura, J.; Matsui, H.; Hou, H.A.; Chou, W.C.; Nagamachi, A.; Kawabata, K.C.; Togami, K.; Nagase, R.; Horikawa, S.; et al. SETBP1 mutations drive leukemic transformation in ASXL1-mutated MDS. Leukemia 2015, 29, 847–857. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Chen, T.C.; Lin, C.C.; Yuan, C.T.; Hsu, C.L.; Hou, H.A.; Kao, C.J.; Chuang, P.H.; Chen, Y.R.; Chou, W.C.; et al. Phf6-null hematopoietic stem cells have enhanced self-renewal capacity and oncogenic potentials. Blood Adv. 2019, 3, 2355–2367. [Google Scholar] [CrossRef] [Green Version]
- McRae, H.M.; Garnham, A.L.; Hu, Y.; Witkowski, M.T.; Corbett, M.A.; Dixon, M.P.; May, R.E.; Sheikh, B.N.; Chiang, W.; Kueh, A.J.; et al. PHF6 regulates hematopoietic stem and progenitor cells and its loss synergizes with expression of TLX3 to cause leukemia. Blood 2019, 133, 1729–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef]
- Loizou, E.; Banito, A.; Livshits, G.; Ho, Y.J.; Koche, R.P.; Sanchez-Rivera, F.J.; Mayle, A.; Chen, C.C.; Kinalis, S.; Bagger, F.O.; et al. A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1. Cancer Discov. 2019, 9, 962–979. [Google Scholar] [CrossRef] [Green Version]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Komeno, Y.; Huang, Y.J.; Qiu, J.; Lin, L.; Xu, Y.; Zhou, Y.; Chen, L.; Monterroza, D.D.; Li, H.; DeKelver, R.C.; et al. SRSF2 Is Essential for Hematopoiesis, and Its Myelodysplastic Syndrome-Related Mutations Dysregulate Alternative Pre-mRNA Splicing. Mol. Cell Biol. 2015, 35, 3071–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Kon, A.; Yamazaki, S.; Nannya, Y.; Kataoka, K.; Ota, Y.; Nakagawa, M.M.; Yoshida, K.; Shiozawa, Y.; Morita, M.; Yoshizato, T.; et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood 2018, 131, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Fei, D.L.; Zhen, T.; Durham, B.; Ferrarone, J.; Zhang, T.; Garrett, L.; Yoshimi, A.; Abdel-Wahab, O.; Bradley, R.K.; Liu, P.; et al. Impaired hematopoiesis and leukemia development in mice with a conditional knock-in allele of a mutant splicing factor gene U2af1. Proc. Natl. Acad. Sci. USA 2018, 115, e10437–e10446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadugu, B.A.; Heard, A.; Srivatsan, S.N.; Alberti, M.O.; Ndonwi, M.; Grieb, S.; Bradley, J.; Shao, J.; Ahmed, T.; Shirai, C.L.; et al. U2AF1 is a haplo-essential gene required for cancer cell survival. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mupo, A.; Seiler, M.; Sathiaseelan, V.; Pance, A.; Yang, Y.; Agrawal, A.A.; Iorio, F.; Bautista, R.; Pacharne, S.; Tzelepis, K.; et al. Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia 2017, 31, 720–727. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconte, V.; Tabarroki, A.; Zhang, L.; Parker, Y.; Hasrouni, E.; Mahfouz, R.; Isono, K.; Koseki, H.; Sekeres, M.A.; Saunthararajah, Y.; et al. Splicing factor 3b subunit 1 (Sf3b1) haploinsufficient mice display features of low risk Myelodysplastic syndromes with ring sideroblasts. J. Hematol. Oncol. 2014, 7, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isono, K.; Mizutani-Koseki, Y.; Komori, T.; Schmidt-Zachmann, M.S.; Koseki, H. Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev. 2005, 19, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Tomaszewski, J.M.; Vazquez, M.; Verschoor, S.; et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef]
- Remeseiro, S.; Cuadrado, A.; Carretero, M.; Martinez, P.; Drosopoulos, W.C.; Canamero, M.; Schildkraut, C.L.; Blasco, M.A.; Losada, A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 2012, 31, 2076–2089. [Google Scholar] [CrossRef]
- De Koninck, M.; Lapi, E.; Badia-Careaga, C.; Cossio, I.; Gimenez-Llorente, D.; Rodriguez-Corsino, M.; Andrada, E.; Hidalgo, A.; Manzanares, M.; Real, F.X.; et al. Essential Roles of Cohesin STAG2 in Mouse Embryonic Development and Adult Tissue Homeostasis. Cell Rep. 2020, 32, 108014. [Google Scholar] [CrossRef]
- Wang, T.; Glover, B.; Hadwiger, G.; Miller, C.A.; di Martino, O.; Welch, J.S. Smc3 is required for mouse embryonic and adult hematopoiesis. Exp. Hematol. 2019, 70, 70–84. [Google Scholar] [CrossRef]
- Rau, R.; Magoon, D.; Greenblatt, S.; Li, L.; Annesley, C.; Duffield, A.S.; Huso, D.; McIntyre, E.; Clohessy, J.G.; Reschke, M.; et al. NPMc+ cooperates with Flt3/ITD mutations to cause acute leukemia recapitulating human disease. Exp. Hematol. 2014, 42, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Mupo, A.; Celani, L.; Dovey, O.; Cooper, J.L.; Grove, C.; Rad, R.; Sportoletti, P.; Falini, B.; Bradley, A.; Vassiliou, G.S. A powerful molecular synergy between mutant Nucleophosmin and Flt3-ITD drives acute myeloid leukemia in mice. Leukemia 2013, 27, 1917–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.I.; You, X.; Kong, G.; Ranheim, E.A.; Wang, J.; Du, J.; Liu, Y.; Zhou, Y.; Ryu, M.J.; Zhang, J. Loss of Dnmt3a and endogenous Kras(G12D/+) cooperate to regulate hematopoietic stem and progenitor cell functions in leukemogenesis. Leukemia 2015, 29, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zuber, J.; Diaz-Flores, E.; Lintault, L.; Kogan, S.C.; Shannon, K.; Lowe, S.W. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010, 24, 1389–1402. [Google Scholar] [CrossRef] [Green Version]
