
Ribociclib Induces Broad Chemotherapy Resistance and EGFR Dependency in ESR1 Wildtype and Mutant Breast Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Compounds
2.3. Viability Assay
2.4. Protein Extraction and Immunoblotting
2.5. Antibodies
2.6. Proteome and Phosphoproteome Analysis
2.7. RNA Sequencing
2.8. Senescence-Associated β-Galactosidase Staining
2.9. Drug Screening
3. Results
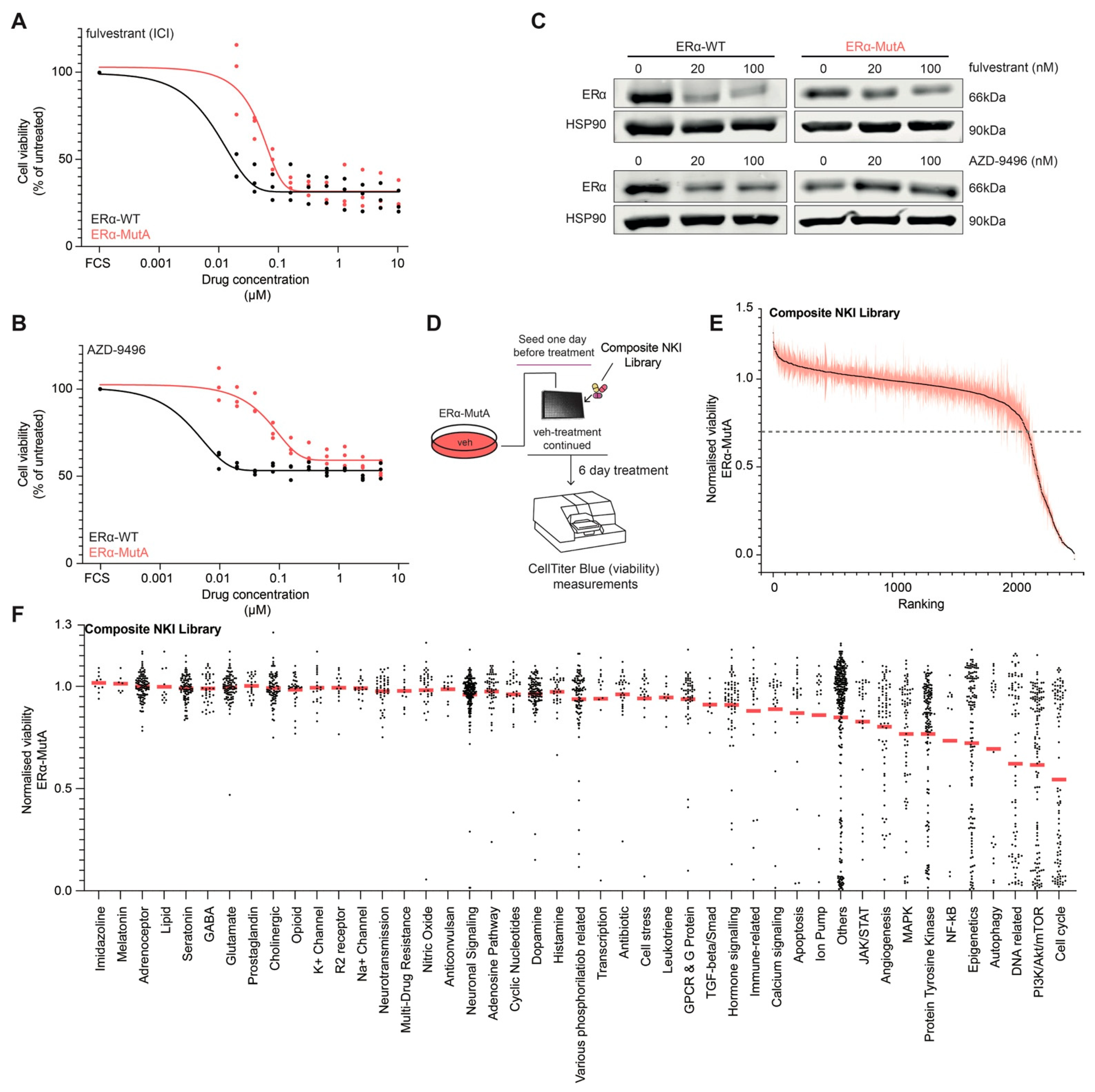
3.1. Drug Screening on ERα Mutant MCF-7 Cells
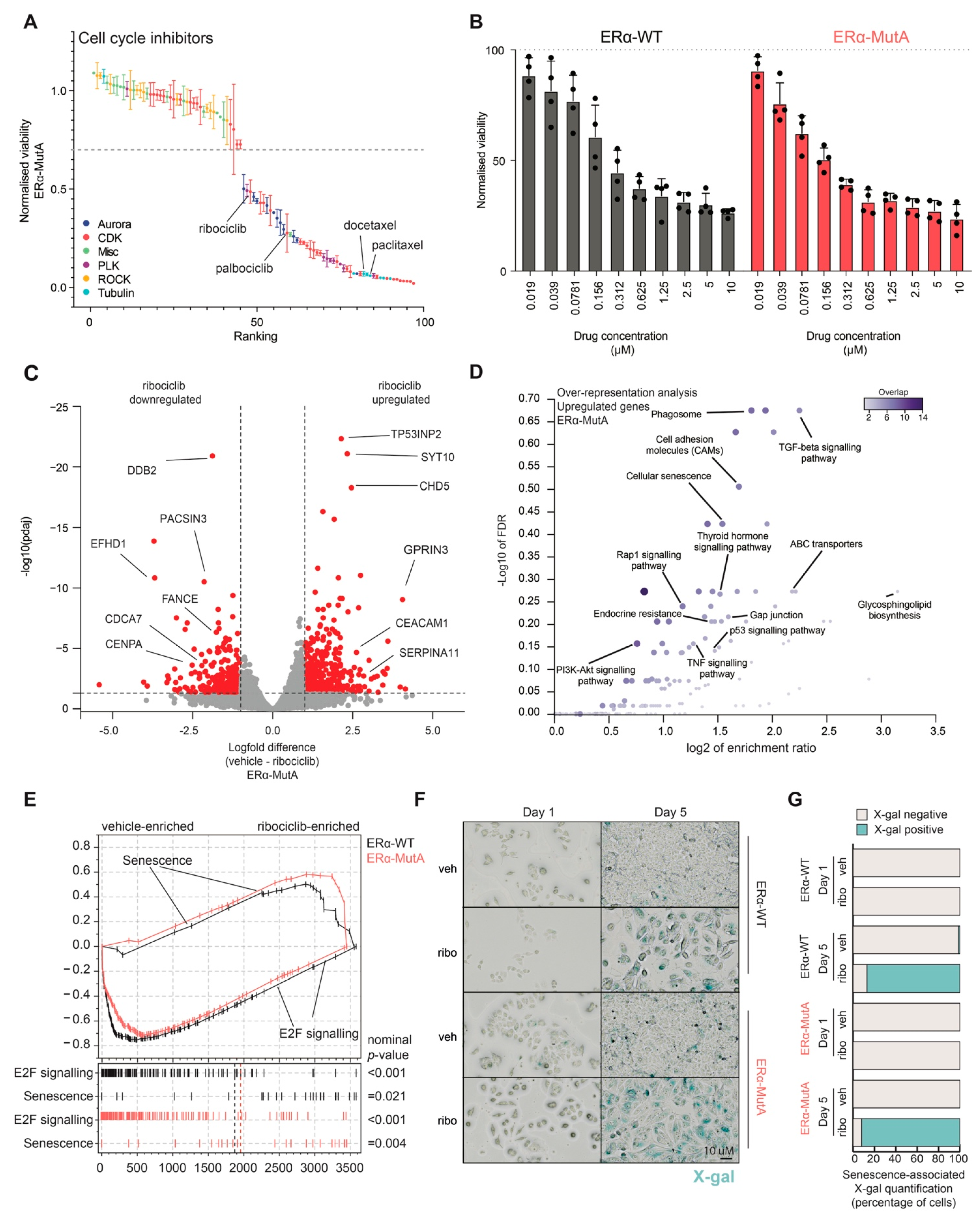
3.2. CDK4/6 Inhibitor Ribociclib Induces Senescence in ERα-Mut and ERα-WT Breast Cancer Models
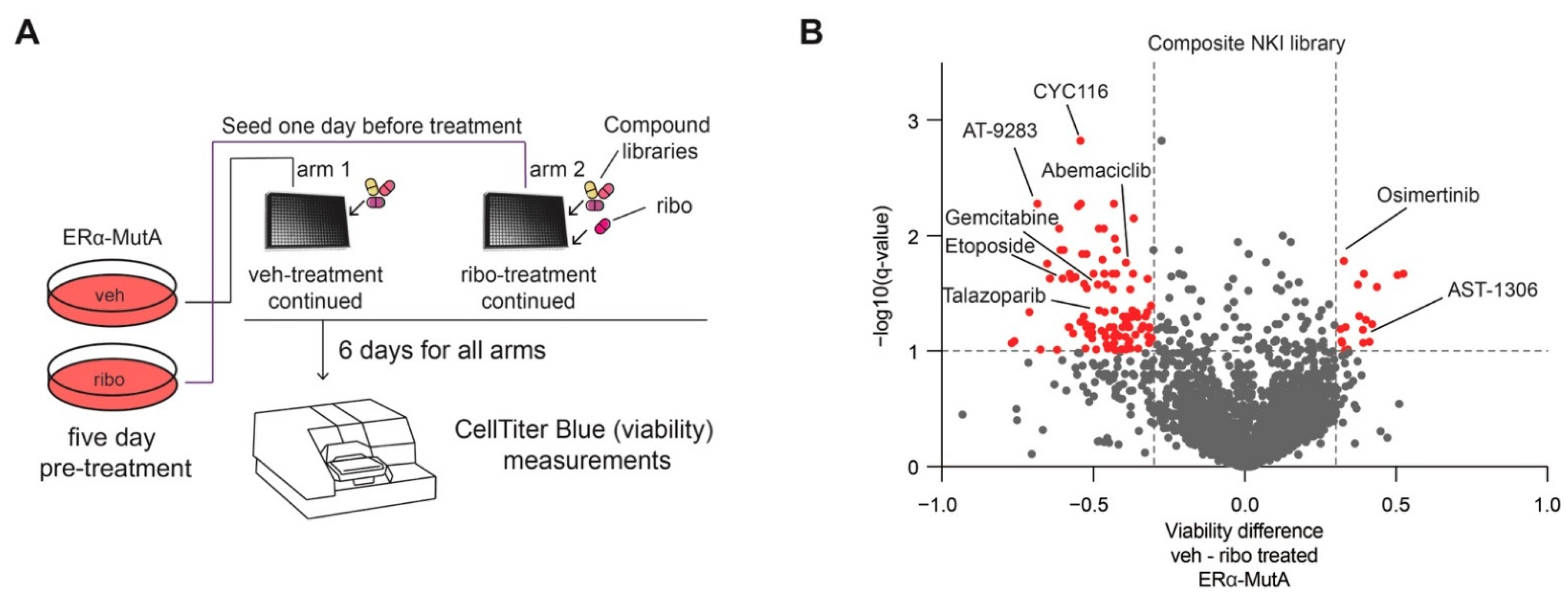
3.3. CDK4/6 Inhibition Induces Broad-Spectrum Drug Resistance and EGFR Dependence
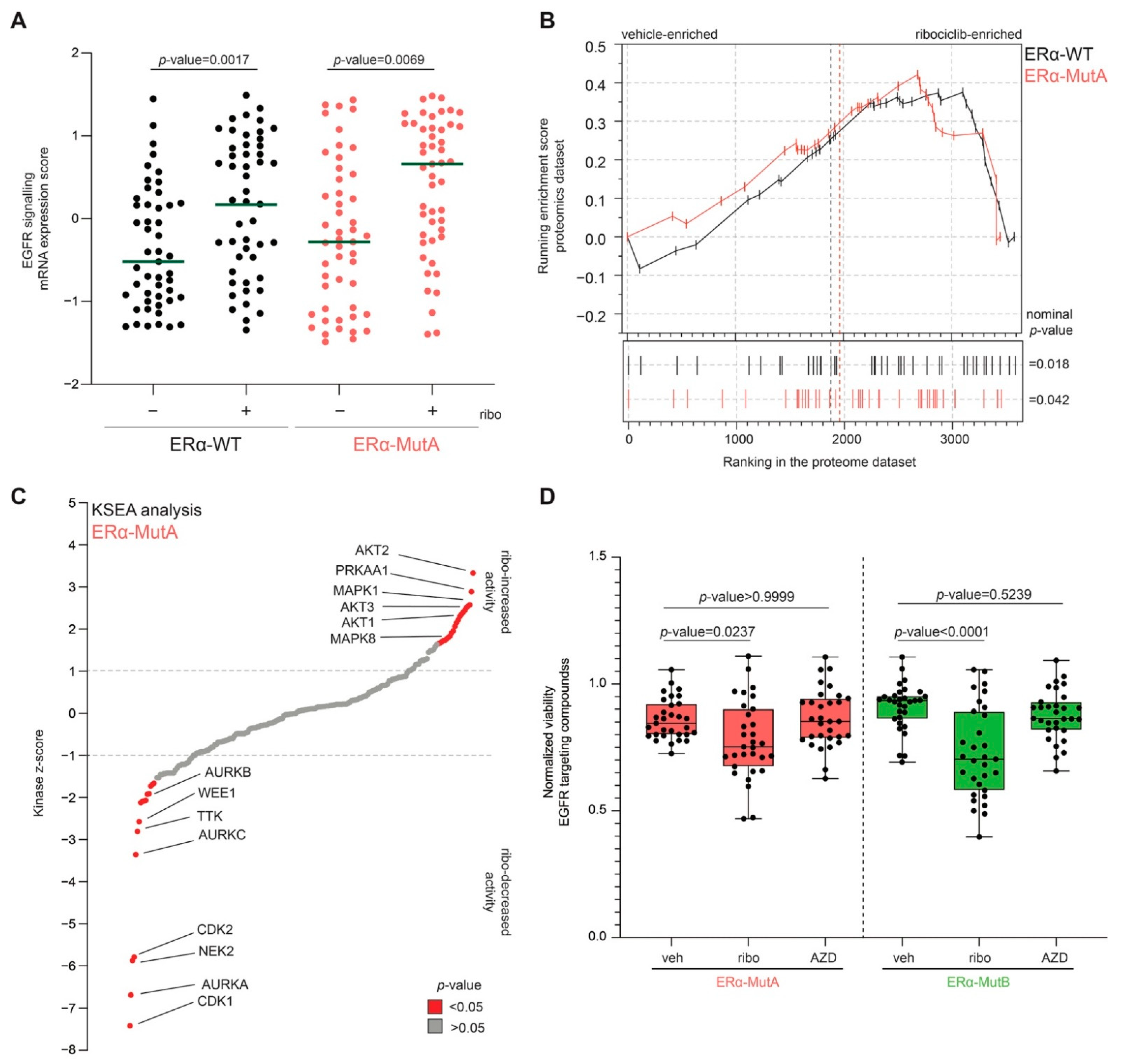
3.4. CDK4/6 Inhibitors Increase EGFR Pathway Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M.M. Current medical treatment of estrogen receptor-positive breast cancer. World J. Biol. Chem. 2015, 6, 231. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.-C.; Massard, C.; Levy, C.; Arnedos, M.; Lacroix-Triki, M. Mutational profile of metastatic breast cancers: A retrospective analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef] [PubMed]
- Pejerrey, S.M.; Dustin, D.; Kim, J.-A.; Gu, G.; Rechoum, Y.; Fuqua, S.A.W. The impact of ESR1 mutations on the treatment of metastatic breast cancer. Horm. Cancer 2018, 9, 215–228. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 2016, 5, e12792. [Google Scholar] [CrossRef] [PubMed]
- Anghel, S.I.; Perly, V.; Melançon, G.; Barsalou, A.; Chagnon, S.; Rosenauer, A.; Miller Jr, W.H.; Mader, S. Aspartate 351 of estrogen receptor α is not crucial for the antagonist activity of antiestrogens. J. Biol. Chem. 2000, 275, 20867–20872. [Google Scholar] [CrossRef] [Green Version]
