
YB1 Is a Major Contributor to Health Disparities in Triple Negative Breast Cancer

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Lines and Reagents
2.3. Antibodies and Reagents
2.4. Immunoblotting
2.5. RNA Extraction and Quantitative Real Time Reverse Transcription PCR (qt-RT-PCR)
2.6. Colony Formation Assay
2.7. Generation of Cisplatin- and Doxorubicin-Resistant TNBC Cells
2.8. sgRNA Preparation and Electroporation for Generating eYb1KO Cell Lines
2.9. Generation of Yb-1 Knockout Cell Lines Using Electroporation
2.10. Animal Experiments
2.11. Human TNBC Specimens
2.12. Analyses of RNA-Seq Data from the TCGA Datasets
2.13. Bioinformatic Methods for Datamining of TCGA PanCancer Dataset
2.14. Other Statistical Analyses
3. Results
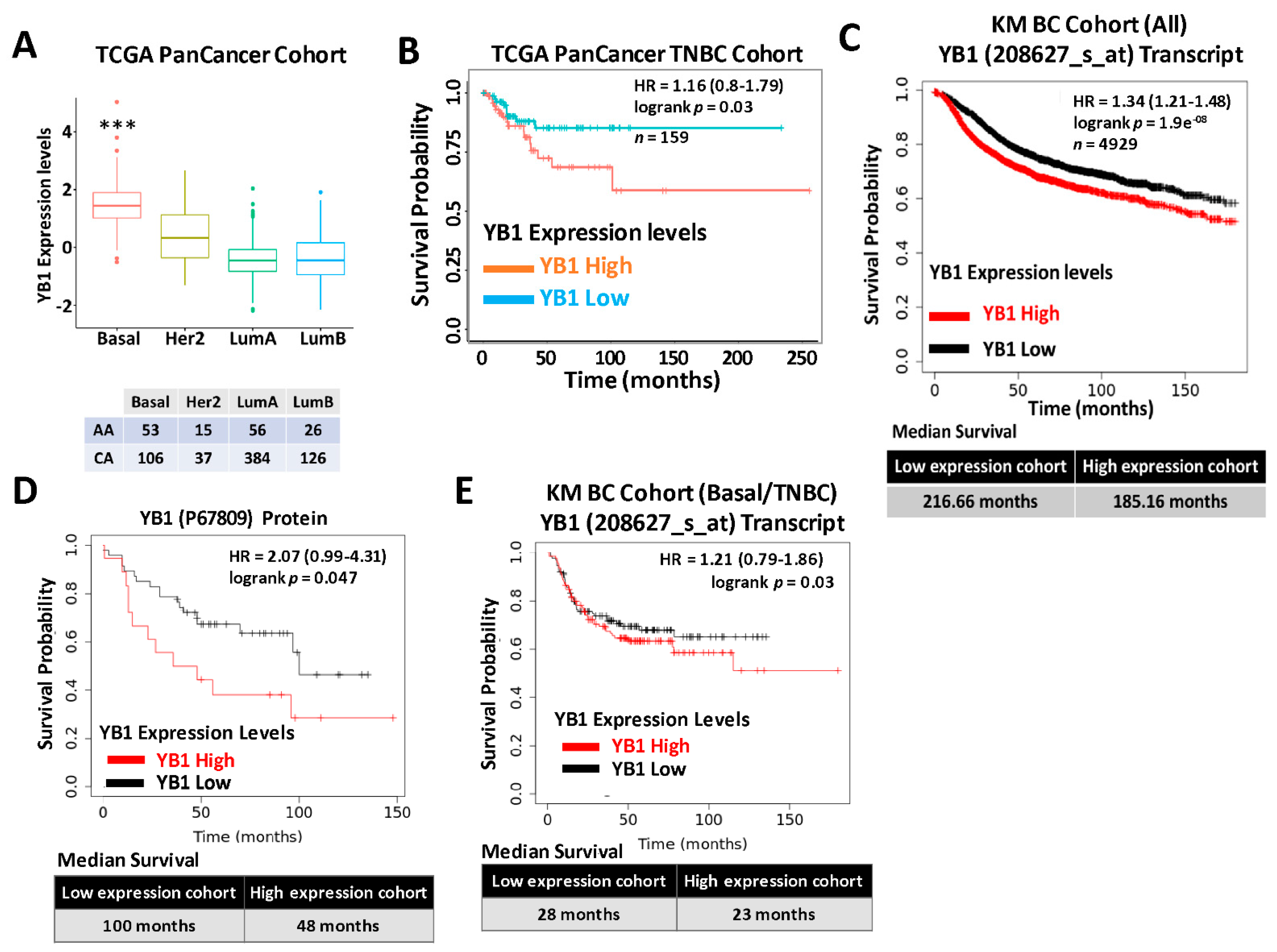
3.1. YB1 Expression Levels Are Increased in TNBC Cell Lines and Tumors and Are Associated with Poor Survival and Worst Patients’ Outcomes
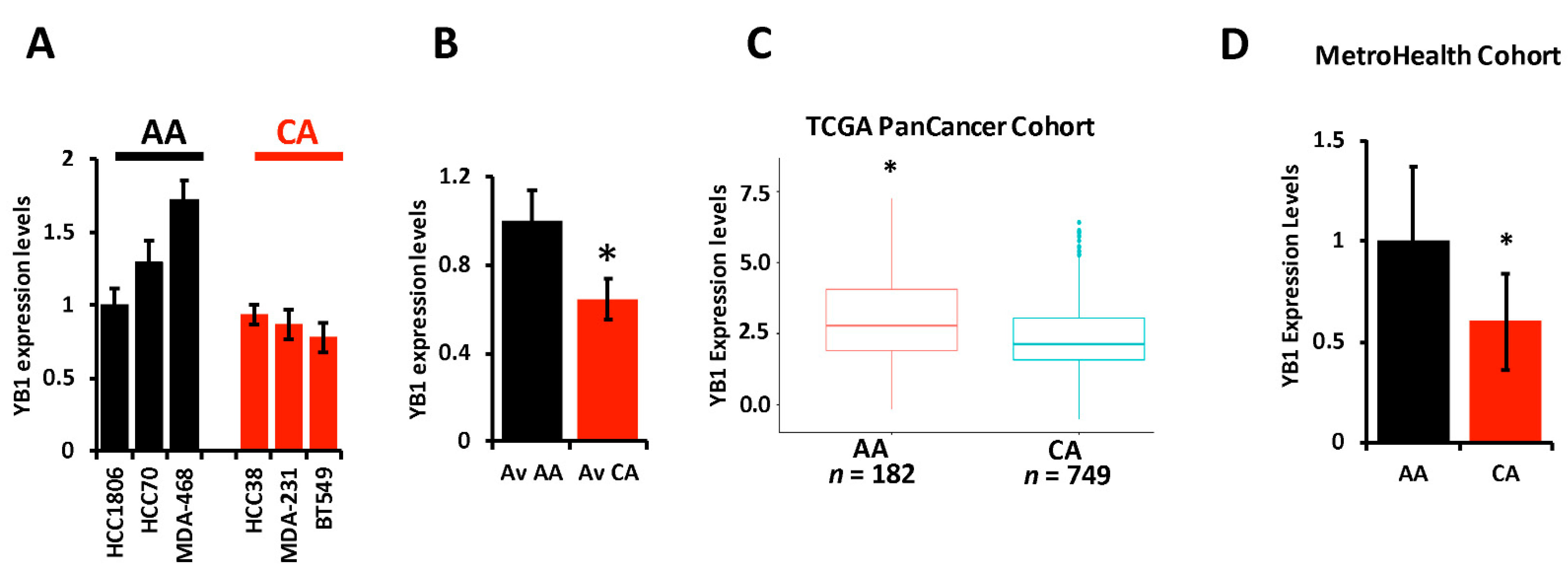
3.2. YB1 Is Disparately Highly Expressed in AA vs. CA TNBC Cell Lines and Tumors and Correlates with Poor Outcome in AA TNBC
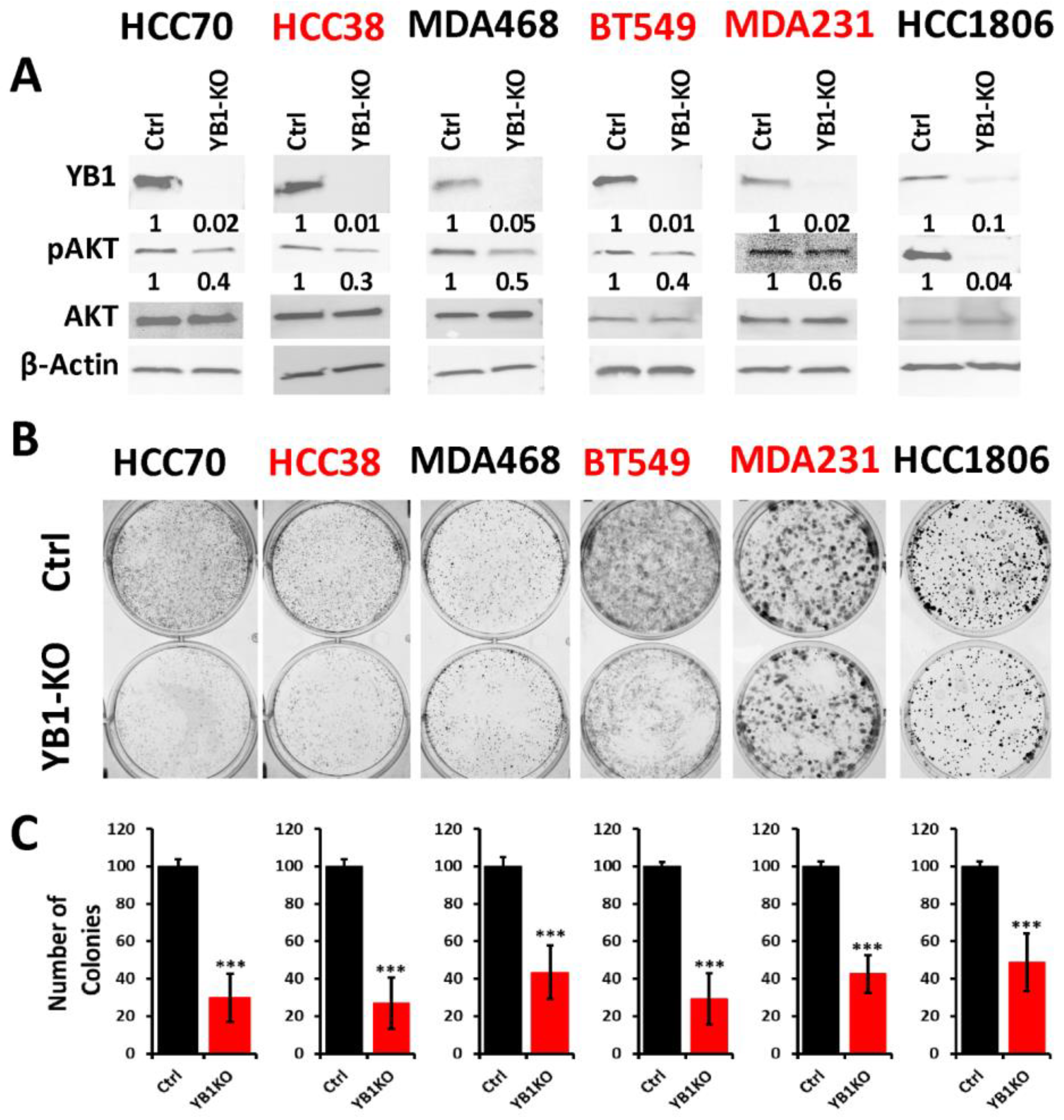
3.3. Loss of YB1 Inhibits the Oncogenic Behavior of AA TNBC Cell Lines In Vitro
3.4. Loss of YB1 Inhibits Tumor Growth and Metastasis of AA TNBC Cell Lines In Vivo
3.5. YB1 Is Associated with the Cancer Stem Cell Phenotype in AA TNBC
3.6. YB1 Is Associated with Chemoresistance in AA TNBC Both In Vitro and In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, J.; Gray, W.H.; Lehmann, B.D.; Bauer, J.A.; Shyr, Y.; Pietenpol, J.A. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inform. 2012, 11, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, T.J.; Carey, L.A. Gene-expression analysis and the basal-like breast cancer subtype. Future Oncol. 2007, 3, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.P.; Winer, E.P.; Foulkes, W.D.; Garber, J.; Perou, C.M.; Richardson, A.; Sledge, G.W.; Carey, L.A. Triple-negative breast cancer: Risk factors to potential targets. Clin. Cancer Res. 2008, 14, 8010–8018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Dawood, S.; Broglio, K.; Kau, S.W.; Green, M.C.; Giordano, S.H.; Meric-Bernstam, F.; Buchholz, T.A.; Albarracin, C.; Yang, W.T.; Hennessy, B.T.; et al. Triple receptor-negative breast cancer: The effect of race on response to primary systemic treatment and survival outcomes. J. Clin. Oncol. 