
Locus-Specific Methylation of GSTP1, RNF219, and KIAA1539 Genes with Single Molecule Resolution in Cell-Free DNA from Healthy Donors and Prostate Tumor Patients: Application in Diagnostics

 ,
,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Blood Collection
2.2. DNA Extraction and Characterization
2.3. Bisulfite Conversion
2.4. TaqMan Real-Time PCR
2.5. Amplification of the Selected Loci
2.6. Preparation of the Sequencing Libraries
2.7. NGS Data and Statistical Analysis
- Patients with PCa versus HD and BPH patients;
- HD versus PCa and BPH patients;
- BPH patients versus HD and PCa patients.
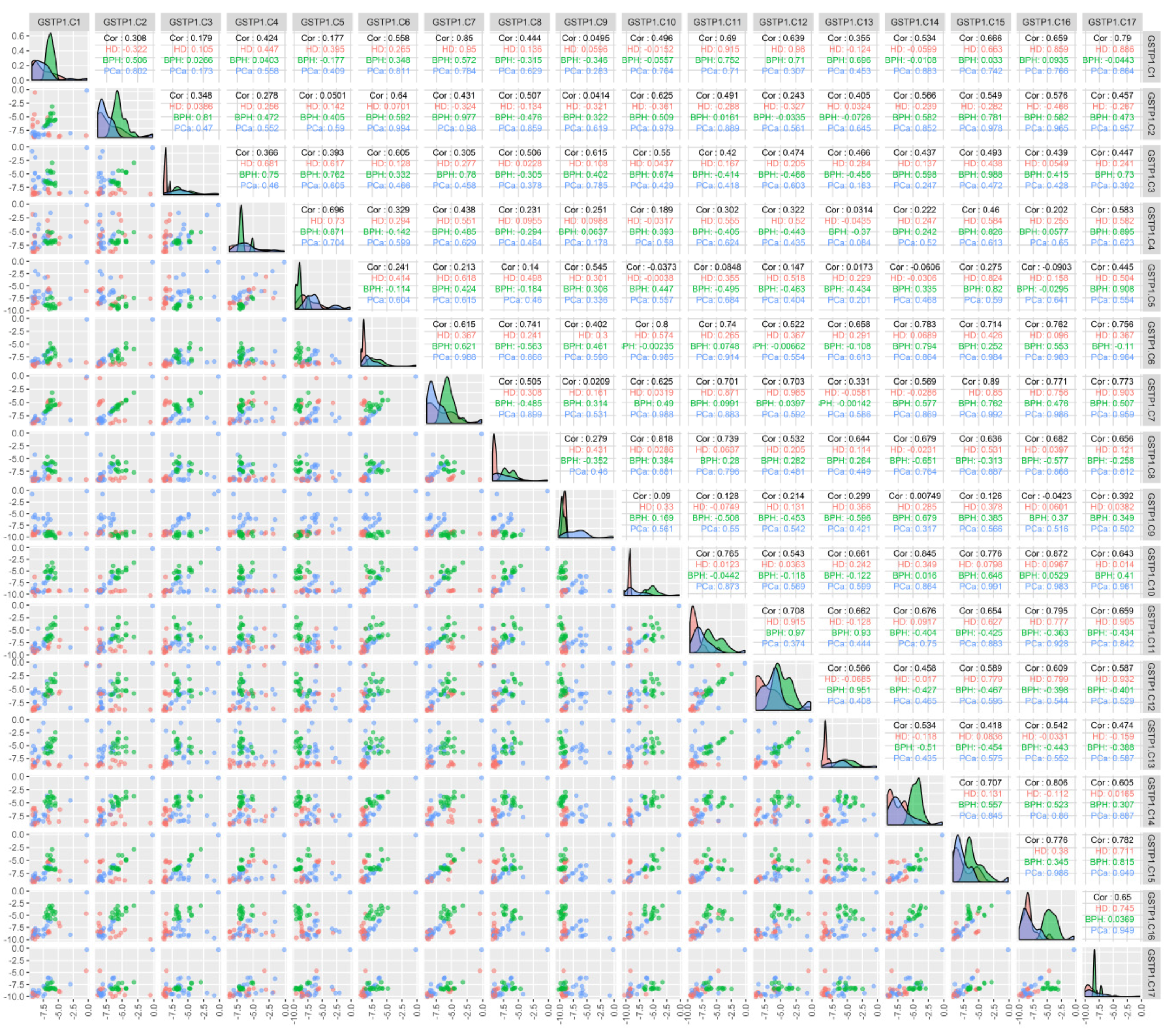
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.; Torre, L.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society. Cancer Facts & Figures 2015; American Cancer Society: Atlanta, GA, USA, 2015. [Google Scholar]
- Hsing, A.; Chokkalingam, A. Prostate cancer epidemiology. Front. Biosci. 2006, 11, 1388–1413. [Google Scholar] [CrossRef] [PubMed]
- Bill-Axelson, A.; Holmberg, L.; Filén, F.; Ruutu, M.; Garmo, H.; Busch, C.; Nordling, S.; Häggman, M.; Andersson, S.; Bratell, S.; et al. Scandinavian Prostate Cancer Group Study Number 4. Radical prostatectomy versus watchful waiting in localized prostate cancer: The Scandinavian prostate cancer group-4 randomized trial. J. Natl. Cancer Inst. 2008, 100, 1144–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.; Lipsitz, R.; Miller, T.; Janakiraman, S.; U.S. Preventive Services Task Force. Benefits and harms of prostate-specific antigen screening for prostate cancer: An evidence update for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2008, 149, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Rosario, D.; Lane, J.; Metcalfe, C.; Donovan, J.; Doble, A.; Goodwin, L.; Davis, M.; Catto, J.; Avery, K.; Neal, D.; et al. Short term outcomes of prostate biopsy in men tested for cancer by prostate specific antigen: Prospective evaluation within Protect study. BMJ 2012, 344, d7894. [Google Scholar] [CrossRef] [Green Version]
- Loeba, S.; Bjurlina, M.; Nicholsonb, J.; Tammelac, T.; Pensond, D.; Cartere, H.; Carrollf, P.; Etzionig, R. Overdiagnosis and Overtreatment of Prostate Cancer. Eur. Urol. 2014, 65, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Moyer, V.; U.S. Preventive Services Task Force. Screening for prostate cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2012, 157, 120–134. [Google Scholar] [CrossRef] [Green Version]
- Hessels, D.; Klein Gunnewiek, J.; van Oort, I.; Karthaus, H.; van Leenders, G.; van Balken, B.; Kiemeney, L.; Witjes, J.; Schalken, J. DD3 (PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur. Urol. 2003, 44, 8–15. [Google Scholar] [CrossRef]
- Filella, X.; Foj, L.; Milà, M.; Augé, J.; Molina, R.; Jiménez, W. PCA3 in the detection and management of early prostate cancer. Tumour Biol. 2013, 34, 1337–1347. [Google Scholar] [CrossRef]