- Eng, F.C.; Lee, H.S.; Ferrara, J.; Willson, T.M.; White, J.H. Probing the structure and function of the estrogen receptor ligand binding domain by analysis of mutants with altered transactivation characteristics. Mol. Cell. Biol. 1997, 17, 4644–4653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, F.C.S.; Barsalou, A.; Akutsu, N.; Mercier, I.; Zechel, C.; Mader, S.; White, J.H. Different classes of coactivators recognize distinct but overlapping binding sites on the estrogen receptor ligand binding domain. J. Biol. Chem. 1998, 273, 28371–28377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, K.E.; Ekena, K.; Thomas, J.A.; Lazennec, G.; Katzenellenbogen, B.S. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol. 1996, 10, 1388–1398. [Google Scholar] [PubMed] [Green Version]
- Zhao, C.; Koide, A.; Abrams, J.; Deighton-Collins, S.; Martinez, A.; Schwartz, J.A.; Koide, S.; Skafar, D.F. Mutation of Leu-536 in human estrogen receptor-α alters the coupling between ligand binding, transcription activation, and receptor conformation. J. Biol. Chem. 2003, 278, 27278–27286. [Google Scholar] [CrossRef] [Green Version]
- Harrod, A.; Fulton, J.; Nguyen, V.T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.-F.; Metodieva, G.; de Giorgio, A.; Williams, R.L.; Santos, D.B. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef] [Green Version]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 2018, 33, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Skafar, D.F. Mutations of tyrosine 537 in the human estrogen receptor-α selectively alter the receptor’s affinity for estradiol and the kinetics of the interaction. Biochemistry 2002, 41, 4209–4217. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef]
- Padrão, N.A.; Peralta, I.M.; Zwart, W. Targeting mutated Estrogen Receptor alpha: Rediscovering old and identifying new therapeutic strategies in metastatic breast cancer treatment. Curr. Opin. Endocr. Metab. Res. 2020, 15, 43–48. [Google Scholar] [CrossRef]
- Infante, J.R.; Cassier, P.A.; Gerecitano, J.F.; Witteveen, P.O.; Chugh, R.; Ribrag, V.; Chakraborty, A.; Matano, A.; Dobson, J.R.; Crystal, A.S. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2016, 22, 5696–5705. [Google Scholar] [CrossRef] [Green Version]