2009, 27, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albain, K.S.; Unger, J.M.; Crowley, J.J.; Coltman, C.A., Jr.; Hershman, D.L. Racial disparities in cancer survival among randomized clinical trials patients of the Southwest Oncology Group. J. Natl. Cancer Inst. 2009, 101, 984–992. [Google Scholar] [CrossRef] [Green Version]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkrekshi, A.; Wang, W.; Rana, P.S.; Markovic, V.; Sossey-Alaoui, K. A comprehensive review of the functions of YB-1 in cancer stemness, metastasis and drug resistance. Cell Signal. 2021, 85, 110073. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Iida, M.; Kosnopfel, C.; Abbariki, M.; Menegakis, A.; Fehrenbacher, B.; Maier, J.; Schaller, M.; Brucker, S.Y.; Wheeler, D.L.; et al. Blocking Y-Box Binding Protein-1 through Simultaneous Targeting of PI3K and MAPK in Triple Negative Breast Cancers. Cancers 2020, 12, 2795. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.; Zhao, K.; Lin, Q.; Li, H.; Xue, X.; Ge, W.; He, H.; Liu, D.; Xie, H.; et al. A novel LncRNA HITT forms a regulatory loop with HIF-1alpha to modulate angiogenesis and tumor growth. Cell Death Differ. 2020, 27, 1431–1446. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Kosnopfel, C.; Sinnberg, T.; Sauer, B.; Niessner, H.; Schmitt, A.; Makino, E.; Forschner, A.; Hailfinger, S.; Garbe, C.; Schittek, B. Human melanoma cells resistant to MAPK inhibitors can be effectively targeted by inhibition of the p90 ribosomal S6 kinase. Oncotarget 2017, 8, 35761–35775. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.L.; Fu, X.; Huang, J.; Jia, T.T.; Zong, F.Y.; Mu, S.R.; Zhu, H.; Yan, Y.; Qiu, S.; Wu, Q.; et al. Genome-wide analysis of YB-1-RNA interactions reveals a novel role of YB-1 in miRNA processing in glioblastoma multiforme. Nucleic Acids Res. 2015, 43, 8516–8528. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Tetteh, P.W.; Merz, P.R.; Dickes, E.; Abukiwan, A.; Hotz-Wagenblatt, A.; Holland-Cunz, S.; Sinnberg, T.; Schittek, B.; Schadendorf, D.; et al. miR-137 inhibits the invasion of melanoma cells through downregulation of multiple oncogenic target genes. J. Invest. Derm. 2013, 133, 768–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evdokimova, V.; Tognon, C.; Ng, T.; Ruzanov, P.; Melnyk, N.; Fink, D.; Sorokin, A.; Ovchinnikov, L.P.; Davicioni, E.; Triche, T.J.; et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 2009, 15, 402–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratford, A.L.; Fry, C.J.; Desilets, C.; Davies, A.H.; Cho, Y.Y.; Li, Y.; Dong, Z.; Berquin, I.M.; Roux, P.P.; Dunn, S.E. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evdokimova, V.; Ruzanov, P.; Anglesio, M.S.; Sorokin, A.V.; Ovchinnikov, L.P.; Buckley, J.; Triche, T.J.; Sonenberg, N.; Sorensen, P.H. Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol. Cell. Biol. 2006, 26, 277–292. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.W.; Kucab, J.; Wu, J.; Lee, C.; Cheang, M.C.; Yorida, E.; Turbin, D.; Dedhar, S.; Nelson, C.; Pollak, M.; et al. Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene 2005, 24, 4281–4292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evdokimova, V.; Ruzanov, P.; Imataka, H.; Raught, B.; Svitkin, Y.; Ovchinnikov, L.P.; Sonenberg, N. The major mRNA-associated protein YB-1 is a potent 5’ cap-dependent mRNA stabilizer. EMBO J. 2001, 20, 5491–5502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceman, S.; Nelson, R.; Warren, S.T. Identification of mouse YB1/p50 as a component of the FMRP-associated mRNP particle. Biochem. Biophys. Res. Commun. 2000, 279, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargou, R.C.; Jurchott, K.; Wagener, C.; Bergmann, S.; Metzner, S.; Bommert, K.; Mapara, M.Y.; Winzer, K.J.; Dietel, M.; Dorken, B.; et al. Nuclear localization and increased levels of transcription factor YB-1 in primary human breast cancers are associated with intrinsic MDR1 gene expression. Nat. Med. 1997, 3, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Royer-Pokora, B.; Fietze, E.; Jurchott, K.; Hildebrandt, B.; Trost, D.; Leenders, F.; Claude, J.C.; Theuring, F.; Bargou, R.; et al. YB-1 provokes breast cancer through the induction of chromosomal instability that emerges from mitotic failure and centrosome amplification. Cancer Res. 2005, 65, 4078–4087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bledzka, K.; Schiemann, B.; Schiemann, W.P.; Fox, P.; Plow, E.F.; Sossey-Alaoui, K. The WAVE3-YB1 interaction regulates cancer stem cells activity in breast cancer. Oncotarget 2017, 8, 104072–104089. [Google Scholar] [CrossRef]
