
Pan-Genomic Sequencing Reveals Actionable CDKN2A/2B Deletions and Kataegis in Anaplastic Thyroid Carcinoma

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Material
2.2. DNA Extraction and Tissue Representativity Testing
2.3. Whole-Genome Sequencing (WGS)
2.4. RNA Sequencing
2.5. WGS Data Analysis
2.6. RNA Sequencing Data analysis
2.7. Mutational Signatures and Kataegis
2.8. Fusion Gene Validation
2.9. Gene Ontology Analyses
3. Results
3.1. Whole-Genome Sequencing Quality Parameters
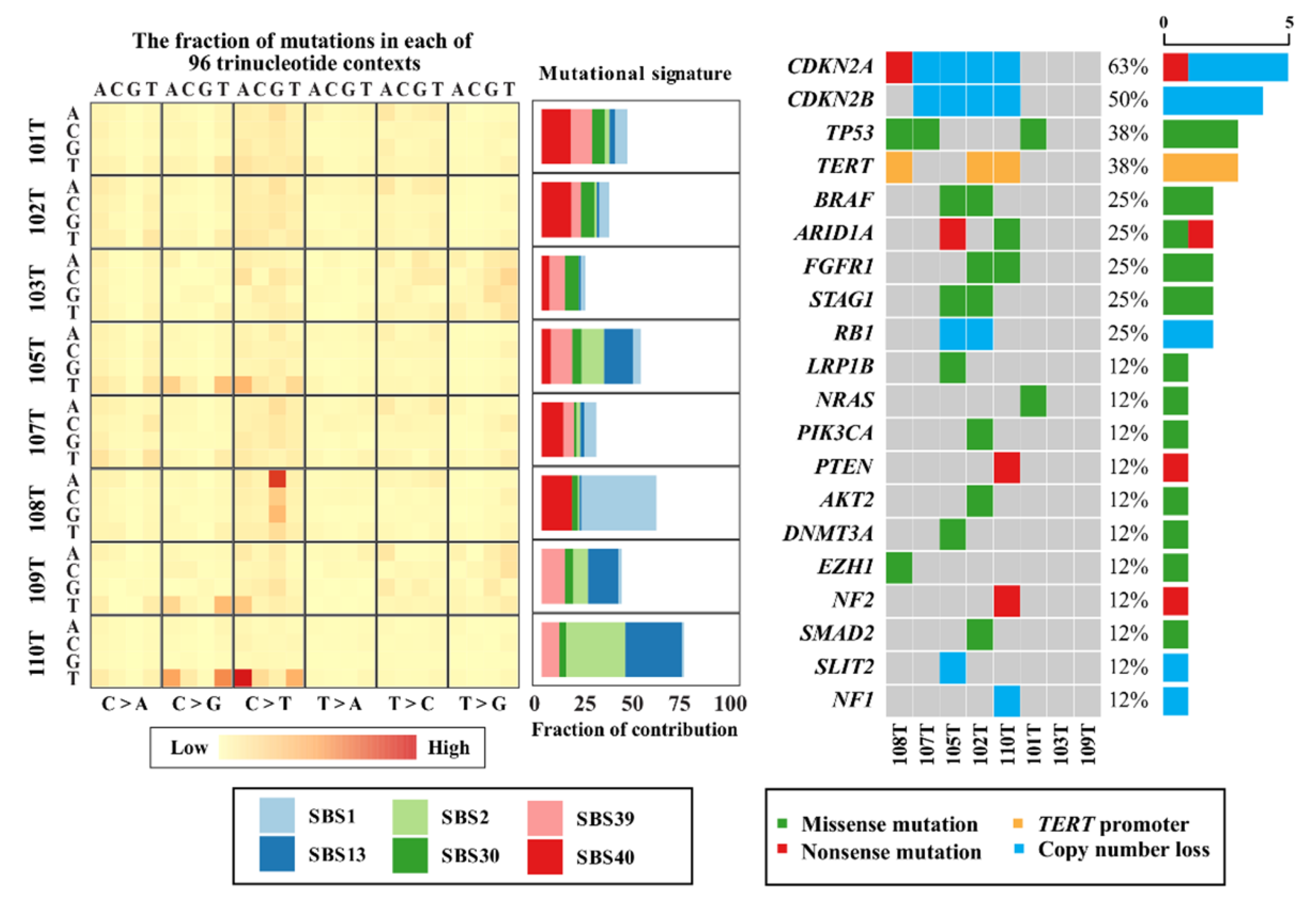
3.2. Somatic Mutational Overview
3.3. Aberrations in Genes Commonly Mutated in Anaplastic Thyroid Cancer
3.4. CDKN2A/B
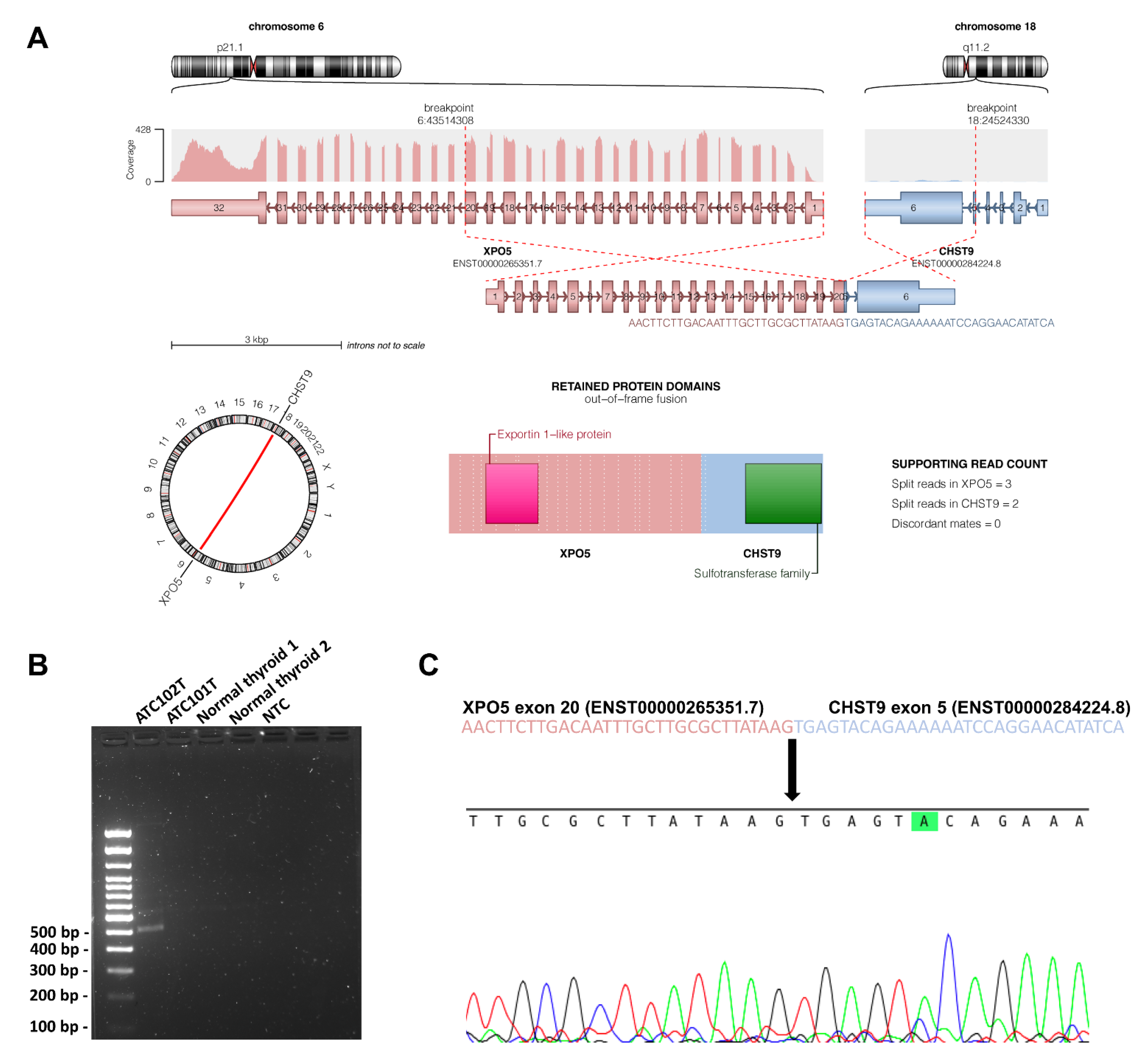
3.5. Novel Genes of Interest
3.6. Mutational Signatures and Kataegis
3.7. Non-Coding Mutations in Introns, Promoters, and Enhancers
3.8. Structural Variants
3.9. Potential Therapeutic Targets
3.10. Transcriptome Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Capdevila, J.; Mayor, R.; Mancuso, F.M.; Iglesias, C.; Caratù, G.; Matos, I.; Zafón, C.; Hernando, J.; Petit, A.; Nuciforo, P.; et al. Early evolutionary divergence between papillary and anaplastic thyroid cancers. Ann. Oncol. 2018, 29, 1454–1460. [Google Scholar] [CrossRef]
- Dong, W.; Nicolson, N.G.; Choi, J.; Barbieri, A.L.; Kunstman, J.W.; Azar, S.A.; Knight, J.; Bilguvar, K.; Mane, S.M.; Lifton, R.P.; et al. Clonal evolution analysis of paired anaplastic and well-differentiated thyroid carcinomas reveals shared common ancestor. Genes Chromosomes Cancer 2018, 57, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Kondo, T.; Ebina, A.; Sato, Y.; Akaishi, J.; Hino, R.; Yamamoto, N.; Mochizuki, K.; Nakazawa, T.; Yokomichi, H.; et al. Molecular alterations of coexisting thyroid papillary carcinoma and anaplastic carcinoma: Identification of TERT mutation as an independent risk factor for transformation. Mod. Pathol. 2017, 30, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.O.; Backman, S.; Wang, N.; Stenman, A.; Crona, J.; Thutkawkorapin, J.; Ghaderi, M.; Tham, E.; Stålberg, P.; Zedenius, J.; et al. Whole-genome sequencing of synchronous thyroid carcinomas identifies aberrant DNA repair in thyroid cancer dedifferentiation. J. Pathol. 2020, 250, 183–194. [Google Scholar] [CrossRef]
- Juhlin, C.C. Aberrant DNA repair as a potential contributor for the clonal evolution in subsets of anaplastic thyroid carcinomas arising through dedifferentiation: Implications for future therapeutic algorithms? Cancer Drug Resist. 2020, 3, 992–1000. [Google Scholar] [CrossRef]