- Haese, A.; de la Taille, A.; van Poppel, H.; Marberger, M.; Stenzl, A.; Mulders, P.; Huland, H.; Abbou, C.; Remzi, M.; Tinzl, M.; et al. Clinical utility of the PCA3 urine assay in European men scheduled for repeat biopsy. Eur. Urol. 2008, 54, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Deras, I.; Aubin, S.; Blasé, A.; Day, J.; Koo, S.; Partin, A.; Ellis, W.; Marks, L.; Fradet, Y.; Rittenhouse, H.; et al. PCA3: A molecular urine assay for predicting prostate biopsy outcome. J. Urol. 2008, 179, 1587–1592. [Google Scholar] [CrossRef]
- Dijkstra, S.; Mulders, P.; Schalken, J. Clinical use of novel urine and blood based prostate cancer biomarkers: A review. Clin. Biochem. 2014, 47, 889–896. [Google Scholar] [CrossRef]
- Lo, Y.; Han, D.; Jiang, P.; Chiu, R. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021, 372, eaaw3616. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Jiang, G.; Woods, C.; Müller, H.; Fiegl, H.; Goebel, G.; Marth, C.; Müller-Holzner, E.; Zeimet, A.; Laird, P.; et al. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004, 64, 4472–4480. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.; Broaddus, R.; Houlihan, P.; Issa, J.; Hamilton, S.; Rashid, A. CpG island methylation in aberrant crypt foci of the colorectum. Am. J. Pathol. 2002, 160, 1823–1830. [Google Scholar] [CrossRef] [Green Version]
- Plass, C.; Pfister, S.; Lindroth, A.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef]
- Shen, S.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.; Chadwick, D.; Zuzarte, P.; Borgida, A.; Wang, T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Giovannucci, E.; Welge, J.; Mallick, P.; Tang, W.; Ho, S. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: A meta-analysis. Br. J. Cancer 2011, 105, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.; Xiong, W.; Bunker, A.; Vaughn, C.; Furtado, L.; Roberts, W.; Fang, J.; Samowitz, W.; Heichman, K. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, B.; Liebenberg, V.; Dietrich, D.; Schlegel, T.; Kneip, C.; Seegebarth, A.; Flemming, N.; Seemann, S.; Distler, J.; Lewin, J.; et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer 2010, 10, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, F.; Schmidt, K.; Choti, M.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jiang, P.; Chan, K.; Wong, J.; Cheng, Y.; Liang, R.; Chan, W.; Ma, E.; Chan, S.; Cheng, S.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [Green Version]
- Korshunova, Y.; Maloney, R.; Lakey, N.; Citek, R.; Bacher, B.; Budiman, A.; Ordway, J.; McCombie, W.; Leon, J.; Jeddeloh, J.; et al. Massively parallel bisulphite pyrosequencing reveals the molecular complexity of breast cancer-associated cytosine-methylation patterns obtained from tissue and serum DNA. Genome Res. 2008, 18, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikeska, T.; Candiloro, I.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Oxnard, G.; Klein, E.; Swanton, C.; Seiden, M.; CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Affinito, O.; Scala, G.; Palumbo, D.; Florio, E.; Monticelli, A.; Miele, G.; Avvedimento, V.; Usiello, A.; Chiariotti, L.; Cocozza, S. Modeling DNA methylation by analyzing the individual configurations of single molecules. Epigenetics 2016, 11, 881–888. [Google Scholar] [CrossRef]