- Cutts, B.A.; Sjogren, A.K.; Andersson, K.M.; Wahlstrom, A.M.; Karlsson, C.; Swolin, B.; Bergo, M.O. Nf1 deficiency cooperates with oncogenic K-RAS to induce acute myeloid leukemia in mice. Blood 2009, 114, 3629–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, M.J.; So, J.; Rogers, A.J.; Gregory, G.; Li, J.; Zethoven, M.; Gearhart, M.D.; Bardwell, V.J.; Johnstone, R.W.; Vervoort, S.J.; et al. Bcor loss perturbs myeloid differentiation and promotes leukaemogenesis. Nat. Commun. 2019, 10, 1347. [Google Scholar] [CrossRef]
- Zhang, J.; Kong, G.; Rajagopalan, A.; Lu, L.; Song, J.; Hussaini, M.; Zhang, X.; Ranheim, E.A.; Liu, Y.; Wang, J.; et al. p53−/− synergizes with enhanced NrasG12D signaling to transform megakaryocyte-erythroid progenitors in acute myeloid leukemia. Blood 2017, 129, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Yang, Y.; Shang, S.; Wu, S.; Zhang, W.; Peng, L.; Huang, T.; Zhang, R.; Ren, R.; Mi, J.; et al. Cooperation of Dnmt3a R878H with Nras G12D promotes leukemogenesis in knock-in mice: A pilot study. BMC Cancer 2019, 19, 1072. [Google Scholar] [CrossRef]
- Dovey, O.M.; Cooper, J.L.; Mupo, A.; Grove, C.S.; Lynn, C.; Conte, N.; Andrews, R.M.; Pacharne, S.; Tzelepis, K.; Vijayabaskar, M.S.; et al. Molecular synergy underlies the co-occurrence patterns and phenotype of NPM1-mutant acute myeloid leukemia. Blood 2017, 130, 1911–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Liu, Y.; Cai, F.; Patrick, M.; Zmajkovic, J.; Cao, H.; Zhang, Y.; Tasdogan, A.; Chen, M.; Qi, L.; et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov. 2019, 9, 1228–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; He, F.; Bai, J.; Yamamoto, S.; Chen, S.; Zhang, L.; Sheng, M.; Zhang, L.; Guo, Y.; Man, N.; et al. Chromatin regulator Asxl1 loss and Nf1 haploinsufficiency cooperate to accelerate myeloid malignancy. J. Clin. Investig. 2018, 128, 5383–5398. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Wang, X.Q.D.; Su, J.; Putluri, N.; Zhou, T.; Qu, Y.; Jeong, M.; Guzman, A.; Rosas, C.; et al. Dnmt3a loss and Idh2 neomorphic mutations mutually potentiate malignant hematopoiesis. Blood 2020, 135, 845–856. [Google Scholar] [CrossRef]
- Loberg, M.A.; Bell, R.K.; Goodwin, L.O.; Eudy, E.; Miles, L.A.; SanMiguel, J.M.; Young, K.; Bergstrom, D.E.; Levine, R.L.; Schneider, R.K.; et al. Sequentially inducible mouse models reveal that Npm1 mutation causes malignant transformation of Dnmt3a-mutant clonal hematopoiesis. Leukemia 2019, 33, 1635–1649. [Google Scholar] [CrossRef] [PubMed]
- Sportoletti, P.; Sorcini, D.; Guzman, A.G.; Reyes, J.M.; Stella, A.; Marra, A.; Sartori, S.; Brunetti, L.; Rossi, R.; Papa, B.D.; et al. Bcor deficiency perturbs erythro-megakaryopoiesis and cooperates with Dnmt3a loss in acute erythroid leukemia onset in mice. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- D’Altri, T.; Wilhelmson, A.S.; Schuster, M.B.; Wenzel, A.; Kalvisa, A.; Pundhir, S.; Meldgaard Hansen, A.; Porse, B.T. The ASXL1-G643W variant accelerates the development of CEBPA mutant acute myeloid leukemia. Haematologica 2021, 106, 1000–1007. [Google Scholar] [CrossRef]
- Edling, C.E.; Hallberg, B. c-Kit—A hematopoietic cell essential receptor tyrosine kinase. Int. J. Biochem. Cell Biol. 2007, 39, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Beghini, A.; Peterlongo, P.; Ripamonti, C.B.; Larizza, L.; Cairoli, R.; Morra, E.; Mecucci, C. C-kit mutations in core binding factor leukemias. Blood 2000, 95, 726–727. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Mrózek, K.; Chen, H.; Kittles, R.A.; Vukosavljevic, T.; Perrotti, D.; Vardiman, J.W.; Carroll, A.J.; et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): A Cancer and Leukemia Group B Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3904–3911. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Kawashima, N.; Atsuta, Y.; Sugiura, I.; Sawa, M.; Dobashi, N.; Yokoyama, H.; Doki, N.; Tomita, A.; Kiguchi, T.; et al. Prospective evaluation of prognostic impact of KIT mutations on acute myeloid leukemia with RUNX1-RUNX1T1 and CBFB-MYH11. Blood Adv. 2020, 4, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Nick, H.J.; Kim, H.G.; Chang, C.W.; Harris, K.W.; Reddy, V.; Klug, C.A. Distinct classes of c-Kit-activating mutations differ in their ability to promote RUNX1-ETO-associated acute myeloid leukemia. Blood 2012, 119, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.M.; Duque, J.; Shizuru, J.A.; Lübbert, M. Complementing mutations in core binding factor leukemias: From mouse models to clinical applications. Oncogene 2008, 27, 5759–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Melenhorst, J.J.; Alemu, L.; Kirby, M.; Anderson, S.; Kench, M.; Hoogstraten-Miller, S.; Brinster, L.; Kamikubo, Y.; Gilliland, D.G.; et al. KIT with D816 mutations cooperates with CBFB-MYH11 for leukemogenesis in mice. Blood 2012, 119, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowy, D.R.; Willumsen, B.M. Function and regulation of ras. Annu. Rev. Biochem. 1993, 62, 851–891. [Google Scholar] [CrossRef]