- Alobuia, W.; Gillis, A.; Kebebew, E. Contemporary Management of Anaplastic Thyroid Cancer. Curr. Treat. Options Oncol. 2020, 21, 78. [Google Scholar] [CrossRef]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosove, M.H.; Peddi, P.F.; Glaspy, J.A. BRAF V600E inhibition in anaplastic thyroid cancer. N. Engl. J. Med. 2013, 368, 684–685. [Google Scholar] [CrossRef]
- Stenman, A.; Hellgren, L.S.; Jatta, K.; Hysek, M.; Zemmler, M.; Altena, R.; Nilsson, I.-L.; Bränström, R.; Zedenius, J.; Juhlin, C.C. Metastatic Anaplastic Thyroid Carcinoma in Complete Remission: Morphological, Molecular, and Clinical Work-Up of a Rare Case. Endocr. Pathol. 2020, 31, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Kunstman, J.W.; Juhlin, C.C.; Goh, G.; Brown, T.C.; Stenman, A.; Healy, J.M.; Rubinstein, J.C.; Choi, M.; Kiss, N.; Nelson-Williams, C.; et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum. Mol. Genet. 2015, 24, 2318–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Fuchs, T.; Dogan, S.; Landa, I.; Katabi, N.; Fagin, J.A.; Tuttle, R.M.; Sherman, E.; Gill, A.J.; Ghossein, R. Dissecting Anaplastic Thyroid Carcinoma: A Comprehensive Clinical, Histologic, Immunophenotypic, and Molecular Study of 360 Cases. Thyroid 2020, 30, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Ravi, N.; Yang, M.; Mylona, N.; Wennerberg, J.; Paulsson, K. Global RNA Expression and DNA Methylation Patterns in Primary Anaplastic Thyroid Cancer. Cancers 2020, 12, 680. [Google Scholar] [CrossRef] [Green Version]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]
- Wala, J.A.; Bandopadhayay, P.; Greenwald, N.F.; O’Rourke, R.; Sharpe, T.; Stewart, C.; Schumacher, S.; Li, Y.; Weischenfeldt, J.; Yao, X.; et al. SvABA: Genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018, 28, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, D.L.; Schröder, J.; Penington, J.S.; Do, H.; Molania, R.; Dobrovic, A.; Speed, T.P. Papenfuss AT. GRIDSS: Sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. [Google Scholar] [CrossRef] [Green Version]
- Reisle, C.; Mungall, K.L.; Choo, C.; Paulino, D.; Bleile, D.W.; Muhammadzadeh, A.; Mungall, A.J.; Moore, R.A.; Shlafman, I.; Coope, R.; et al. MAVIS: Merging, annotation, validation, and illustration of structural variants. Bioinformatics 2019, 35, 515–517. [Google Scholar] [CrossRef]