- Ricketts, C.; Linehan, W. Intratumoral heterogeneity in kidney cancer. Nat. Genet. 2014, 46, 214–215. [Google Scholar] [CrossRef] [Green Version]
- Gerashchenko, T.; Denisov, E.; Litviakov, N.; Zavyalova, M.; Vtorushin, S.; Tsyganov, M.; Perelmuter, V.; Cherdyntseva, N. Intratumor heterogeneity: Nature and biological significance. Biochemistry 2013, 78, 1201–1215. [Google Scholar] [CrossRef]
- Cheow, L.; Courtois, E.; Tan, Y.; Viswanathan, R.; Xing, Q.; Tan, R.; Tan, D.; Robson, P.; Loh, Y.; Quake, S.; et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat. Methods 2016, 13, 833–836. [Google Scholar] [CrossRef]
- Sheffield, N.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schönegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef]
- Kerjean, A.; Dupont, J.; Vasseur, C.; Le Tessier, D.; Cuisset, L.; Pàldi, A.; Jouannet, P.; Jeanpierre, M. Establishment of the paternal methylation imprint of the human H19 and MEST/PEG1 genes during spermatogenesis. Hum. Mol. Genet. 2000, 9, 2183–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryzgunova, O.; Laktionov, P.; Skvortsova, T.; Bondar, A.; Morozkin, E.; Lebedeva, A.; Krause, H.; Miller, K.; Vlassov, V. Efficacy of bisulfite modification and recovery of human genomic and circulating DNA using commercial kits. Comput. Biol. Bioinform. 2013, 1, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Bryzgunova, O.; Morozkin, E.; Yarmoschuk, S.; Vlassov, V.; Laktionov, P. Methylation–specific sequencing of GSTP1 gene promoter in circulating/extracellular DNA from blood and urine of healthy donors and prostate cancer patients. Ann. N. Y. Acad. Sci. 2008, 1137, 222–225. [Google Scholar] [CrossRef]
- Lee, J.; Han, J.; Jang, A.; Kim, J.; Hong, S.; Myung, S. DNA Methylation-Mediated Downregulation of DEFB1 in Prostate Cancer Cells. PLoS ONE 2016, 11, e0166664. [Google Scholar]
- Heitzer, E.; Haque, I.; Roberts, C.; Speicher, M. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- van der Vaart, M.; Pretorius, P. Is the role of circulating DNA as a biomarker of cancer being prematurely overrated? Clin. Biochem. 2010, 43, 26–36. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.; Goodman, S.; David, K.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [Green Version]
- Warnecke, P.; Stirzaker, C.; Song, J.; Grunau, C.; Melki, J.; Clark, S. Identification and resolution of artifacts in bisulfite sequencing. Methods 2002, 27, 101–107. [Google Scholar] [CrossRef]
- Tamkovich, S.; Bryzgunova, O. Protease Activity and Cell-Free DNA in Blood Plasma of Healthy Donors and Breast Cancer Patients. J. Immunoass. Immunochem. 2016, 37, 141–153. [Google Scholar] [CrossRef]
- Bryzgunova, O.; Bondar, A.; Morozkin, E.; Mileyko, V.; Vlassov, V.; Laktionov, P. A reliable method to concentrate circulating DNA. Anal. Biochem. 2011, 408, 354–356. [Google Scholar] [CrossRef]
