
Clinicopathological and Molecular Prognostic Classifier for Intermediate/High-Risk Clear Cell Renal Cell Carcinoma

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Clinical Features of the Cohort
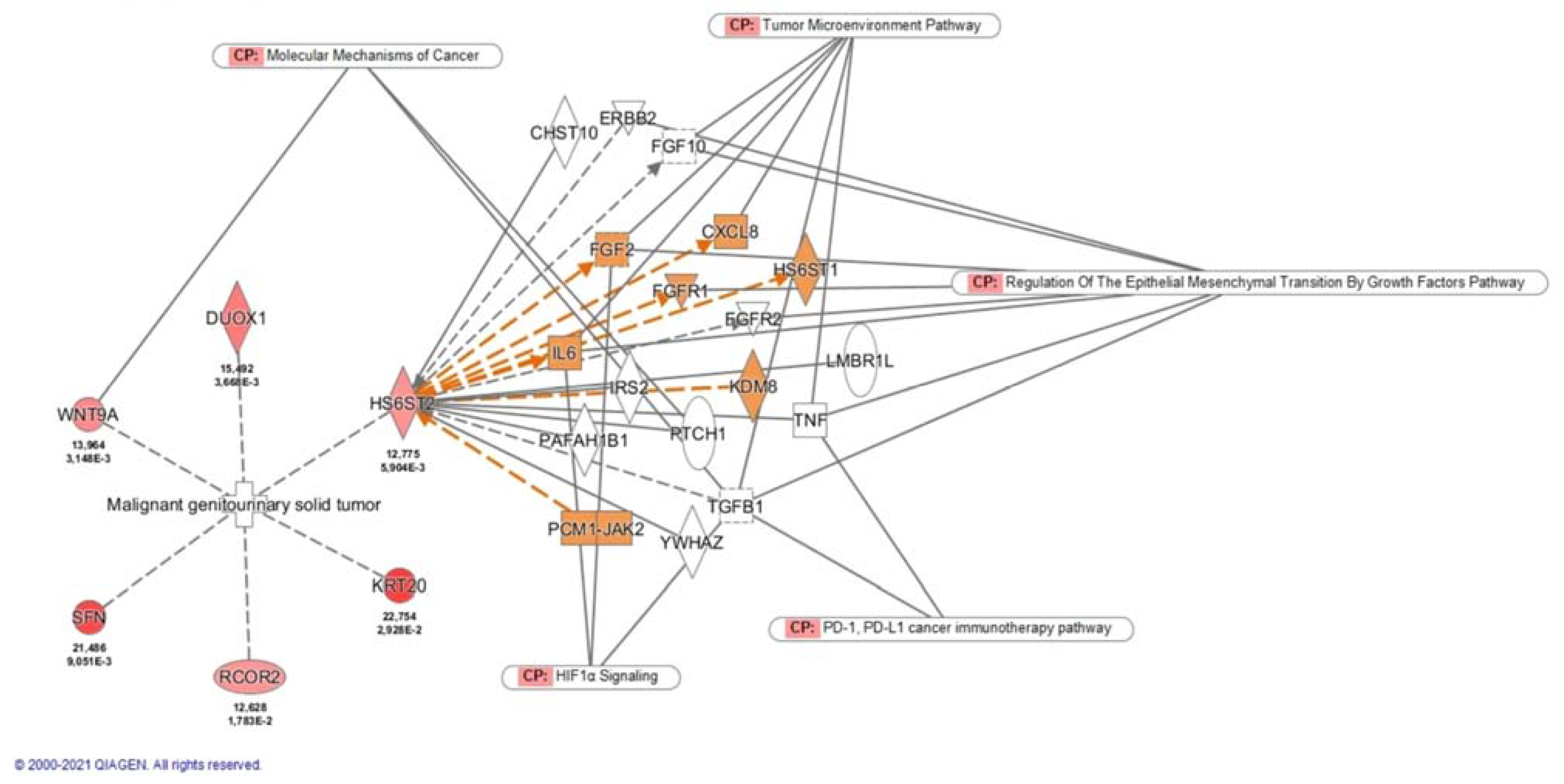
2.2. Biomarker Discovery Phase
2.3. Biomarker Validation Phase
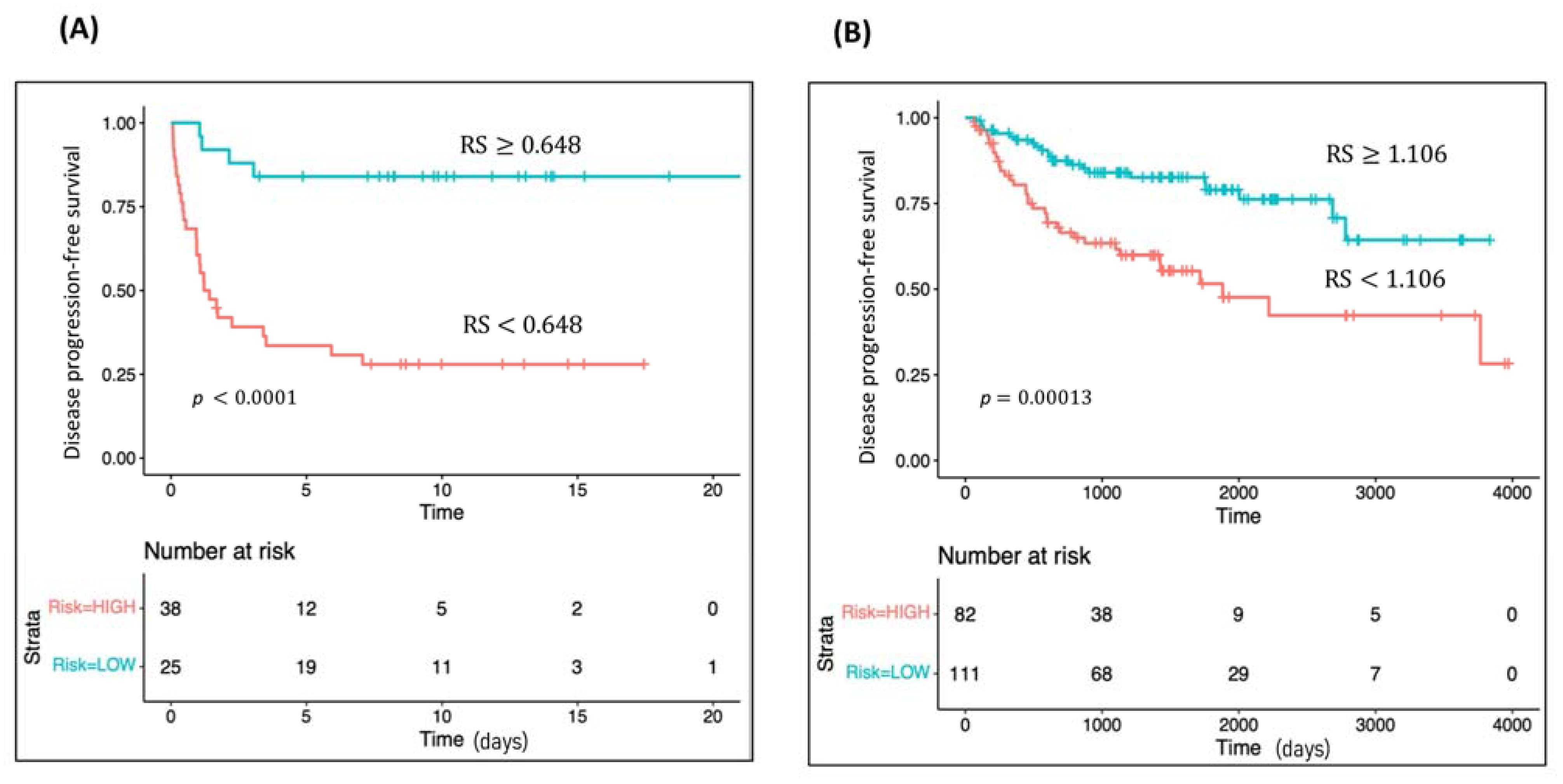
2.4. Survival Analyses
2.5. Classifier Development Phase
2.6. Classifier In Silico Validation
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Tissue Specimens and RNA Isolation
4.3. Biomarker Discovery Phase—RNA Sequencing
4.3.1. Library Preparation and Sequencing Method
4.3.2. Read Alignment and Differential Gene Expression Analysis
4.4. Biomarker Validation Phase
4.4.1. nCounter Elements Gene Expression Analysis
4.4.2. Survival Analysis
4.5. Classifier Development Phase
4.6. Pathway Enrichment Analysis
4.7. Classifier In Silico Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghatalia, P.; Gordetsky, J.; Kuo, F.; Dulaimi, E.; Cai, K.Q.; Devarajan, K.; Bae, S.; Naik, G.; Chan, T.A.; Uzzo, R.; et al. Prognostic impact of immune gene expression signature and tumor infiltrating immune cells in localized clear cell renal cell carcinoma. J. Immunother. Cancer 2019, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Sharma, T.; Tajzler, C.; Kapoor, A. Is There a Role for Adjuvant Therapy After Surgery in “High Risk for Recurrence” Kidney Cancer? An Update on Current Concepts. Curr. Oncol. 2018, 25, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Janzen, N.K.; Kim, H.L.; Figlin, R.A.; Belldegrun, A.S. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol. Clin. N. Am. 2003, 30, 843–852. [Google Scholar] [CrossRef]
- Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Lohse, C.M.; Frank, I.; Kwon, E.D.; Weaver, A.L.; Parker, A.S.; Zincke, H. Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003, 97, 1663–1671. [Google Scholar] [CrossRef]
- Frank, I.G.O.R.; Blute, M.L.; Cheville, J.C.; Lohse, C.M.; Weaver, A.L.; Zincke, H. An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: The SSIGN score. J. Urol. 2002, 168, 2395–2400. [Google Scholar] [CrossRef]