- Mayrhofer, M.; DiLorenzo, S.; Isaksson, A. Patchwork: Allele-specific copy number analysis of whole-genome sequenced tumor tissue. Genome Biol. 2013, 14, R24. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Imai-Matsushima, A.; Paul, L.; Seitz, A.; Meyer, T.F.; Garcia-Alcalde, F. InFusion: Advancing Discovery of Fusion Genes and Chimeric Transcripts from Deep RNA-Sequencing Data. PLoS ONE 2016, 11, e0167417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Breast Cancer Working Group of the International Cancer Genome Consortium. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [PubMed] [Green Version]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.S.; Lorch, J.H.; Alexander, E.K.; Nehs, M.A.; Nowak, J.A.; Hornick, J.L.; Barletta, J.A. Clinicopathologic Features of Mismatch Repair-Deficient Anaplastic Thyroid Carcinomas. Thyroid 2019, 29, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Höti, N.; Yang, S.; Aiyetan, P.; Kumar, B.; Hu, Y.; Clark, D.; Eroglu, A.U.; Shah, P.; Johnson, T.; Chowdery, W.H.; et al. Overexpression of Exportin-5 Overrides the Inhibitory Effect of miRNAs Regulation Control and Stabilize Proteins via Posttranslation Modifications in Prostate Cancer. Neoplasia 2017, 19, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Shigeyasu, K.; Okugawa, Y.; Toden, S.; Boland, C.R.; Goel, A. Exportin-5 Functions as an Oncogene and a Potential Therapeutic Target in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 1312–1322. [Google Scholar] [CrossRef] [Green Version]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, G.; Nappi, T.C.; Salerno, P.; Jiang, Y.; Garbi, C.; Ugolini, C.; Miccoli, P.; Basolo, F.; Castellone, M.D.; Cirafici, A.M.; et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007, 67, 10148–10158. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lee, J.E.; Riemondy, K.; Yu, Y.; Marquez, S.M.; Lai, E.C.; Yi, R. XPO5 promotes primary miRNA processing independently of RanGTP. Nat. Commun. 2020, 11, 1845. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; Barve, M.A.; Chiorean, E.G.; LoRusso, P.; Courtney, K.D.; Qi, D.; Bullington, J.; Sardone, M.; Chen, J.; Brooks, C.; et al. 564P Updated results of a phase I study of Felezonexor (SL-801), a novel XPO-1 reversible inhibitor, in patients with relapsed/refractory solid tumors. Ann. Oncol. 2020, 31, S485–S486. [Google Scholar] [CrossRef]