- Caporaso, J.; Lauber, C.; Walters, W.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.; Lawrence, M.; Risbridger, G.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, K.; Masle-Farquhar, E.; Luu, P.; Song, J.; Qu, W.; Zotenko, E.; Gould, C.; Du, Q.; Peters, T.; Colino-Sanguino, Y.; et al. DNA Hypermethylation Encroachment at CpG Island Borders in Cancer Is Predisposed by H3K4 Monomethylation Patterns. Cancer Cell 2019, 35, 297–314.e8. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Yegnasubramanian, S.; De Marzo, A.; Nelson, W. Prostate Cancer Epigenetics: From Basic Mechanisms to Clinical Implications. Cold Spring Harb. Perspect. Med. 2019, 9, a030445. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Jiang, P.; Chan, C.; Sun, K.; Wong, J.; Hui, E.; Chan, S.; Chan, W.; Hui, D.; Ng, S.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef] [Green Version]
- Mohler, J.; Antonarakis, E.; Armstrong, A.; D’Amico, A.; Davis, B.; Dorff, T.; Eastham, J.; Enke, C.; Farrington, T.; Higano, C.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [Green Version]
- Rigau, M.; Olivan, M.; Garcia, M.; Sequeiros, T.; Montes, M.; Colás, E.; Llauradó, M.; Planas, J.; Torres, I.d.; Morote, J.; et al. The Present and Future of Prostate Cancer Urine Biomarkers. Int. J. Mol. Sci. 2013, 14, 12620–12649. [Google Scholar] [CrossRef] [Green Version]
- Batra, J.; Girdhani, S.; Hlatky, L. A Quest to Identify Prostate Cancer Circulating Biomarkers with a Bench-to-Bedside Potential. J. Biomark. 2014, 2014, 321680. [Google Scholar] [CrossRef] [Green Version]
- Martignano, F.; Gurioli, G.; Salvi, S.; Calistri, D.; Costantini, M.; Gunelli, R.; De Giorgi, U.; Foca, F.; Casadio, V. GSTP1 Methylation and Protein Expression in Prostate Cancer: Diagnostic Implications. Dis. Markers 2016, 2016, 4358292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Su, X.; Long, Y.; Zhou, D.; Zhang, X.; Ye, Z.; Ma, J.; Tang, T.; Wang, F.; He, C. A systematic review and quantitative assessment of methylation biomarkers in fecal DNA and colorectal cancer and its precursor, colorectal adenoma. Mutat. Res. 2019, 779, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Cortese, R.; Kwan, A.; Lalonde, E.; Bryzgunova, O.; Bondar, A.; Wu, Y.; Gordevicius, J.; Park, M.; Oh, G.; Kaminsky, Z.; et al. Epigenetic markers of prostate cancer in plasma circulating DNA. Hum. Mol. Genet. 2012, 21, 3619–3631. [Google Scholar] [CrossRef] [Green Version]
- Gurioli, G.; Martignano, F.; Salvi, S.; Costantini, M.; Gunelli, R.; Casadio, V. GSTP1 methylation in cancer: A liquid biopsy biomarker? Clin. Chem. Lab. Med. 2018, 56, 702–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Dhanasekaran, S.; Prensner, J.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep sequencing reveals distinct patterns of DNAmethylation in prostate cancer. Genome Res. 2011, 21, 1028–1041. [Google Scholar] [CrossRef] [Green Version]
- Bryzgunova, O.; Laktionov, P. Generation of Blood Circulating DNAs: Sources, Features of Struction and Circulation. Biochem. Suppl. Ser. B Biomed. Chem. 2014, 8, 203–219. [Google Scholar] [CrossRef]