- Rugo, H.S.; Turner, N.C.; Finn, R.S.; Joy, A.A.; Verma, S.; Harbeck, N.; Masuda, N.; Im, S.-A.; Huang, X.; Kim, S. Palbociclib plus endocrine therapy in older women with HR+/HER2–advanced breast cancer: A pooled analysis of randomised PALOMA clinical studies. Eur. J. Cancer 2018, 101, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Rossi, L.; McCartney, A.; Risi, E.; De Santo, I.; Migliaccio, I.; Malorni, L.; Biganzoli, L.; Di Leo, A. Cyclin-dependent kinase 4/6 inhibitors in neoadjuvant endocrine therapy of hormone receptor-positive breast cancer. Clin. Breast Cancer 2019, 19, 392–398. [Google Scholar] [CrossRef]
- Xu, B.; Fan, Y. CDK4/6 inhibition in early-stage breast cancer: How far is it from becoming standard of care? Lancet Oncol. 2021, 22, 159–160. [Google Scholar] [CrossRef]
- Prekovic, S.; van Royen, M.E.; Voet, A.R.D.; Geverts, B.; Houtman, R.; Melchers, D.; Zhang, K.Y.J.; Van den Broeck, T.; Smeets, E.; Spans, L. The effect of F877L and T878A mutations on androgen receptor response to enzalutamide. Mol. Cancer Ther. 2016, 15, 1702–1712. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Post, H.; Penning, R.; Fitzpatrick, M.A.; Garrigues, L.B.; Wu, W.; MacGillavry, H.D.; Hoogenraad, C.C.; Heck, A.J.R.; Altelaar, A.F.M. Robust, sensitive, and automated phosphopeptide enrichment optimized for low sample amounts applied to primary hippocampal neurons. J. Proteome Res. 2017, 16, 728–737. [Google Scholar] [CrossRef]
- Wiredja, D.D.; Koyutürk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef]
- Prekovic, S.; Schuurman, K.; Mayayo-Peralta, I.; Manjón, A.G.; Buijs, M.; Yavuz, S.; Wellenstein, M.D.; Barrera, A.; Monkhorst, K.; Huber, A.; et al. Glucocorticoid receptor triggers a reversible drug-tolerant dormancy state with acquired therapeutic vulnerabilities in lung cancer. Nat. Commun. 2021. Accepted. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J. Cancer Metastasis Treat. 2019, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Byers, K.F. Ribociclib and Abemaciclib: CDK4/6 Inhibitors for the Treatment of Hormone Receptor–Positive Metastatic Breast Cancer. J. Adv. Pract. Oncol. 2021, 12, 100. [Google Scholar] [PubMed]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef]
- Wagner, V.; Gil, J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene 2020, 39, 5165–5176. [Google Scholar] [CrossRef]
- Iyengar, M.; O’Hayer, P.; Cole, A.; Sebastian, T.; Yang, K.; Coffman, L.; Buckanovich, R.J. CDK4/6 inhibition as maintenance and combination therapy for high grade serous ovarian cancer. Oncotarget 2018, 9, 15658. [Google Scholar] [CrossRef] [Green Version]
- Kishino, E.; Ogata, R.; Saitoh, W.; Koike, Y.; Ohta, Y.; Kanomata, N.; Kurebayashi, J. Anti-cell growth and anti-cancer stem cell activity of the CDK4/6 inhibitor palbociclib in breast cancer cells. Breast Cancer 2020, 27, 415–425. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D. Multi-Omics Profiling Establishes the Polypharmacology of FDA Approved CSK4/6 Inhibitors and Its Impact on Drug Response. Cell Chem. Biol. 2018. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent cells in cancer therapy: Friends or foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.C.; Lee, S.S.; Abraham, J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Sem. Oncol. 2017, 44, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.I.; Chen, J.; Cosgrove, P.A.; O’Dea, A.; Sharma, P.; Ma, C.; Trivedi, M.; Kalinsky, K.; Wisinski, K.B.; O’Regan, R. Serial single-cell genomics reveals convergent subclonal evolution of resistance as patients with early-stage breast cancer progress on endocrine plus CDK4/6 therapy. Nat. Cancer 2021, 2, 658–671. [Google Scholar] [CrossRef]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine resistance in hormone receptor positive breast cancer–from mechanism to therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Toi, M.; Neven, P.; Sohn, J.; Grischke, E.