- Ficarra, V.; Schips, L.; Guillè, F.; Li, G.; De La Taille, A.; Galetti, T.P.; Cindolo, L.; Novara, G.; Zigeuner, R.E.; Bratti, E.; et al. Multiinstitutional European validation of the 2002 TNM staging system in conventional and papillary localized renal cell carcinoma. Cancer 2005, 104, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, S.A.; Lasky, L.C.; Limas, C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am. J. Surg. Pathol. 1982, 6, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Kwa, M.; Makris, A.; Esteva, F.J. Clinical utility of gene-expression signatures in early stage breast cancer. Nat. Rev. Clin. Oncol. 2017, 14, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.; Goddard, A.; Knezevic, D.; Maddala, T.; Zhou, M.; Aydin, H.; Campbell, S.; Elson, P.; Koscielny, S.; Lopatin, M.; et al. A 16-gene assay to predict recurrence after surgery in localised renal cell carcinoma: Development and validation studies. Lancet Oncol. 2015, 16, 676–685. [Google Scholar] [CrossRef]
- Brooks, S.A.; Brannon, A.R.; Parker, J.S.; Fisher, J.C.; Sen, O.; Kattan, M.W.; Hakimi, A.A.; Hsieh, J.J.; Choueiri, T.K.; Tamboli, P.; et al. ClearCode34: A Prognostic Risk Predictor for Localized Clear Cell Renal Cell Carcinoma. Eur. Urol. 2014, 66, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Patard, J.-J.; Kim, H.L.; Lam, J.S.; Dorey, F.J.; Pantuck, A.J.; Zisman, A.; Ficarra, V.; Han, K.-R.; Cindolo, L.; De La Taille, A.; et al. Use of the University of California Los Angeles Integrated Staging System to Predict Survival in Renal Cell Carcinoma: An International Multicenter Study. J. Clin. Oncol. 2004, 22, 3316–3322. [Google Scholar] [CrossRef]
- Ljungberg, B.; Albiges, L.; Bensalah, K.; Bex, A.; Giles, R.H.; Hora, M. EAU Guidelines. Edn. presented at the EAU Annual Congress Amsterdam 2020. Eur. Urol. 2020, 67, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Motzer, R.J.; Pandha, H.S.; George, D.J.; Pantuck, A.J.; Patel, A.; Chang, Y.