- Bowen, D.T.; Frew, M.E.; Hills, R.; Gale, R.E.; Wheatley, K.; Groves, M.J.; Langabeer, S.E.; Kottaridis, P.D.; Moorman, A.V.; Burnett, A.K.; et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood 2005, 106, 2113–2119. [Google Scholar] [CrossRef] [Green Version]
- Bacher, U.; Haferlach, T.; Schoch, C.; Kern, W.; Schnittger, S. Implications of NRAS mutations in AML: A study of 2502 patients. Blood 2006, 107, 3847–3853. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Z.; Li, T.; Li, Y.; Wang, W.; Hao, Q.; Xie, X.; Wan, D.; Jiang, Z.; Wang, C.; et al. Mutational spectrum and prognosis in NRAS-mutated acute myeloid leukemia. Sci. Rep. 2020, 10, 12152. [Google Scholar] [CrossRef]
- Li, Q.; Haigis, K.M.; McDaniel, A.; Harding-Theobald, E.; Kogan, S.C.; Akagi, K.; Wong, J.C.; Braun, B.S.; Wolff, L.; Jacks, T.; et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood 2011, 117, 2022–2032. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, Y.; Li, Z.; Du, J.; Ryu, M.J.; Taylor, P.R.; Fleming, M.D.; Young, K.H.; Pitot, H.; Zhang, J. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood 2010, 116, 5991–6002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A.; Silver, D.P.; Jackson, E.L.; Chang, S.; Mercer, K.L.; Grochow, R.; Hock, H.; Crowley, D.; et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004, 5, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Schmoellerl, J.; Barbosa, I.A.M.; Eder, T.; Brandstoetter, T.; Schmidt, L.; Maurer, B.; Troester, S.; Pham, H.T.T.; Sagarajit, M.; Ebner, J.; et al. CDK6 is an essential direct target of NUP98 fusion proteins in acute myeloid leukemia. Blood 2020, 136, 387–400. [Google Scholar] [CrossRef]
- Kim, W.I.; Matise, I.; Diers, M.D.; Largaespada, D.A. RAS oncogene suppression induces apoptosis followed by more differentiated and less myelosuppressive disease upon relapse of acute myeloid leukemia. Blood 2009, 113, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Radtke, I.; Pardee, T.S.; Zhao, Z.; Rappaport, A.R.; Luo, W.; McCurrach, M.E.; Yang, M.M.; Dolan, M.E.; Kogan, S.C.; et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009, 23, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Eisfeld, A.K.; Kohlschmidt, J.; Mrózek, K.; Mims, A.; Walker, C.J.; Blachly, J.S.; Nicolet, D.; Orwick, S.; Maharry, S.E.; Carroll, A.J.; et al. NF1 mutations are recurrent in adult acute myeloid leukemia and confer poor outcome. Leukemia 2018, 32, 2536–2545. [Google Scholar] [CrossRef]
- Le, D.T.; Kong, N.; Zhu, Y.; Lauchle, J.O.; Aiyigari, A.; Braun, B.S.; Wang, E.; Kogan, S.C.; Le Beau, M.M.; Parada, L.; et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood 2004, 103, 4243–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.; Morgan, K.; Hasz, D.E.; Wiesner, S.M.; Lauchle, J.O.; Geurts, J.L.; Diers, M.D.; Le, D.T.; Kogan, S.C.; Parada, L.F.; et al. Beta common receptor inactivation attenuates myeloproliferative disease in Nf1 mutant mice. Blood 2007, 109, 1687–1691. [Google Scholar] [CrossRef] [Green Version]
- Chan, R.J.; Feng, G.S. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2007, 109, 862–867. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, K.S.; Rosário, M.; Birchmeier, C.; Birchmeier, W. The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res. 2010, 106, 53–89. [Google Scholar] [CrossRef]
- Pandey, R.; Saxena, M.; Kapur, R. Role of SHP2 in hematopoiesis and leukemogenesis. Curr. Opin. Hematol. 2017, 24, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Alfayez, M.; Issa, G.C.; Patel, K.P.; Wang, F.; Wang, X.; Short, N.J.; Cortes, J.E.; Kadia, T.; Ravandi, F.; Pierce, S.; et al. The Clinical impact of PTPN11 mutations in adults with acute myeloid leukemia. Leukemia 2021, 35, 691–700. [Google Scholar] [CrossRef]
- Chan, G.; Cheung, L.S.; Yang, W.; Milyavsky, M.; Sanders, A.D.; Gu, S.; Hong, W.X.; Liu, A.X.; Wang, X.; Barbara, M.; et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood 2011, 117, 4253–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarnawsky, S.P.; Yu, W.M.; Qu, C.K.; Chan, R.J.; Yoder, M.C. Hematopoietic-restricted Ptpn11E76K reveals indolent MPN progression in mice. Oncotarget 2018, 9, 21831–21843. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Liu, X.; Yu, W.M.; Meyerson, H.J.; Guo, C.; Gerson, S.L.; Qu, C.K. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J. Exp. Med. 2011, 208, 1977–1988. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, W.; Mysliwski, M.; Serio, J.; Ropa, J.; Abulwerdi, F.A.; Chan, R.J.; Patel, J.P.; Tallman, M.S.; Paietta, E.; et al. Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia 2015, 29, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.F.; Liang, S.T.; Huang, Y.J.; Liang, K.H.; Yen, T.H.; Liang, D.C.; Shih, L.Y. Cooperation of MLL/AF10(OM-LZ) with PTPN11 activating mutation induced monocytic leukemia with a shorter latency in a mouse bone marrow transplantation model. Int. J. Cancer 2017, 140, 1159–1172. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Mayle, A.; Yang, L.; Rodriguez, B.; Zhou, T.; Chang, E.; Curry, C.V.; Challen, G.A.; Li, W.; Wheeler, D.; Rebel, V.I.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Rodriguez, B.; Mayle, A.; Park, H.J.; Lin, X.; Luo, M.; Jeong, M.; Curry, C.V.; Kim, S.B.; Ruau, D.; et al. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer Cell 2016, 29, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Celik, H.; Mallaney, C.; Kothari, A.; Ostrander, E.L.; Eultgen, E.; Martens, A.; Miller, C.A.; Hundal, J.; Klco, J.M.; Challen, G.A. Enforced differentiation of Dnmt3a-null bone marrow leads to failure with c-Kit mutations driving leukemic transformation. Blood 2015, 125, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Richine, B.M.; Virts, E.L.; Jideonwo-Auman, V.N.; Chan, R.J.; Kapur, R. Rapid development of myeloproliferative neoplasm in mice with Ptpn11(D61Y) mutation and haploinsufficient for Dnmt3a. Oncotarget 2018, 9, 6055–6061. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Pronier, E.; Almire, C.; Mokrani, H.; Vasanthakumar, A.; Simon, A.; da Costa Reis Monte Mor, B.; Massé, A.; Le Couédic, J.P.; Pendino, F.; Carbonne, B.; et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 2011, 118, 2551–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Gonzalez-Avalos, E.; Chawla, A.; Jeong, M.; Lopez-Moyado, I.F.; Li, W.; Goodell, M.A.; Chavez, L.; Ko, M.; Rao, A. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat. Commun. 2015, 6, 10071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, R.; Sakata-Yanagimoto, M.; Maie, K.; Oshima, M.; Ishihara, M.; Suehara, Y.; Fukumoto, K.; Nakajima-Takagi, Y.; Matsui, H.; Kato, T.; et al. Molecular pathogenesis of progression to myeloid leukemia from TET-insufficient status. Blood Adv. 2020, 4, 845–854. [Google Scholar] [CrossRef]
- Kunimoto, H.; Meydan, C.; Nazir, A.; Whitfield, J.; Shank, K.; Rapaport, F.; Maher, R.; Pronier, E.; Meyer, S.C.; Garrett-Bakelman, F.E.; et al. Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell 2018, 33, 44–59. [Google Scholar] [CrossRef] [Green Version]
- Muto, T.; Sashida, G.; Oshima, M.; Wendt, G.R.; Mochizuki-Kashio, M.; Nagata, Y.; Sanada, M.; Miyagi, S.; Saraya, A.; Kamio, A.; et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 2013, 210, 2627–2639. [Google Scholar] [CrossRef]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Montalban-Bravo, G.; DiNardo, C.D. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018, 14, 979–993. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattih, B.; Shirvani, A.; Klement, P.; Garrido, A.M.; Gabdoulline, R.; Liebich, A.; Brandes, M.; Chaturvedi, A.; Seeger, T.; Thol, F.; et al. IDH1/2 mutations in acute myeloid leukemia patients and risk of coronary artery disease and cardiac dysfunction—a retrospective propensity score analysis. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Goparaju, R.; Schwarzer, A.; Görlich, K.; Schottmann, R.; Struys, E.A.; Jansen, E.E.; et al. Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia 2016, 30, 1708–1715. [Google Scholar] [CrossRef] [Green Version]
- Ogawara, Y.; Katsumoto, T.; Aikawa, Y.; Shima, Y.; Kagiyama, Y.; Soga, T.; Matsunaga, H.; Seki, T.; Araki, K.; Kitabayashi, I. IDH2 and NPM1 Mutations Cooperate to Activate Hoxa9/Meis1 and Hypoxia Pathways in Acute Myeloid Leukemia. Cancer Res. 2015, 75, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Safaei, S.; Baradaran, B.; Hagh, M.F.; Alivand, M.R.; Talebi, M.; Gharibi, T.; Solali, S. Double sword role of EZH2 in leukemia. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 98, 626–635. [Google Scholar] [CrossRef]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun 2014, 5, 4177. [Google Scholar] [CrossRef] [Green Version]
- Neff, T.; Sinha, A.U.; Kluk, M.J.; Zhu, N.; Khattab, M.H.; Stein, L.; Xie, H.; Orkin, S.H.; Armstrong, S.A. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 5028–5033. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Miyagi, S.; Sashida, G.; Chiba, T.; Yuan, J.; Mochizuki-Kashio, M.; Suzuki, Y.; Sugano, S.; Nakaseko, C.; Yokote, K.; et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 2012, 120, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Xu, M.; Yang, F.-C. The Role of ASXL1/2 and Their Associated Proteins in Malignant Hematopoiesis. Curr. Stem Cell Rep. 2020, 6, 6–15. [Google Scholar] [CrossRef]
- Chou, W.C.; Huang, H.H.; Hou, H.A.; Chen, C.Y.; Tang, J.L.; Yao, M.; Tsay, W.; Ko, B.S.; Wu, S.J.; Huang, S.Y.; et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010, 116, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, S.; Fujino, T.; Goyama, S.; Kitamura, T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. 2019, 76, 2511–2523. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micol, J.B.; Duployez, N.; Boissel, N.; Petit, A.; Geffroy, S.; Nibourel, O.; Lacombe, C.; Lapillonne, H.; Etancelin, P.; Figeac, M.; et al. Frequent ASXL2 mutations in acute myeloid leukemia patients with t(8;21)/RUNX1-RUNX1T1 chromosomal translocations. Blood 2014, 124, 1445–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Micol, J.-B.; Pastore, A.; Inoue, D.; Duployez, N.; Kim, E.; Lee, S.C.-W.; Durham, B.H.; Chung, Y.R.; Cho, H.; Zhang, X.J.; et al. ASXL2 is essential for haematopoiesis and acts as a haploinsufficient tumour suppressor in leukemia. Nat. Commun. 2017, 8, 15429. [Google Scholar] [CrossRef] [Green Version]
- Jeong, E.G.; Lee, S.H.; Yoo, N.J.; Lee, S.H. Absence of nucleophosmin 1 (NPM1) gene mutations in common solid cancers. APMIS 2007, 115, 341–346. [Google Scholar] [CrossRef]