-M.; Llombart-Cussac, A.; Soliman, H.; Litchfield, L.M.; Wijayawardana, S.; Forrester, T. Clinical significance of PIK3CA and ESR1 mutations in ctDNA and FFPE samples from the MONARCH 2 study of abemaciclib plus fulvestrant. In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Preusser, M.; De Mattos-Arruda, L.; Thill, M.; Criscitiello, C.; Bartsch, R.; Ruhstaller, T.; de Azambuja, E.; Zielinski, C.C. CDK4/6 inhibitors in the treatment of patients with breast cancer: Summary of a multidisciplinary round-table discussion. ESMO Open 2018, 3, e000368. [Google Scholar] [CrossRef] [Green Version]
- Bashour, S.I.; Doostan, I.; Keyomarsi, K.; Valero, V.; Ueno, N.T.; Brown, P.H.; Litton, J.K.; Koenig, K.B.; Karuturi, M.; Abouharb, S. Rapid breast cancer disease progression following cyclin dependent kinase 4 and 6 inhibitor discontinuation. J. Cancer 2017, 8, 2004. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor–positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Guo, X.; Wang, M.; Li, Y.; Sun, X.; Li, Q. The application and prospect of CDK4/6 inhibitors in malignant solid tumors. J. Hematol. Oncol. 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D. Multiomics profiling establishes the polypharmacology of FDA-approved CDK4/6 inhibitors and the potential for differential clinical activity. Cell Chem. Biol. 2019, 26, 1067–1080. [Google Scholar] [CrossRef]
- Endo, H.; Inoue, M. Dormancy in cancer. Cancer Sci. 2019, 110, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-Y.; Nam, J.-S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, J. Mechanisms governing metastatic dormancy in breast cancer. Semin. Cancer Biol. 2017, 44, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, A.C.; Adam, A.P.; Aguirre-Ghiso, J.A. Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle 2006, 5, 1799–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; del Camino Menéndez, M.