-H.; Escudier, B.; Donskov, F.; Magheli, A.; et al. Adjuvant Sunitinib in High-Risk Renal-Cell Carcinoma after Nephrectomy. N. Engl. J. Med. 2016, 375, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Haas, N.B.; Donskov, F.; Gross-Goupil, M.; Varlamov, S.; Kopyltsov, E.; Lee, J.L.; Melichar, B.; Rini, B.I.; Choueiri, T.K.; et al. Randomized Phase III Trial of Adjuvant Pazopanib Versus Placebo after Nephrectomy in Patients with Localized or Locally Advanced Renal Cell Carcinoma. J. Clin. Oncol. 2017, 35, 3916–3923. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ASCO 2021: Results from the KEYNOTE-564 Study of Adjuvant Pembrolizumab in Resected Clear-Cell Renal Cell Carcinoma (RCC). Available online: https://ascopost.com/issues/june-25-2021/keynote-564-adjuvant-pembrolizumab-extends-disease-free-survival-in-high-risk-renal-cell-carcinoma/ (accessed on 12 August 2021).
- Palumbo, C.; Mazzone, E.; Mistretta, F.A.; Knipper, S.; Perrotte, P.; Shariat, S.F.; Saad, F.; Kapoor, A.; Lattouf, J.-B.; Simeone, C.; et al. A Plea for Optimizing Selection in Current Adjuvant Immunotherapy Trials for High-Risk Nonmetastatic Renal Cell Carcinoma According to Expected Cancer-specific Mortality. Clin. Genitourin. Cancer 2020, 18, 314–321. [Google Scholar] [CrossRef]
- Wu, J.; Jin, S.; Gu, W.; Wan, F.; Zhang, H.; Shi, G.; Qu, Y.; Ye, D. Construction and Validation of a 9-Gene Signature for Predicting Prognosis in Stage III Clear Cell Renal Cell Carcinoma. Front. Oncol. 2019, 9, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; He, J.; Du, J.; Zhang, R.-X.; Yao, H.-B.; Shao, Q.-S. Overexpression of HS6ST2 is associated with poor prognosis in patients with gastric cancer. Oncol. Lett. 2017, 14, 6191–6197. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [Green Version]
- Lundin, L.; Larsson, H.; Kreuger, J.; Kanda, S.; Lindahl, U.; Salmivirta, M.; Claesson-Welsh, L. Selectively Desulfated Heparin Inhibits Fibroblast Growth Factor-induced Mitogenicity and Angiogenesis. J. Biol. Chem. 2000, 275, 24653–24660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Stringer, S.E.; Rusch, M.A.; Selleck, S.B.; Ekker, S.C. A unique role for 6-O sulfation modification in zebrafish vascular development. Dev. Biol. 2005, 284, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Q.; Peng, Y.