- Bryzgunova, O.; Zaporozhchenko, I.; Lekchnov, E.; Amelina, E.; Konoshenko, M.; Yarmoschuk, S.; Pashkovskaya, O.; Zheravin, A.; Pak, S.; Rykova, E.; et al. Data analysis algorithm for the development of extracellular miRNA-based diagnostic systems for prostate cancer. PLoS ONE 2019, 14, e0215003. [Google Scholar]
- Kitchen, M.; Bryan, R.; Emes, R.; Luscombe, C.; Cheng, K.; Zeegers, M.; James, N.; Gommersall, L.; Fryer, A. HumanMethylation450K Array-Identified Biomarkers Predict Tumour Recurrence/Progression at Initial Diagnosis of High-risk Non-muscle Invasive Bladder Cancer. Biomark. Cancer 2018, 10, 1179299X17751920. [Google Scholar] [CrossRef] [PubMed]
- Pisanic, T.R.; Athamanolap, P.; Poh, W.; Chen, C.; Hulbert, A.; Brock, M.; Herman, J.; Wang, T. DREAMing: A simple and ultrasensitive method for assessing intratumor epigenetic heterogeneity directly from liquid biopsies. Nucleic Acids Res. 2015, 43, e154. [Google Scholar] [CrossRef] [PubMed]
- Vinci, G.; Buffat, C.; Simoncini, S.; Boubred, F.; Ligi, I.; Dumont, F.; Le Bonniec, B.; Fournier, T.; Vaiman, D.; Dignat-George, F.; et al. Gestational age-related patterns of AMOT methylation are revealed in preterm infant endothelialprogenitors. PLoS ONE 2017, 12, e0186321. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Koh, I.; Choi, N.; Kim, B.; Han, B.; Lee, S. Differential DNA methylation of MSI2 and its correlation with diabetic traits. PLoS ONE 2017, 12, e0177406. [Google Scholar]
- Giarraputo, J.; DeLoach, J.; Padbury, J.; Uzun, A.; Marsit, C.; Hawes, K.; Lester, B. Medical morbidities and DNA methylation of NR3C1 in preterm infants. Pediatr. Res. 2017, 81, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wu, M.; Xiao, H.; Lee, M.; Levin, L.; Leung, Y.; Ho, S. Methylation of a Single Intronic CpG Mediates Expression Silencing of the PMP24 Gene in Prostate Cancer. Prostate 2010, 70, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fackler, M.; Lopez Bujanda, Z.; Umbricht, C.; Teo, W.; Cho, S.; Zhang, Z.; Visvanathan, K.; Jeter, S.; Argani, P.; Wang, C.; et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res. 2014, 74, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Warr, A.; Robert, C.; Hume, D.; Archibald, A.; Deeb, N.; Watson, M. Exome Sequencing: Current and Future Perspectives. G3 (Bethesda) 2015, 5, 1543–1550. [Google Scholar] [CrossRef] [Green Version]
- Adamowicz, M.; Maratou, K.; Aitman, T. Multiplexed DNA Methylation Analysis of Target Regions Using Microfluidics (Fluidigm). Methods Mol. Biol 2018, 1708, 349–363. [Google Scholar]
- How Kit, A.; Nielsen, H.; Tost, J. DNA methylation based biomarkers: Practical considerations and applications. Biochimie 2012, 94, 2314–2337. [Google Scholar] [CrossRef] [PubMed]
- Levenson, V. DNA methylation as a universal biomarker. Expert Rev. Mol. Diagn. 2010, 10, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Koochekpour, S. Genetic and epigenetic changes in human prostate cancer. Iran. Red Crescent Med. J. 2011, 13, 80–98. [Google Scholar]