- Rau, R.; Brown, P. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: Towards definition of a new leukaemia entity. Hematol. Oncol. 2009, 27, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Zarka, J.; Short, N.J.; Kanagal-Shamanna, R.; Issa, G.C. Nucleophosmin 1 Mutations in Acute Myeloid Leukemia. Genes 2020, 11, 649. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Falini, B.; Bolli, N.; Shan, J.; Martelli, M.P.; Liso, A.; Pucciarini, A.; Bigerna, B.; Pasqualucci, L.; Mannucci, R.; Rosati, R.; et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 2006, 107, 4514–4523. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, M.; Tiacci, E.; Bergomas, R.; Bigerna, B.; Venturini, E.; Minardi, S.P.; Meani, N.; Diverio, D.; Bernard, L.; Tizzoni, L.; et al. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood 2005, 106, 899–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rucker, F.G.; Dohner, K.; McGeehan, G.M.; et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef]
- Dzama, M.M.; Steiner, M.; Rausch, J.; Sasca, D.; Schönfeld, J.; Kunz, K.; Taubert, M.C.; McGeehan, G.M.; Chen, C.W.; Mupo, A.; et al. Synergistic targeting of FLT3 mutations in AML via combined menin-MLL and FLT3 inhibition. Blood 2020, 136, 2442–2456. [Google Scholar] [CrossRef] [PubMed]
- Avellino, R.; Delwel, R. Expression and regulation of C/EBPα in normal myelopoiesis and in malignant transformation. Blood 2017, 129, 2083–2091. [Google Scholar] [CrossRef]
- Wouters, B.J.; Löwenberg, B.; Erpelinck-Verschueren, C.A.; van Putten, W.L.; Valk, P.J.; Delwel, R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009, 113, 3088–3091. [Google Scholar] [CrossRef] [Green Version]
- Taskesen, E.; Bullinger, L.; Corbacioglu, A.; Sanders, M.A.; Erpelinck, C.A.; Wouters, B.J.; van der Poel-van de Luytgaarde, S.C.; Damm, F.; Krauter, J.; Ganser, A.; et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: Further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 2011, 117, 2469–2475. [Google Scholar] [CrossRef] [PubMed]
- Leroy, H.; Roumier, C.; Huyghe, P.; Biggio, V.; Fenaux, P.; Preudhomme, C. CEBPA point mutations in hematological malignancies. Leukemia 2005, 19, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Iwasaki-Arai, J.; Iwasaki, H.; Fenyus, M.L.; Dayaram, T.; Owens, B.M.; Shigematsu, H.; Levantini, E.; Huettner, C.S.; Lekstrom-Himes, J.A.; et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 2004, 21, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Okhovat, M.; Coblentz, C.; Carratt, S.A.; Foley, A.; Schonrock, Z.; Smith, B.M.; Nevonen, K.; Davis, B.; Garcia, B.; et al. Myeloid lineage enhancers drive oncogene synergy in CEBPA/CSF3R mutant acute myeloid leukemia. Nat. Commun. 2019, 10, 5455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlsson, E.; Hasemann, M.S.; Willer, A.; Lauridsen, F.K.; Rapin, N.; Jendholm, J.; Porse, B.T. Initiation of MLL-rearranged AML is dependent on C/EBPα. J. Exp. Med. 2014, 211, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, C.; Wang, J.; Miao, H.; Bronstein, J.; Nawer, H.; Xu, T.; Figueroa, M.; Muntean, A.G.; Hess, J.L. C/EBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 9899–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, H.; Kobayashi, S.S. Targeting transcription factors in acute myeloid leukemia. Int. J. Hematol. 2019, 109, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Okuda, T.; Nishimura, M.; Nakao, M.; Fujita, Y. RUNX1/AML1: A central player in hematopoiesis. Int. J. Hematol. 2001, 74, 252–257. [Google Scholar] [CrossRef]
- Growney, J.D.; Shigematsu, H.; Li, Z.; Lee, B.H.; Adelsperger, J.; Rowan, R.; Curley, D.P.; Kutok, J.L.; Akashi, K.; Williams, I.R.; et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005, 106, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood 2011, 117, 2348–2357. [Google Scholar] [CrossRef] [Green Version]
- Chin, D.W.; Watanabe-Okochi, N.; Wang, C.Q.; Tergaonkar, V.; Osato, M. Mouse models for core binding factor leukemia. Leukemia 2015, 29, 1970–1980. [Google Scholar] [CrossRef]
- Bera, R.; Chiu, M.C.; Huang, Y.J.; Lin, T.H.; Kuo, M.C.; Shih, L.Y. RUNX1 mutations promote leukemogenesis of myeloid malignancies in ASXL1-mutated leukemia. J. Hematol. Oncol. 2019, 12, 104. [Google Scholar] [CrossRef] [Green Version]
- Goyama, S.; Schibler, J.; Cunningham, L.; Zhang, Y.; Rao, Y.; Nishimoto, N.; Nakagawa, M.; Olsson, A.; Wunderlich, M.; Link, K.A.; et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J. Clin. Investig. 2013, 123, 3876–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, M.D.; León, J. Myc roles in hematopoiesis and leukemia. Genes Cancer 2010, 1, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Astolfi, A.; Fiore, M.; Melchionda, F.; Indio, V.; Bertuccio, S.N.; Pession, A. BCOR involvement in cancer. Epigenomics 2019, 11, 835–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, V.; Tiacci, E.; Holmes, A.B.; Kohlmann, A.; Martelli, M.P.; Kern, W.; Spanhol-Rosseto, A.; Klein, H.U.; Dugas, M.; Schindela, S.; et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood 2011, 118, 6153–6163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.R.; Achille, N.J.; Kuntimaddi, A.; Boulton, A.M.; Leach, B.I.; Zhang, S.; Zeleznik-Le, N.J.; Bushweller, J.H. BCOR Binding to MLL-AF9 Is Essential for Leukemia via Altered EYA1, SIX, and MYC Activity. Blood Cancer Discov. 2020, 1, 162–177. [Google Scholar] [CrossRef] [PubMed]