-B.; Li, J.; Ding, K.; Chen, L.-J.; Shao, C.-H.; Zhang, L.-J.; Li, P. Silencing of hHS6ST2 inhibits progression of pancreatic cancer through inhibition of Notch signalling. Biochem. J. 2011, 436, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, C.L.; Rushton, G.; Jayson, G.C.; Avizienyte, E. Ovarian Cancer Cell Heparan Sulfate 6-O-Sulfotransferases Regulate an Angiogenic Program Induced by Heparin-Binding Epidermal Growth Factor (EGF)-Like Growth Factor/EGF Receptor Signaling. J. Biol. Chem. 2014, 289, 10488–10501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, M.; Pisano, A.; Abonante, S.; Maggiolini, M.; Musti, A.M. GPER activates Notch signaling in breast cancer cells and cancer-associated fibroblasts (CAFs). Int. J. Biochem. Cell Biol. 2014, 46, 56–67. [Google Scholar] [CrossRef]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Jain, S.; Azad, A.K.; Xu, X.; Yu, H.C.; Xu, Z.; Godbout, R.; Fu, Y. Notch and TGFβ form a positive regulatory loop and regulate EMT in epithelial ovarian cancer cells. Cell. Signal. 2016, 28, 838–849. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between notch, HIF-1α and GPER in breast cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef] [Green Version]
- Pollari, S.; Käkönen, R.S.; Mohammad, K.S.; Rissanen, J.P.; Halleen, J.M.; Wärri, A.; Nissinen, L.; Pihlavisto, M.; Marjamäki, A.; Perälä, M.; et al. Heparin-like Polysaccharides Reduce Osteolytic Bone Destruction and Tumor Growth in a Mouse Model of Breast Cancer Bone Metastasis. Mol. Cancer Res. 2012, 10, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Hatabe, S.; Kimura, H.; Arao, T.; Kato, H.; Hayashi, H.; Nagai, T.; Matsumoto, K.; De Velasco, M.; Fujita, Y.; Yamanouchi, G.; et al. Overexpression of heparan sulfate 6-O-sulfotransferase-2 in colorectal cancer. Mol. Clin. Oncol. 2013, 1, 845–850. [Google Scholar] [CrossRef] [Green Version]