- Darb-Esfahani, S.; Loibl, S.; Muller, B.M.; Roller, M.; Denkert, C.; Komor, M.; Schluns, K.; Blohmer, J.U.; Budczies, J.; Gerber, B.; et al. Identification of biology-based breast cancer types with distinct predictive and prognostic features: Role of steroid hormone and HER2 receptor expression in patients treated with neoadjuvant anthracycline/taxane-based chemotherapy. Breast Cancer Res. 2009, 11, R69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, J.; Astanehe, A.; Lee, C.; Fotovati, A.; Hu, K.; Dunn, S.E. The expression of activated Y-box binding protein-1 serine 102 mediates trastuzumab resistance in breast cancer cells by increasing CD44+ cells. Oncogene 2010, 29, 6294–6300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Seki, N.; Namoto-Matsubayashi, R.; Takahashi, H.; Inoue, Y.; Toh, U.; Kage, M.; Shirouzu, K. YB-1 prevents apoptosis via the mTOR/STAT3 pathway in HER-2-overexpressing breast cancer cells. Future Oncol. 2009, 5, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Ito, K.; Izumi, H.; Kimura, M.; Sano, M.; Nakagomi, H.; Maeno, K.; Hama, Y.; Shingu, K.; Tsuchiya, S.; et al. Increased nuclear localization of transcription factor Y-box binding protein 1 accompanied by up-regulation of P-glycoprotein in breast cancer pretreated with paclitaxel. Clin. Cancer Res. 2005, 11, 8837–8844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guay, D.; Evoy, A.A.; Paquet, E.; Garand, C.; Bachvarova, M.; Bachvarov, D.; Lebel, M. The strand separation and nuclease activities associated with YB-1 are dispensable for cisplatin resistance but overexpression of YB-1 in MCF7 and MDA-MB-231 breast tumor cells generates several chemoresistance signatures. Int. J. Biochem. Cell Biol. 2008, 40, 2492–2507. [Google Scholar] [CrossRef] [PubMed]
- Habibi, G.; Leung, S.; Law, J.H.; Gelmon, K.; Masoudi, H.; Turbin, D.; Pollak, M.; Nielsen, T.O.; Huntsman, D.; Dunn, S.E. Redefining prognostic factors for breast cancer: YB-1 is a stronger predictor of relapse and disease-specific survival than estrogen receptor or HER-2 across all tumor subtypes. Breast Cancer Res. 2008, 10, R86. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Kamijo, S.; Izumi, H.; Kohno, K.; Amano, J.; Ito, K. Alteration of Y-box binding protein-1 expression modifies the response to endocrine therapy in estrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2012, 133, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Janz, M.; Harbeck, N.; Dettmar, P.; Berger, U.; Schmidt, A.; Jurchott, K.; Schmitt, M.; Royer, H.D. Y-box