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell 2020, 37, 340–353. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Dean, J.L.; Rivadeneira, D.B.; Yu, J.E.; Reed, C.A.; Gao, E.; Farber, J.L.; Force, T.; Koch, W.J.; Knudsen, E.S. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle 2012, 11, 2747–2755. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.L.; McClendon, A.K.; Knudsen, E.S. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem. 2012, 287, 29075–29087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Nair, P.N.; De Armond, D.T.; Adamo, M.L.; Strodel, W.E.; Freeman, J.W. Aberrant expression and activation of insulin-like growth factor-1 receptor (IGF-1R) are mediated by an induction of IGF-1R promoter activity and stabilization of IGF-1R mRNA and contributes to growth factor independence and increased survival of the panc. Oncogene 2001, 20, 8203–8214. [Google Scholar] [CrossRef] [Green Version]
- Szaszi, B.; Palinkas, A.; Palfi, B.; Szollosi, A.; Aczel, B. A Systematic Scoping Review of the Choice Architecture Movement: Toward Understanding When and Why Nudges Work. J. Behav. Decis. Mak. 2018, 31, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Riebl, S.K.; Estabrooks, P.A.; Dunsmore, J.C.; Savla, J.; Frisard, M.I.; Dietrich, A.M.; Peng, Y.; Zhang, X.; Davy, B.M. A systematic literature review and meta-analysis: The Theory of Planned Behavior’s application to understand and predict nutrition-related behaviors in youth. Eat. Behav. 2015, 18, 160–178. [Google Scholar] [CrossRef]
- Orchinik, M.; Murray, T.F.; Moore, F.L. A corticosteroid receptor in neuronal membranes. Science 1991, 252, 1848–1851. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, Z.; Wong, G.; Pectasides, E.; Nagaraja, A.; Stachler, M.; Zhang, H.; Chen, T.; Zhang, H.; Liu, J. Bin CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat. Commun. 