- McNerney, M.E.; Brown, C.D.; Wang, X.; Bartom, E.T.; Karmakar, S.; Bandlamudi, C.; Yu, S.; Ko, J.; Sandall, B.P.; Stricker, T.; et al. CUX1 is a haploinsufficient tumor suppressor gene on chromosome 7 frequently inactivated in acute myeloid leukemia. Blood 2013, 121, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aly, M.; Ramdzan, Z.M.; Nagata, Y.; Balasubramanian, S.K.; Hosono, N.; Makishima, H.; Visconte, V.; Kuzmanovic, T.; Adema, V.; Nazha, A.; et al. Distinct clinical and biological implications of CUX1 in myeloid neoplasms. Blood Adv. 2019, 3, 2164–2178. [Google Scholar] [CrossRef] [PubMed]
- An, N.; Khan, S.; Imgruet, M.K.; Gurbuxani, S.K.; Konecki, S.N.; Burgess, M.R.; McNerney, M.E. Gene dosage effect of CUX1 in a murine model disrupts HSC homeostasis and controls the severity and mortality of MDS. Blood 2018, 131, 2682–2697. [Google Scholar] [CrossRef]
- Stieglitz, E.; Troup, C.B.; Gelston, L.C.; Haliburton, J.; Chow, E.D.; Yu, K.B.; Akutagawa, J.; Taylor-Weiner, A.N.; Liu, Y.L.; Wang, Y.-D.; et al. Subclonal mutations in SETBP1 confer a poor prognosis in juvenile myelomonocytic leukemia. Blood 2015, 125, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Thol, F.; Suchanek, K.J.; Koenecke, C.; Stadler, M.; Platzbecker, U.; Thiede, C.; Schroeder, T.; Kobbe, G.; Kade, S.; Löffeld, P.; et al. SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia 2013, 27, 2072–2075. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshida, K.; Nguyen, N.; Przychodzen, B.; Sanada, M.; Okuno, Y.; Ng, K.P.; Gudmundsson, K.O.; Vishwakarma, B.A.; Jerez, A.; et al. Somatic SETBP1 mutations in myeloid malignancies. Nat. Genet. 2013, 45, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Lower, K.M.; Turner, G.; Kerr, B.A.; Mathews, K.D.; Shaw, M.A.; Gedeon, A.K.; Schelley, S.; Hoyme, H.E.; White, S.M.; Delatycki, M.B.; et al. Mutations in PHF6 are associated with Börjeson-Forssman-Lehmann syndrome. Nat. Genet. 2002, 32, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Patel, J.; Abdel-Wahab, O.; Lobry, C.; Hedvat, C.V.; Balbin, M.; Nicolas, C.; Payer, A.R.; Fernandez, H.F.; Tallman, M.S.; et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia 2011, 25, 130–134. [Google Scholar] [CrossRef]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Todd, M.A.; Ivanochko, D.; Picketts, D.J. PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein. Genes 2015, 6, 325–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King-Underwood, L.; Little, S.; Baker, M.; Clutterbuck, R.; Delassus, S.; Enver, T.; Lebozer, C.; Min, T.; Moore, A.; Schedl, A.; et al. Wt1 is not essential for hematopoiesis in the mouse. Leuk. Res. 2005, 29, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.Y.; Brownstein, D.; Mjoseng, H.; Lee, W.C.; Buza-Vidas, N.; Nerlov, C.; Jacobsen, S.E.; Perry, P.; Berry, R.; Thornburn, A.; et al. Acute multiple organ failure in adult mice deleted for the developmental regulator Wt1. PLoS Genet. 2011, 7, e1002404. [Google Scholar] [CrossRef] [Green Version]
- Miwa, H.; Beran, M.; Saunders, G.F. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409. [Google Scholar]
- King-Underwood, L.; Renshaw, J.; Pritchard-Jones, K. Mutations in the Wilms’ tumor gene WT1 in leukemias. Blood 1996, 87, 2171–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menssen, H.D.; Renkl, H.J.; Rodeck, U.; Maurer, J.; Notter, M.; Schwartz, S.; Reinhardt, R.; Thiel, E. Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 1995, 9, 1060–1067. [Google Scholar] [PubMed]
- Becker, H.; Marcucci, G.; Maharry, K.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Whitman, S.P.; Paschka, P.; Holland, K.B.; Schwind, S.; et al. Mutations of the Wilms tumor 1 gene (WT1) in older patients with primary cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. Blood 2010, 116, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard-Jones, K.; Fleming, S.; Davidson, D.; Bickmore, W.; Porteous, D.; Gosden, C.; Bard, J.; Buckler, A.; Pelletier, J.; Housman, D.; et al. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 1990, 346, 194–197. [Google Scholar] [CrossRef]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Figueroa, M.E. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica 2016, 101, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 2019, 58, 875–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ellison, F.M.; Keyvanfar, K.; Omokaro, S.O.; Desierto, M.J.; Eckhaus, M.A.; Young, N.S. Enrichment of hematopoietic stem cells with SLAM and LSK markers for the detection of hematopoietic stem cell function in normal and Trp53 null mice. Exp. Hematol. 2008, 36, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- TeKippe, M.; Harrison, D.E.; Chen, J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp. Hematol. 2003, 31, 521–527. [Google Scholar] [CrossRef]