- Ema, A.; Waraya, M.; Yamashita, K.; Kokubo, K.; Kobayashi, H.; Hoshi, K.; Shinkai, Y.; Kawamata, H.; Nakamura, K.; Nishimiya, H.; et al. Identification of EGFR expression status association with metastatic lymph node density (ND) by expression microarray analysis of advanced gastric cancer. Cancer Med. 2015, 4, 90–100. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, Q.; Fu, X.; Yu, B.; Shao, Y.; Yang, H.; Guan, M.; Huang, X.; Zhang, W.; Wan, J. Examination of copy number variations of CHST9 in multiple types of hematologic malignancies. Cancer Genet. Cytogenet. 2010, 203, 176–179. [Google Scholar] [CrossRef]
- Yin, X.; Bi, R.; Ma, P.; Zhang, S.; Zhang, Y.; Sun, Y.; Zhang, Y.; Jing, Y.; Yu, M.; Wang, W.; et al. Multiregion whole-genome sequencing depicts intratumor heterogeneity and punctuated evolution in ovarian clear cell carcinoma. J. Med. Genet. 2020, 57, 605–609. [Google Scholar] [CrossRef]
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet. 2020, 52, 884–890. [Google Scholar] [CrossRef]
- Kim, S.T.; Klempner, S.J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, K.-M.; Lee, J. Correlating programmed death ligand 1 (PD-L1) expression, mismatch repair deficiency, and outcomes across tumor types: Implications for immunotherapy. Oncotarget 2017, 8, 77415–77423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chintakuntlawar, A.V.; Rumilla, K.M.; Smith, C.Y.; Jenkins, S.M.; Foote, R.L.; Kasperbauer, J.L.; Morris, J.C.; Ryder, M.; Alsidawi, S.; Hilger, C.; et al. Expression of PD-1 and PD-L1 in Anaplastic Thyroid Cancer Patients Treated With Multimodal Therapy: Results From a Retrospective Study. J. Clin. Endocrinol. Metab. 2017, 102, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Wirth, L.J.; Ernst, T.; Aix, S.P.; Lin, C.-C.; Ramlau, R.; Butler, M.O.; Delord, J.-P.; Gelderblom, H.; Ascierto, P.A.; et al. PD-1 Blockade in Anaplastic Thyroid Carcinoma. J. Clin. Oncol. 2020, 38, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Segerhammar, I.; Larsson, C.; Nilsson, I.-L.; Bäckdahl, M.; Höög, A.; Wallin, G.; Foukakis, T.; Zedenius, J. Anaplastic carcinoma of the thyroid gland: Treatment and outcome over 13 years at one institution. J. Surg. Oncol. 2012, 106, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Haase, J.; Misiak, D.; Bauer, M.; Pazaitis, N.; Braun, J.; Pötschke, R.; Mensch, A.; Bell, J.L.; Dralle, H.; Siebolts, U.; et al. IGF2BP1 is the first positive marker for anaplastic thyroid carcinoma diagnosis. Mod. Pathol. 2021, 34, 32–41. [Google Scholar] [CrossRef]