2017, 8, 1–12. [Google Scholar]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Megias, D.; Blanco-Aparicio, C.; Hernández-Encinas, E.; Rovira, M.; Pietrocola, F.; Serrano, M. Lysosomal trapping of palbociclib and its functional implications. Oncogene 2019, 38, 3886–3902. [Google Scholar] [CrossRef] [Green Version]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Ménard, L.; Floc’h, N.; Martin, M.J.; Cross, D.A.E. Reactivation of mutant-EGFR degradation through clathrin inhibition overcomes resistance to EGFR tyrosine kinase inhibitors. Cancer Res. 2018, 78, 3267–3279. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayayo-Peralta, I.; Faggion, B.; Hoekman, L.; Morris, B.; Lieftink, C.; Goldsbrough, I.; Buluwela, L.; Siefert, J.C.; Post, H.; Altelaar, M.; et al. Ribociclib Induces Broad Chemotherapy Resistance and EGFR Dependency in ESR1 Wildtype and Mutant Breast Cancer. Cancers 2021, 13, 6314. https://doi.org/10.3390/cancers13246314
Mayayo-Peralta I, Faggion B, Hoekman L, Morris B, Lieftink C, Goldsbrough I, Buluwela L, Siefert JC, Post H, Altelaar M, et al. Ribociclib Induces Broad Chemotherapy Resistance and EGFR Dependency in ESR1 Wildtype and Mutant Breast Cancer. Cancers. 2021; 13(24):6314. https://doi.org/10.3390/cancers13246314
Chicago/Turabian StyleMayayo-Peralta, Isabel, Beatrice Faggion, Liesbeth Hoekman, Ben Morris, Cor Lieftink, Isabella Goldsbrough, Lakjaya Buluwela, Joseph C. Siefert, Harm Post, Maarten Altelaar, and et al. 2021. "Ribociclib Induces Broad Chemotherapy Resistance and EGFR Dependency in ESR1 Wildtype and Mutant Breast Cancer" Cancers 13, no. 24: 6314. https://doi.org/10.3390/cancers13246314
APA StyleMayayo-Peralta, I., Faggion, B., Hoekman, L., Morris, B., Lieftink, C., Goldsbrough, I., Buluwela, L., Siefert, J. C., Post, H., Altelaar, M., Beijersbergen, R., Ali, S., Zwart, W., & Prekovic, S. (2021). Ribociclib Induces Broad Chemotherapy Resistance and EGFR Dependency in ESR1 Wildtype and Mutant Breast Cancer. Cancers, 13(24), 6314. https://doi.org/10.3390/cancers13246314