factor YB-1 predicts drug resistance and patient outcome in breast cancer independent of clinically relevant tumor biologic factors HER2, uPA and PAI-1. Int. J. Cancer 2002, 97, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Dhillon, J.; Wang, M.Y.; Gao, Y.; Hu, K.; Park, E.; Astanehe, A.; Hung, M.C.; Eirew, P.; Eaves, C.J.; et al. Targeting YB-1 in HER-2 overexpressing breast cancer cells induces apoptosis via the mTOR/STAT3 pathway and suppresses tumor growth in mice. Cancer Res. 2008, 68, 8661–8666. [Google Scholar] [CrossRef] [Green Version]
- Lettau, K.; Zips, D.; Toulany, M. Simultaneous Targeting of RSK and AKT Efficiently Inhibits YB-1-Mediated Repair of Ionizing Radiation-Induced DNA Double-Strand Breaks in Breast Cancer Cells. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Nair, S.; Shyamasundar, S.; Chua, P.J.; Muniasamy, U.; Matsumoto, K.; Gunaratne, J.; Bay, B.H. Silencing Y-box binding protein-1 inhibits triple-negative breast cancer cell invasiveness via regulation of MMP1 and beta-catenin expression. Cancer Lett. 2019, 452, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Shyamasundar, S.; Gunaratne, J.; Scully, O.J.; Matsumoto, K.; Bay, B.H. YBX1 gene silencing inhibits migratory and invasive potential via CORO1C in breast cancer in vitro. BMC Cancer 2017, 17, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stope, M.B.; Weiss, M.; Koensgen, D.; Popp, S.L.; Joffroy, C.; Mustea, A.; Buck, M.B.; Knabbe, C. Y-box Binding Protein-1 Enhances Oncogenic Transforming Growth Factor beta Signaling in Breast Cancer Cells via Triggering Phospho-Activation of Smad2. Anticancer Res. 2017, 37, 6745–6748. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, S.; He, S.; Huo, Q.; Hu, Y.; Xie, N. YB-1 interplays with ERalpha to regulate the stemness and differentiation of ER-positive breast cancer stem cells. Theranostics 2020, 10, 3816–3832. [Google Scholar] [CrossRef] [PubMed]
- Davuluri, G.; Augoff, K.; Schiemann, W.P.; Plow, E.F.; Sossey-Alaoui, K. WAVE3-NFkappaB interplay is essential for the survival and invasion of cancer cells. PLoS ONE 2014, 9, e110627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davuluri, G.; Schiemann, W.P.; Plow, E.F.; Sossey-Alaoui, K. Loss of WAVE3 sensitizes triple-negative breast cancers to chemotherapeutics by inhibiting the STAT-HIF-1alpha-mediated angiogenesis. JAKSTAT 2014, 3, e1009276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sossey-Alaoui, K.; Safina, A.; Li, X.; Vaughan, M.M.; Hicks, D.G.; Bakin, A.V.; Cowell, J.K. Down-regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am. J. Pathol. 