- Abe, I.; Lam, A.K.-Y. Anaplastic Thyroid Carcinoma: Current Issues in Genomics and Therapeutics. Curr. Oncol. Rep. 2021, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, C.C.; Sadow, P.M.; Daniels, G.H.; Wirth, L.J. Progress in Treating Advanced Thyroid Cancers in the Era of Targeted Therapy. Thyroid 2021, 31, 1451–1462. [Google Scholar] [CrossRef]
- Ravi, N.; Yang, M.; Gretarsson, S.; Jansson, C.; Mylona, N.; Sydow, S.R.; Woodward, E.L.; Ekblad, L.; Wennerberg, J.; Paulsson, K. Identification of Targetable Lesions in Anaplastic Thyroid Cancer by Genome Profiling. Cancers 2019, 11, 402. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Sex | Age at Surgery | Outcome | Follow-Up Time (Months) | Tumor Size (cm) | Synchronous WDTC | Tumor Cell Content (%) * |
|---|---|---|---|---|---|---|---|
| 101 | F | 77 | DOD | 3 | 7 | PTC | 90 |
| 102 | M | 82 | DOD | 1 | 4 | PTC | 50 |
| 103 | F | 72 | DOD | 1 | 10 | No | 50 |
| 105 | F | 78 | DOC | >200 | 8.5 | No | 90 |
| 107 | F | 89 | DOD | 0.5 | 8 | PTC | 90 |
| 108 | F | 81 | DOD | 4 | 5 | No | 50 |
| 109 | M | 70 | DOD | 3 | 11 | PTC | 50 |
| 110 | F | 92 | DOD | 1 | 6.5 | No | 90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stenman, A.; Yang, M.; Paulsson, J.O.; Zedenius, J.; Paulsson, K.; Juhlin, C.C. Pan-Genomic Sequencing Reveals Actionable CDKN2A/2B Deletions and Kataegis in Anaplastic Thyroid Carcinoma. Cancers 2021, 13, 6340. https://doi.org/10.3390/cancers13246340
Stenman A, Yang M, Paulsson JO, Zedenius J, Paulsson K, Juhlin CC. Pan-Genomic Sequencing Reveals Actionable CDKN2A/2B Deletions and Kataegis in Anaplastic Thyroid Carcinoma. Cancers. 2021; 13(24):6340. https://doi.org/10.3390/cancers13246340
Chicago/Turabian StyleStenman, Adam, Minjun Yang, Johan O. Paulsson, Jan Zedenius, Kajsa Paulsson, and C. Christofer Juhlin. 2021. "Pan-Genomic Sequencing Reveals Actionable CDKN2A/2B Deletions and Kataegis in Anaplastic Thyroid Carcinoma" Cancers 13, no. 24: 6340. https://doi.org/10.3390/cancers13246340
APA StyleStenman, A., Yang, M., Paulsson, J. O., Zedenius, J., Paulsson, K., & Juhlin, C. C. (2021). Pan-Genomic Sequencing Reveals Actionable CDKN2A/2B Deletions and Kataegis in Anaplastic Thyroid Carcinoma. Cancers, 13(24), 6340. https://doi.org/10.3390/cancers13246340