- Tirado-Magallanes, R.; Rebbani, K.; Lim, R.; Pradhan, S.; Benoukraf, T. Whole genome DNA methylation: Beyond genes silencing. Oncotarget 2017, 8, 5629–5637. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, T.; Bryzgunova, O.; Lebedeva, A.; Mak, V.; Vlassov, V.; Laktionov, P. Methylated Cell-Free DNA In Vitro and In Vivo. In Circulating Nucleic Acids in Plasma and Serum; Springer Science+Business Media B.V.: Dordrecht, The Netherlands, 2011; pp. 185–194. [Google Scholar] [CrossRef]
- Lin, X.; Tascilar, M.; Lee, W.; Vles, W.; Lee, B.; Veeraswamy, R.; Asgari, K.; Freije, D.; van Rees, B.; Gage, W.; et al. GSTP1 CpG island hypermethylation is responsible for the absence of GSTP1 expression in human prostate cancer cells. Am. J. Pathol. 2001, 159, 1815–1826. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Groups | ||
|---|---|---|---|
| Prostate Cancer Patients | Benign Prostatic Hyperplasia Patients | Healthy Donors | |
| (n = 20) | (n = 17) | (n = 18) | |
| Age | |||
| Mean ± SD | 67.6 ± 7.3 | 66.6 ± 7.5 | 61.7 ± 6.6 |
| Range | 55–77 | 54–79 | 53–74 |
| Total PSA (ng/mL) | |||
| Mean ± SD | 17.5 ± 12.3 | 10.7 ± 8.5 | 1.2 ± 0.7 |
| Range | 4.8–48.7 | 0.6–41.5 | 0.2–2.3 |
| Tumor stage | |||
| T2bNxMx | 7 | N/A | N/A |
| T2cNxMx | 6 | ||
| T3aNxMx | 4 | ||
| T3bNxMx | 3 | ||
| Gleason scale | |||
| Unknown | 1 | N/A | N/A |
| 4–5 | 1 | ||
| 5 | 2 | ||
| 5–6 | 3 | ||
| 6 | 7 | ||
| 7 | 4 | ||
| 8 | 2 | ||
| Target’s Name | Primer’s Sequence (without Barcodes) | Primers/Probe Concentration (nM) | Length of PCR Product (bp) | Length of PCR Product without Barcodes (bp) | CG Number | 1× Buffer Composition | PCR Conditions | ||
|---|---|---|---|---|---|---|---|---|---|
| LINE1-For LINE1-Rev LINE1-Probe | 5′-AATGGAAGATGAAATGAATGAAATGA-3′ | 600/ 300 | - | 155 | - | BioMaster qPCR Mix from Biolabmix (Novosibirsk, Russia) | 95 °C for 3 min, (95 °C for 15 s, and 60 °C for 60 s) ×40. | ||
| 5′-TTCCATTCTCCCCATCACTTTCA-3′ | |||||||||
| 5′-FAM-GAGAAGGGAAGTTTAGAGAAAAAAGAAT-FQ-3′ | |||||||||
| RNF219-For RNF219-Rev | 5′-(Y1-12)GTGATTGTGGGTATAGTTATAAAA-3′ | 600 | 177 | 161 | 17 | Hotstart PCR buffer with additional MgCl2 (final concentration: 5 mM), 1 mM dNTPs, and 0.65 units of Hotstart Taq polymerase | 95 °C for 15 min (95 °C for 60 s, 58 °C for 45 s, and 72 °C for 60 s) ×50 | ||
| 5′-(X1-8)ACTACCCCCATCTCCCAAAA-3′ | |||||||||
| KIAA1539-For KIAA1539-Rev | 5′-(X1-8)AGGAAGGAGGAGATAAAGTGAT-3′ | 600 | 105 | 89 | 5 | ||||
| 5′-(Y1-12)CCCCTCTAAACTTATCATCACA-3′ | |||||||||
| GSTP1-For GSTP1-Rev | 5′-(Y1-12)ATTTGGGAAAGAGGGAAAGGTT-3′ | 600 | 158 | 142 | 17 | ||||
| 5′-(X1-8)CTCTTCTAAAAAATCC-3′ | |||||||||
| Barcodes | Forward or Reverse Primer the Exact Barcode Used for Each Target | ||
|---|---|---|---|
| RNF219 | KIAA1539 | GSTP1 | |
| X1: TAGATCGC, X2: CTCTCTAT, X3: TATCCTCT, X4: AGAGTAGA, X5: ACTGCATA, X6: AAGGAGTA, X7: CTAAGCCT, X8: CCTCTCTG | Reverse | Forward | Reverse |
| Y1: TCGCCTTA, Y2: CTAGTACG, Y3: TTCTGCCT, Y4: GCTCAGGA, Y5: AGGAGTCC, Y6: CATGCCTA, Y7: GTAGAGAG, Y8: CCTCTCTG, Y9: AGCGTAGC, Y10: CAGCCTCG, Y11: TGCCTCTT, Y12: TCCTCTAC | Forward | Reverse | Forward |