- Pant, V.; Quintas-Cardama, A.; Lozano, G. The p53 pathway in hematopoiesis: Lessons from mouse models, implications for humans. Blood 2012, 120, 5118–5127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Parant, J.M.; Lang, G.; Chau, P.; Chavez-Reyes, A.; El-Naggar, A.K.; Multani, A.; Chang, S.; Lozano, G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat. Genet. 2004, 36, 63–68. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [Green Version]
- Basova, P.; Pospisil, V.; Savvulidi, F.; Burda, P.; Vargova, K.; Stanek, L.; Dluhosova, M.; Kuzmova, E.; Jonasova, A.; Steidl, U.; et al. Aggressive acute myeloid leukemia in PU.1/p53 double-mutant mice. Oncogene 2014, 33, 4735–4745. [Google Scholar] [CrossRef] [Green Version]
- Jyotsana, N.; Heuser, M. Exploiting differential RNA splicing patterns: A potential new group of therapeutic targets in cancer. Expert Opin. Ther. Targets 2018, 22, 107–121. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627; quiz 3699. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamopoulos, S.A.; Batcha, A.M.N.; Jurinovic, V.; Rothenberg-Thurley, M.; Janke, H.; Ksienzyk, B.; Philippou-Massier, J.; Graf, A.; Krebs, S.; Blum, H.; et al. Clinical presentation and differential splicing of SRSF2, U2AF1 and SF3B1 mutations in patients with acute myeloid leukemia. Leukemia 2020, 34, 2621–2634. [Google Scholar] [CrossRef]
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.-W.; Micol, J.-B.; Zhang, X.J.; de Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Yan, M.; Kim, E.; Davis, A.G.; Shima, T.; Miyauchi, S.; Fu, X.-D.; Abdel-Wahab, O.; Zhang, D.-E. RUNX1 Deficiency and SRSF2 Mutation Cooperate to Promote Myelodysplastic Syndrome Development. Blood 2017, 130, 119. [Google Scholar] [CrossRef]
- Wang, E.; Aifantis, I. RNA Splicing and Cancer. Trends Cancer 2020, 6, 631–644. [Google Scholar] [CrossRef]
- Heimbruch, K.E.; Meyer, A.E.; Agrawal, P.; Viny, A.D.; Rao, S. A cohesive look at leukemogenesis: The cohesin complex and other driving mutations in AML. Neoplasia 2021, 23, 337–347. [Google Scholar] [CrossRef]
- Cuartero, S.; Innes, A.J.; Merkenschlager, M. Towards a Better Understanding of Cohesin Mutations in AML. Front. Oncol. 2019, 9, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Wang, X. Roles of cohesin in chromosome architecture and gene expression. Semin. Cell Dev. Biol. 2019, 90, 187–193. [Google Scholar] [CrossRef]
- Han, C.; Gao, X.; Li, Y.; Zhang, J.; Yang, E.; Zhang, L.; Yu, L. Characteristics of Cohesin Mutation in Acute Myeloid Leukemia and Its Clinical Significance. Expert Opin. Ther. Targets 2021, 11. [Google Scholar] [CrossRef]
- Fang, C.; Rao, S.; Crispino, J.D.; Ntziachristos, P. Determinants and role of chromatin organization in acute leukemia. Leukemia 2020, 34, 2561–2575. [Google Scholar] [CrossRef]
- Thol, F.; Bollin, R.; Gehlhaar, M.; Walter, C.; Dugas, M.; Suchanek, K.J.; Kirchner, A.; Huang, L.; Chaturvedi, A.; Wichmann, M.; et al. Mutations in the cohesin complex in acute myeloid leukemia: Clinical and prognostic implications. Blood 2014, 123, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Mintzas, K.; Heuser, M. Emerging strategies to target the dysfunctional cohesin complex in cancer. Expert Opin. Ther. Targets 2019, 23, 525–537. [Google Scholar] [CrossRef]
- Mullenders, J.; Aranda-Orgilles, B.; Lhoumaud, P.; Keller, M.; Pae, J.; Wang, K.; Kayembe, C.; Rocha, P.P.; Raviram, R.; Gong, Y.; et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 2015, 212, 1833–1850. [Google Scholar] [CrossRef] [PubMed]
- Viny, A.D.; Bowman, R.L.; Liu, Y.; Lavallee, V.P.; Eisman, S.E.; Xiao, W.; Durham, B.H.; Navitski, A.; Park, J.; Braunstein, S.; et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell 2019, 25, 682–696. [Google Scholar] [CrossRef]
- Ochi, Y.; Kon, A.; Sakata, T.; Nakagawa, M.M.; Nakazawa, N.; Kakuta, M.; Kataoka, K.; Koseki, H.; Nakayama, M.; Morishita, D.; et al. Combined Cohesin-RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov. 2020, 10, 836–853. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.H.; Hou, H.A.; Tang, J.L.; Kuo, Y.Y.; Chiu, Y.C.; Lin, C.C.; Liu, C.Y.; Tseng, M.H.; Lin, T.Y.; Liu, M.C.; et al. Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer J. 2017, 7, 663. [Google Scholar] [CrossRef] [PubMed]


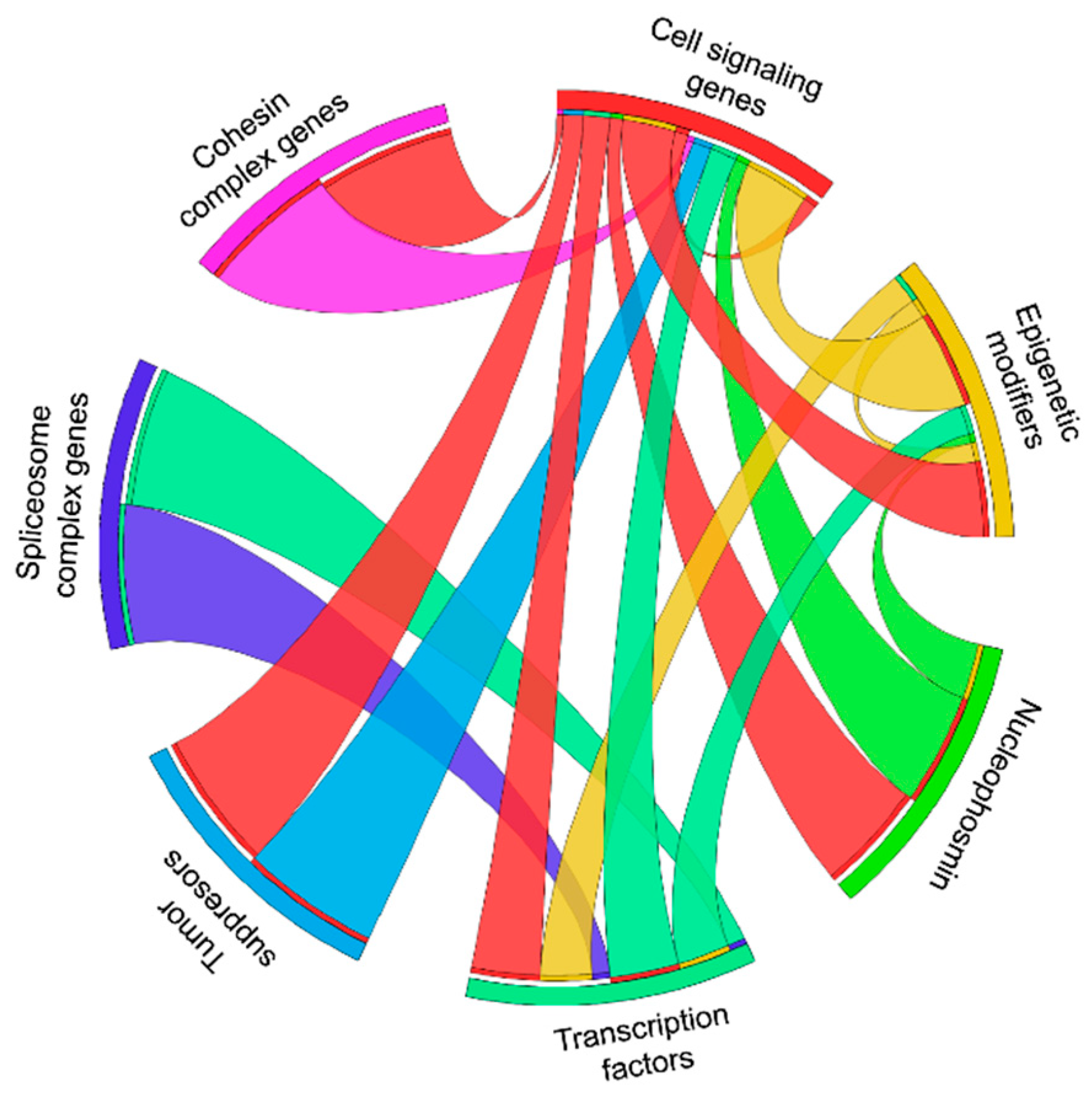{kind=link}
| Group | Genes | Overexpression/ Transgenic Mice Model | Knockin | Knockout |
|---|---|---|---|---|
| Cell signaling genes | FLT3 | FLT3 WT-ND FLT3 ITD -MPN [13] FLT3-ITD (Vav promoter) -MPN and B- or T-lymphoid disorders [15] FLT3 TKD -Lymphoid disorder [14] | FLT3wt/ITD-MPN [17] FLT3/D835Y- MPN, lymphomas and sarcomas [18] Flt3+/ITD and Flt3ITD/ITD -CMML [16] | FLT3−/− -Viable but deficiencies in B lymphoid progenitor [12] |
| KIT | hKIT wt -ND hKIT D816V -ND hybrid C-KIT D816V-MPN [35] HyC-KIT N822K-MPN [36] | NA | c-Kit−/− -Postnatal death [37] | |
| KRAS | KRAS G12D-ND [38] | KRAS G12D- MPN [39,40] | Kras−/− -Embryonic lethal [41,42] | |
| NRAS | NRAS G12D (MSCV promoter) -ND [38,43] NRASD12 -CMML and AML [44] NRAS G12D (hMRP8 promoter) -Hyperkeratotic skin lesions [45] Mo-MuLV Nras G12D -MPNs [46] | Nras G12D -MPN [47] | Nras−/− -Viable and no defect in hematopoiesis [48] | |