2007, 170, 2112–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansakar, U.; Wang, W.; Markovic, V.; Sossey-Alaoui, K. Phosphorylation of the proline-rich domain of WAVE3 drives its oncogenic activity in breast cancer. Sci. Rep. 2021, 11, 3868. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Zhu, J.Y.; Fang, D.; Ding, H.F.; Dong, Z.; Jing, Q.; Su, S.B.; Huang, S. Triple negative breast cancer development can be selectively suppressed by sustaining an elevated level of cellular cyclic AMP through simultaneously blocking its efflux and decomposition. Oncotarget 2016, 7, 87232–87245. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santner, S.J.; Dawson, P.J.; Tait, L.; Soule, H.D.; Eliason, J.; Mohamed, A.N.; Wolman, S.R.; Heppner, G.H.; Miller, F.R. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res. Treat. 2001, 65, 101–110. [Google Scholar] [CrossRef]
- Aslakson, C.J.; Miller, F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52, 1399–1405. [Google Scholar]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-beta-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendt, M.K.; Taylor, M.A.; Schiemann, B.J.; Schiemann, W.P. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Mol. Biol. Cell 2011, 22, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Felts, K.A.; Jiang, N.; Chang, H.W.; Vogt, P.K. Y box-binding protein 1 induces resistance to oncogenic transformation by the phosphatidylinositol 3-kinase pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 12384–12389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honma, K.; Iwao-Koizumi, K.; Takeshita, F.; Yamamoto, Y.; Yoshida, T.; Nishio, K.; Nagahara, S.; Kato, K.; Ochiya, T. RPN2 gene confers docetaxel resistance in breast cancer. Nat. Med. 2008, 14, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Hossain, F.; Danos, D.; Lassak, A.; Scribner, R.; Miele, L. Racial Disparities in Triple Negative Breast Cancer: A Review of the Role of Biologic and Non-biologic Factors. Front. Public Health 2020, 8, 576964. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Daly, B.; Olopade, O.I. A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J. Clin. 2015, 65, 221–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foy, K.C.; Fisher, J.L.; Lustberg, M.B.; Gray, D.M.; DeGraffinreid, C.R.; Paskett, E.D. Disparities in breast cancer tumor characteristics, treatment, time to treatment, and survival probability among African American and white women. NPJ Breast Cancer 2018, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.A. Parsing the Etiology of Breast Cancer Disparities. J. Clin. Oncol. 2016, 34, 1013–1014. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Reipas, K.; Hu, K.; Berns, R.; Firmino, N.; Stratford, A.L.; Dunn, S.E. Inhibition of RSK with the novel small-molecule inhibitor LJI308 overcomes chemoresistance by eliminating cancer stem cells. Oncotarget 2015, 6, 20570–20577. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Race | Basal | Her2 | LumA | LumB | NA | Normal Like | Grand Total |
|---|---|---|---|---|---|---|---|
| AfAm | 53 | 15 | 56 | 26 | 23 | 9 | 182 |
| Asian | 7 | 15 | 20 | 16 | 1 | 1 | 60 |
| NA | 5 | 10 | 39 | 29 | 6 | 1 | 90 |
| Native American | 1 | 1 | |||||
| White | 106 | 37 | 384 | 126 | 71 | 25 | 749 |
| Grand Total | 171 | 78 | 499 | 197 | 101 | 36 | 1082 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, P.S.; Wang, W.; Alkrekshi, A.; Markovic, V.; Khiyami, A.; Chan, R.; Perzynski, A.; Joseph, N.; Sossey-Alaoui, K. YB1 Is a Major Contributor to Health Disparities in Triple Negative Breast Cancer. Cancers 2021, 13, 6262. https://doi.org/10.3390/cancers13246262
Rana PS, Wang W, Alkrekshi A, Markovic V, Khiyami A, Chan R, Perzynski A, Joseph N, Sossey-Alaoui K. YB1 Is a Major Contributor to Health Disparities in Triple Negative Breast Cancer. Cancers. 2021; 13(24):6262. https://doi.org/10.3390/cancers13246262
Chicago/Turabian StyleRana, Priyanka Shailendra, Wei Wang, Akram Alkrekshi, Vesna Markovic, Amer Khiyami, Ricky Chan, Adam Perzynski, Natalie Joseph, and Khalid Sossey-Alaoui. 2021. "YB1 Is a Major Contributor to Health Disparities in Triple Negative Breast Cancer" Cancers 13, no. 24: 6262. https://doi.org/10.3390/cancers13246262
APA StyleRana, P. S., Wang, W., Alkrekshi, A., Markovic, V., Khiyami, A., Chan, R., Perzynski, A., Joseph, N., & Sossey-Alaoui, K. (2021). YB1 Is a Major Contributor to Health Disparities in Triple Negative Breast Cancer. Cancers, 13(24), 6262. https://doi.org/10.3390/cancers13246262