| Gene, Position, and Status (C or T after Conversion) | p-Value × 3 × 1167 | Means (%) | Sensitivity for a 100% Specificity (%) | Specificity for a 100% Sensitivity (%) | Cutoff (%) | CV Accuracy (%) | CV Sensitivity (%) | CV Specificity (%) | CV AUC (% (DeLong’s CI) |
|---|---|---|---|---|---|---|---|---|---|
| PSA | 0.016 | 10.0 | 55.6 | 75.0 | 65.0 | 80.6 | 83.9 (73.5, 94.2) | ||
| GSTP1.C9 + RNF219.C2 + GSTP1.C2 | 100 | 100 | 100 | 100 | 100 | 100 | |||
| GSTP1.C9 + RNF219.C2 + GSTP1.C16 | 100 | 100 | 100 | 100 | 100 | 100 | |||
| GSTP1.C9 + RNF219.C2.T10 | 95.0 | 97.1 | 94.5 | 90.0 | 97.1 | 92.4 (83.2, 100) | |||
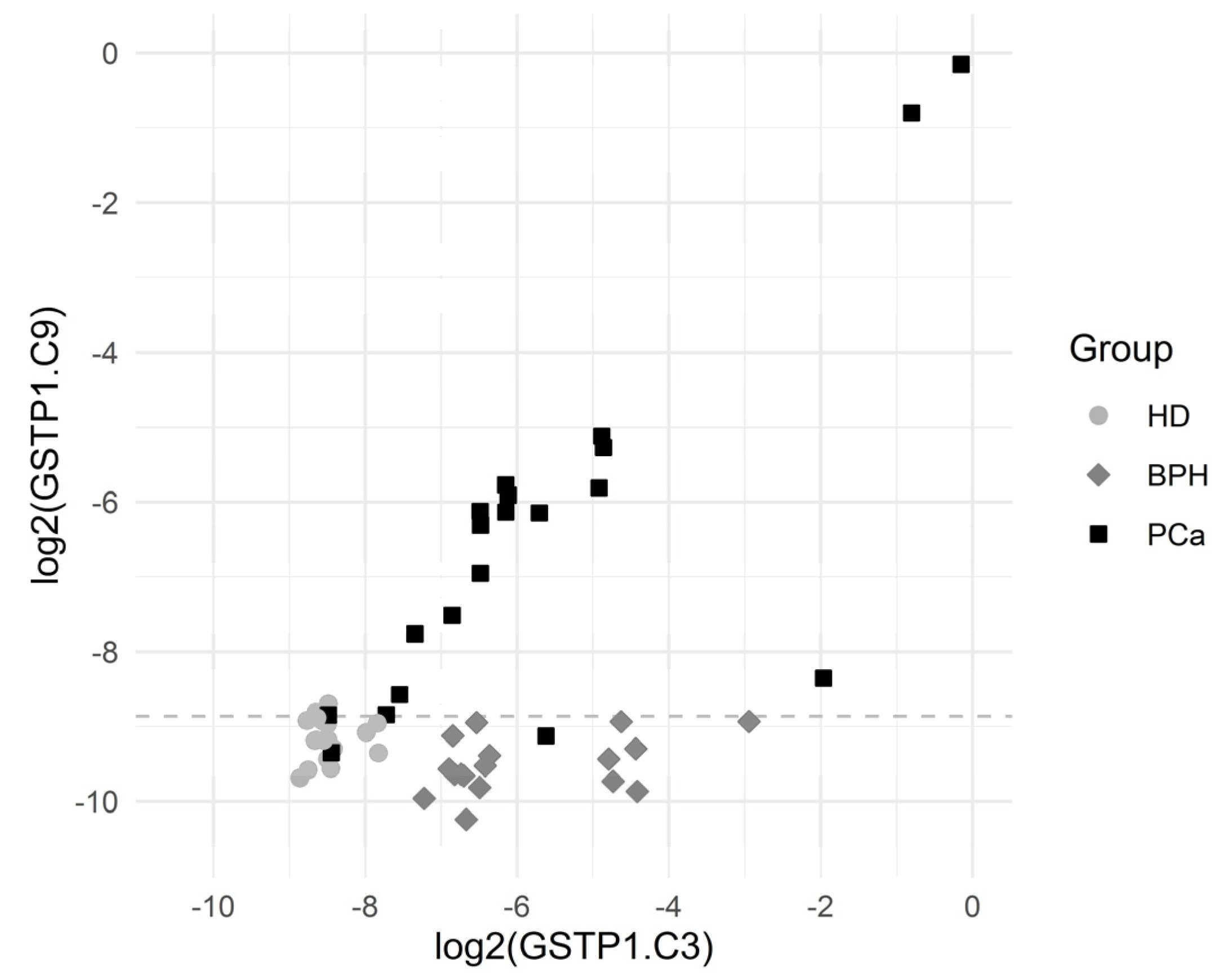
| GSTP1.C3.C9 | 0.00000073 | 8.03/0.0394 | 80.0 | 54.3 | 0.101 | 87.3 | 80.0 | 91.4 | 93.0 (85.1, 100) |
| GSTP1.C9 | 0.00000090 | 8.35/0.160 | 80.0 | 48.6 | 0.215 | 90.9 | 80.0 | 97.1 | 92.6 (83.2, 100) |
| GSTP1.C9.T17 | 0.0000011 | 4.04/0.143 | 80.0 | 37.1 | 0.198 | 89.1 | 80.0 | 94.3 | 93.1 (83.5, 100) |
| GSTP1.T2.C9 | 0.0000011 | 3.71/0.142 | 75.0 | 45.7 | 0.177 | 87.3 | 80.0 | 91.4 | 93.0 (84.7, 100) |
| GSTP1.T1.C9 | 0.0000024 | 3.94/0.142 | 75.0 | 34.3 | 0.199 | 89.1 | 80.0 | 94.3 | 92.1 (82.2, 100) |
| GSTP1.T6.C9 | 0.0000024 | 3.71/0.142 | 70.0 | 51.4 | 0.177 | 87.3 | 80.0 | 91.4 | 92.6 (84.4, 100) |
| GSTP1.T4.C9 | 0.0000029 | 3.71/0.142 | 75.0 | 51.4 | 0.179 | 87.3 | 80.0 | 91.4 | 92.1 (83.1, 100) |
| GSTP1.C9.T16 | 0.0000050 | 4.01/0.140 | 70.0 | 48.6 | 0.190 | 89.1 | 85.0 | 91.4 | 92.0 (83.4, 100) |
| GSTP1.C9.T14 | 0.0000070 | 3.90/0.138 | 70.0 | 34.3 | 0.190 | 87.3 | 85.0 | 88.6 | 91.9 (82.3, 100) |
| GSTP1.T5.C9 | 0.0000084 | 3.67/0.143 | 75.0 | 42.9 | 0.188 | 87.3 | 80.0 | 91.4 | 91.7 (82.9, 100) |
| GSTP1.C9.C13 | 0.0044 | 4.76/0.0197 | 70.0 | 40.0 | 0.0512 | 85.5 | 70.0 | 94.3 | 85.0 (73.0, 97.0) |
| Option Number | Gene, Position, and Status (C or T after Conversion) | p-Calue × 3 × 1167 | Means (%) | Sensitivity for a 100% Specificity (%) | Specificity for a 100% Sensitivity (%) | Cutoff (%; Ratio) | CV Accuracy (%) | CV Sensitivity (%) | CV Specificity (%) | CV AUC (%; DeLong’s CI) |
|---|---|---|---|---|---|---|---|---|---|---|
| PSA | >1 | 0 | 12.8 | 55.4 | 23.5 | 69.2 | 54.9 (38.8, 71.0) | |||
| 1 | RNF219.C1.C2 | 0.000000000071 | 0.00500/0.133 | 100 | 100 | 0.0526 (1.03) | 100 | 100 | 100 | 100 |
| 2 | RNF219.C2.C4 | 0.000000000071 | 0.0101/0.206 | 100 | 100 | 0.0567 (1.20) | 100 | 100 | 100 | 100 |
| 3 | RNF219.C1.C5 | 0.000000000071 | 0.00986/0.136 | 100 | 100 | 0.0526 (1.03) | 100 | 100 | 100 | 100 |
| 4 | RNF219.C2.C5 | 0.000000000071 | 0.00528/0.160 | 100 | 100 | 0.0481 (1.24) | 100 | 100 | 100 | 100 |
| 5 | RNF219.C4.C5 | 0.000000000071 | 0.0199/0.144 | 100 | 100 | 0.0561 (1.31) | 100 | 100 | 100 | 100 |
| 76 | RNF219.C12.C17 | 0.000000000071 | 0.00464/0.135 | 100 | 100 | 0.0508 (1.38) | 100 | 100 | 100 | 100 |
| 77 | RNF219.C13.C17 | 0.000000000071 | 0.00650/0.139 | 100 | 100 | 0.0557 (1.15) | 100 | 100 | 100 | 100 |
| 78 | GSTP1.T7.C16 | 0.00000000010 | 4.10/0.26 | 100 | 100 | 1.32 (1.20) | 100 | 100 | 100 | 100 |
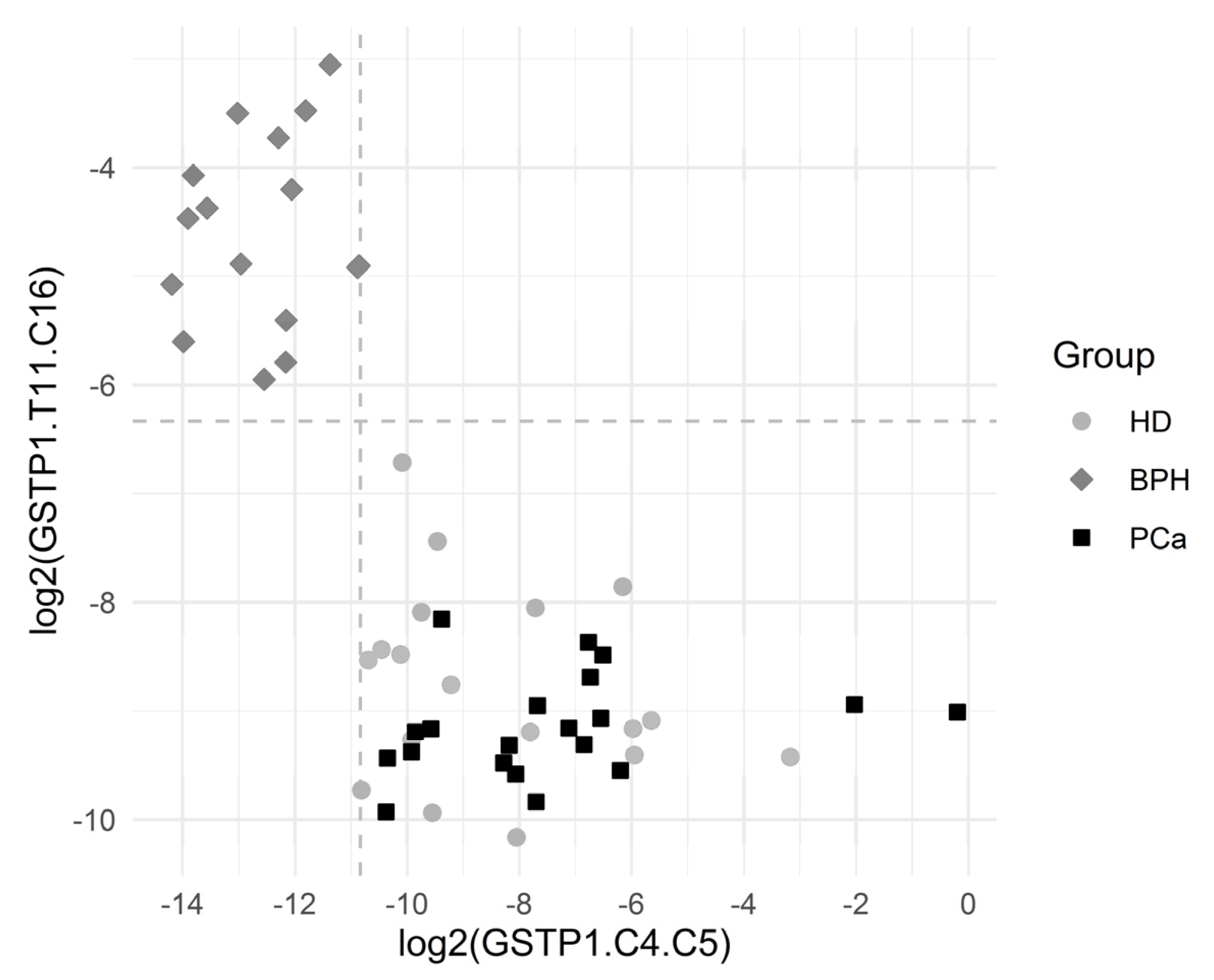
| 79 | GSTP1.T11.C16 | 0.00000000010 | 5.33/0.231 | 100 | 100 | 1.24 (1.70) | 100 | 100 | 100 | 100 |