| NF1 | NA | NA | Nf1−/− -Embryonic lethality [49] Nf1+/− -Various tumors [50] | |
| PTPN11 | PTPN11 wt -ND PTPN11 E76K -JMML PTPN11 D61Y -JMML [51] | Ptpn11 D61Y [52] -MPN Ptpn11 E76K/+ -MPN [53] | Ptpn11−/− -Embryonic lethal [54] | |
| Epigenetic modifiers | DNMT3A | DNMT3A Wt-ND DNMT3A R882H -CMML [55] | Dnmt3a R878H/WT -AML [56] | Dnmt3a−/− -Viable [57] Conditional Dnmt3a−/− -MDS/ MPN [58] |
| TET2 | NA | NA | TET2−/− -wide spectrum myeloid malignancies [59,60] Conditional Tet2+/− -EMH [61] | |
| IDH1 | IDH1 WT-ND IDH1 R132C -ND [62] | IDH1 R132H -EMH and Splenomegaly [63] | Idh1−/− -Viable [64] | |
| IDH2 | IDH2 R140Q -EMH, Splenomegaly [24] | Idh2 R140Q -ND [65,66] | Idh2−/−-Viable [67] | |
| EZH2 | EZH2 wt -MPN [68] | NA | Ezh2−/− -Embryonic lethal [69] Hematopoietic Ezh2−/− -MDS [70] | |
| ASXL1 | C -terminal truncated mutant ASXL1 -MDS [71] Asxl1 Y588X -AML/MDS/MPN [72] | Asxl1 G643fs -ND [73] Asxl1 G643fs/+ -MDS [74] | Asxl1−/− -Embryonic lethal/MDS Asxl1+/− -MDS [75] | |
| ASXL2 | NA | NA | Asxl2−/− -Partial Embryonic lethal/Mild BM disorders/MDS [76,77,78] | |
| Nucleophosmin 1 | NPM1 | NPM1c+ -myeloproliferation [79] NPM1 -ND [79] | Npm1 wt/c+ -MPN some mice [80] late AML onset in some mice [81,82] | Npm1+/− -MDS [83,84] Npm1−/− -Embryonic lethal [83] |
| Transcription factors | CEBPA | NA | CEEBPA K313KK/Lp30 Retxn -AML [85] | CEBPA−/− -Postnatal death [86] CEBPA p42+/− -ND CEBPA p42−/− -AML [87] |
| RUNX1 | RUNX1 D171N and S291fsX300 -MDS [88] | NA | Runx1−/− -Embryonic lethal [89] with hematopoietic defect | |
| MYC | C-Myc -AML [90] N-Myc -AML [91] | NA | C-Myc−/− Embryonic lethal [92] | |
| BCOR | NA | Bcor ΔE4/y -TALL [93] Bcor ΔE9−10/y -TALL [94] | Bcor−/Y -Male embryonic lethality [95] | |
| CUX1 | p75 CUX -MPN [96] | NA | Cux1−/− -Postnatal death [97] | |
| SETBP1 | Setbp1 -Myeloid leukemia [98] SETBP1-D868N Splenomegaly [99] | NA | NA | |
| PHF6 | NA | NA | Conditional hematopoietic knockout -Myelodysplasia-like disease [100] Phf6−/Y -Perinatal lethality in males [101] | |
| Tumor suppresors | WT1 | NA | Wt1+/R394W -MDS [26] | Wt1−/− -Embryonic lethal [102] Wt1fl/+ -T-ALL [27] |
| TP53 | NA | p53 R172H -Lymphoma, leukemia or mix [103] p53 R248Q -T cell/B cell lymphoma, solid tumors [104] | P53−/− -Majorly lymphoma [105,106] | |
| Spliceosome complex | SRSF2 | SRSF2 WT -ND SRSF2 P95H -ND [107] | Srsf2 P95H/wt -Myelodysplasia [108] impaired hematopoietic stem cell functions [109] | Srsf2−/− -Embryonic lethal [107] |
| U2AF1 | U2AF1 S34F -Leukopenia [110] | U2af1 S34F/WT -MDS-like phenotype [111] | U2af1−/− -Embryonic lethal [112] | |
| SF3B1 | NA | Sf3b1 K700E/+ -Anemia [113] Sf3b1+/K700E -MDS [114] | Sf3b1+/− -MDS [115] Sf3b1−/− -Embryonic lethal [116] | |
| Cohesin complex | RAD21 | NA | NA | Rad21−/− -Embryonic lethal [117] |
| STAG1 | NA | NA | Stag1−/− -Embryonic lethal [118] | |
| STAG2 | NA | NA | Stag2−/− -Embryonic lethal [119] | |
| SMC3 | NA | NA | Smc3−/− -Embryonic lethal [120] |
| Group | Genes | Synergistic Genes in the Development of AML in Mice |
|---|---|---|
| Cell signaling genes | FLT3 | SMC3−/+ [19] RUNX1 [20] NPM1c+ [82,121,122] Dnamt3a−/− [22,23] IDH2 R140Q [24] or IDH2 R172K [25] Wt1fl/+ [26,27] TET2−/− [28] Cux1+/− [30] Setbp1 [29] |
| KIT | NA | |
| KRAS | Dnmt3a−/− [123] P53 [124] Nf1 [125] Bcor ΔE9−10 [126] | |
| NRAS | P53−/− [127] Dnmt3a R878H [128] IDH2 R140Q or IDH2 R172K [25] Npm1cA [129] EZH2−/− [130] | |
| NF1 | Asxl1+/− [131] Kras G12D [125] | |
| PTPN11 | NA | |
| Epigenetic modifiers | DNMT3A | Kras G12D/+ [123] FLT3 ITD [22,23] Nras G12D [128,132] IDH2 neomorphic [133] Npm1cA [134] Bcor−/− [135] |
| TET2 | FLT3 ITD [28] | |
| IDH1 | NA | |
| IDH2 | Nras G12D [25] FLT3 ITD [24,25] Dnmt3a−/− [133] | |
| EZH2 | NRAS G12D [130] | |
| ASXL1 | CEBPA [136] Cebpa D/p30 [136] SETBP1 D868N [99] Nf1+/– [131] | |
| ASXL2 | NA | |
| Nucleophosmin 1 | NPM1 | FLT3 ITD [82,121,122] FLT3 TKD [21] NRAS G12D [129] Dnmt3a R878H [134] |
| Transcription factors | CEBPA | Asxl1 G643W [136] |
| RUNX1 | FLT3 ITD [20] U2af1 S34F [111] | |
| MYC | NA | |
| BCOR | Dnamt3a−/− [135] Kras G12D [126] | |
| CUX1 | Flt3 ITD [30] | |
| SETBP1 | ASXL1 MT [99] FLT3 ITD [29] | |
| PHF6 | NA | |
| Tumor suppressors | WT1 | FLT3 ITD [26,27] |
| TP53 | NARS G12D [127] Kras G12D [124] | |
| Spliceosome complex | SRSF2 | NA |
| U2AF1 | Runx1F/F [111] | |
| SF3B1 | NA | |
| Cohesin complex | RAD21 | NA |
| STAG1 | NA | |
| STAG2 | NA | |
| SMC3 | FLT3 ITD [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanty, S.; Heuser, M. Mouse Models of Frequently Mutated Genes in Acute Myeloid Leukemia. Cancers 2021, 13, 6192. https://doi.org/10.3390/cancers13246192
Mohanty S, Heuser M. Mouse Models of Frequently Mutated Genes in Acute Myeloid Leukemia. Cancers. 2021; 13(24):6192. https://doi.org/10.3390/cancers13246192
Chicago/Turabian StyleMohanty, Sagarajit, and Michael Heuser. 2021. "Mouse Models of Frequently Mutated Genes in Acute Myeloid Leukemia" Cancers 13, no. 24: 6192. https://doi.org/10.3390/cancers13246192
APA StyleMohanty, S., & Heuser, M. (2021). Mouse Models of Frequently Mutated Genes in Acute Myeloid Leukemia. Cancers, 13(24), 6192. https://doi.org/10.3390/cancers13246192