| 80 | GSTP1.C4.C5 | 0.00000000010 | 0.0197/3.72 | 100 | 100 | 0.0548 (1.03) | 100 | 100 | 100 | 100 |
| Gene, Position, and Status (C or T after Conversion) | p-Value × 3 × 1167 | Means (%) | Sensitivity for a 100% Specificity (%) | Specificity for a 100% Sensitivity (%) | Cutoff (%; Ratio) | CV Accuracy (%) | CV Sensitivity (%) | CV Specificity (%) | CV AUC (%; DeLong’s CI) |
|---|---|---|---|---|---|---|---|---|---|
| PSA | 0.0000000076 | 26.3 | 97.3 | 98.2 | 100 | 97.3 | 97.3 (92.0, 100) | ||
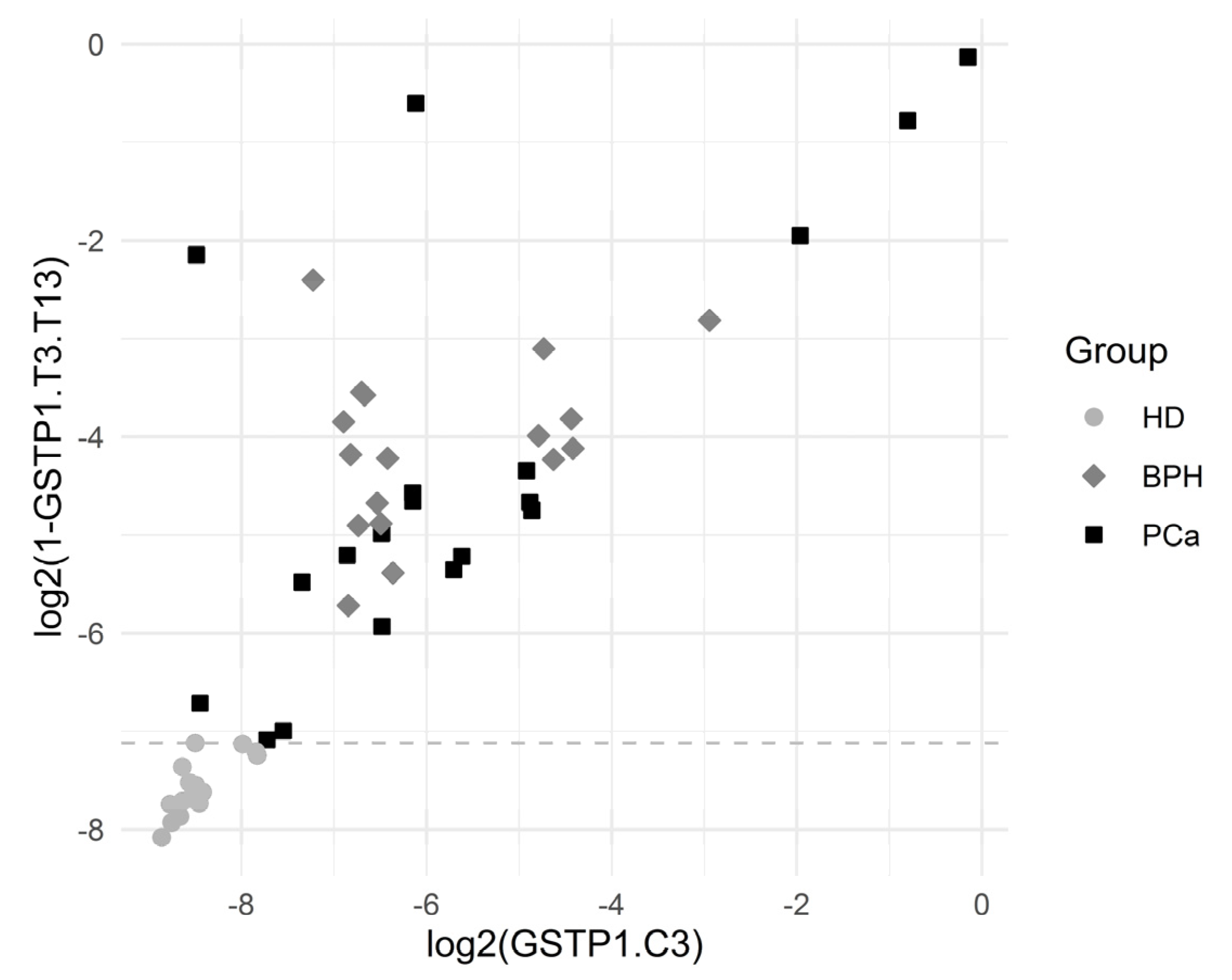
| GSTP1.T3.T13 | 0.000000000049 | 99.5/88.5 | 100 | 100 | 99.3 (1.00) | 100 | 100 | 100 | 100 |
| GSTP1.T8.T13 | 0.00000000019 | 99.6/91.7 | 88.9 | 97.3 | 99.4 | 96.4 | 94.4 | 97.3 | 94.1 (86.1, 100) |
| GSTP1.T9.T13 | 0.0000000068 | 99.6/90.5 | 77.8 | 91.9 | 99.3 | 92.7 | 94.4 | 91.9 | 97.6 (94.3, 100) |
| GSTP1.C13 | 0.000000018 | 0.260/7.48 | 77.8 | 89.2 | 0.506 | 90.9 | 94.4 | 89.2 | 96.8 (93.2, 100) |
| GSTP1.T6.T13 | 0.000000018 | 99.5/91.9 | 77.8 | 89.2 | 99.2 | 90.9 | 94.4 | 89.2 | 97.7 (94.8, 100) |
| GSTP1.T1.C13 | 0.000000025 | 0.238/5.09 | 77.8 | 89.2 | 0.497 | 90.9 | 94.4 | 89.2 | 96.4 (92.4, 100) |
| GSTP1.T1.C6 | 0.000000078 | 0.188/.780 | 77.8 | 64.9 | 0.242 | 89.1 | 88.9 | 89.2 | 95.5 (89.8, 100) |
| GSTP1.C13.T17 | 0.000000078 | 0.241/5.15 | 77.8 | 83.8 | 0.496 | 87.3 | 94.4 | 83.8 | 96.7 (92.7, 100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryzgunova, O.; Bondar, A.; Ruzankin, P.; Laktionov, P.; Tarasenko, A.; Kurilshikov, A.; Epifanov, R.; Zaripov, M.; Kabilov, M.; Laktionov, P. Locus-Specific Methylation of GSTP1, RNF219, and KIAA1539 Genes with Single Molecule Resolution in Cell-Free DNA from Healthy Donors and Prostate Tumor Patients: Application in Diagnostics. Cancers 2021, 13, 6234. https://doi.org/10.3390/cancers13246234
Bryzgunova O, Bondar A, Ruzankin P, Laktionov P, Tarasenko A, Kurilshikov A, Epifanov R, Zaripov M, Kabilov M, Laktionov P. Locus-Specific Methylation of GSTP1, RNF219, and KIAA1539 Genes with Single Molecule Resolution in Cell-Free DNA from Healthy Donors and Prostate Tumor Patients: Application in Diagnostics. Cancers. 2021; 13(24):6234. https://doi.org/10.3390/cancers13246234
Chicago/Turabian StyleBryzgunova, Olga, Anna Bondar, Pavel Ruzankin, Petr Laktionov, Anton Tarasenko, Alexander Kurilshikov, Rostislav Epifanov, Marat Zaripov, Marsel Kabilov, and Pavel Laktionov. 2021. "Locus-Specific Methylation of GSTP1, RNF219, and KIAA1539 Genes with Single Molecule Resolution in Cell-Free DNA from Healthy Donors and Prostate Tumor Patients: Application in Diagnostics" Cancers 13, no. 24: 6234. https://doi.org/10.3390/cancers13246234
APA StyleBryzgunova, O., Bondar, A., Ruzankin, P., Laktionov, P., Tarasenko, A., Kurilshikov, A., Epifanov, R., Zaripov, M., Kabilov, M., & Laktionov, P. (2021). Locus-Specific Methylation of GSTP1, RNF219, and KIAA1539 Genes with Single Molecule Resolution in Cell-Free DNA from Healthy Donors and Prostate Tumor Patients: Application in Diagnostics. Cancers, 13(24), 6234. https://doi.org/10.3390/cancers13246234