- Backen, A.C.; Cole, C.L.; Lau, S.C.; Clamp, A.R.; McVey, R.; Gallagher, J.T.; Jayson, G.C. Heparan sulphate synthetic and editing enzymes in ovarian cancer. Br. J. Cancer 2007, 96, 1544–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulten, H.-J.; Al-Mansouri, Z.; Baghallab, I.; Bagatian, N.; Subhi, O.; Karim, S.; Al-Aradati, H.; Al-Mutawa, A.; Johary, A.; Meccawy, A.A.; et al. Comparison of microarray expression profiles between follicular variant of papillary thyroid carcinomas and follicular adenomas of the thyroid. BMC Genom. 2015, 16, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Q.; Zeng, T.; Liu, S.; Cheng, H.; Ma, L.; Wang, Y. A novel 10 glycolysis-related genes signature could predict overall survival for clear cell renal cell carcinoma. BMC Cancer 2021, 21, 381. [Google Scholar] [CrossRef]
- Liep, J.; Kilic, E.; Meyer, H.A.; Busch, J.; Jung, K.; Rabien, A. Cooperative Effect of miR-141-3p and miR-145-5p in the Regulation of Targets in Clear Cell Renal Cell Carcinoma. PLoS ONE 2016, 11, e0157801. [Google Scholar] [CrossRef] [PubMed]
- FireBrowse. Available online: http://firebrowse.org/ (accessed on 8 October 2021).
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics-Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 15 May 2019).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.I.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Bjerregaard, H.; Pedersen, S.; Kristensen, S.R.; Marcussen, N. Reference Genes for Gene Expression Analysis by Real-time Reverse Transcription Polymerase Chain Reaction of Renal Cell Carcinoma. Diagn. Mol. Pathol. 2011, 20, 212–217. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Characteristics | Discovery Phase Hospital Clinic Barcelona (n = 13) | Validation Phase Institute Valenciano of Oncology (n = 64) |
|---|---|---|
| Gender | ||
| Male | 9 (69.2) | 49 (76.6) |
| Female | 4 (30.8) | 15 (23.4) |
| Age at diagnosis (yr) | 54.85 (36–81) | 58.6 (35–87) |
| Pathological tumor size (cm) | 8.2 (2.5–14) | 8 (3.1–24) |
| ISUP | ||
| ISUP 1 | - | 4 (6.3) |
| ISUP 2 | 2 (15.4) | 20 (31.3) |
| ISUP 3 | 6 (46.2) | 31 (48.4) |
| ISUP 4 | 5 (38.4) | 9 (14) |
| Tumor stage | ||
| pT1 | 5 (38.4) | 9 (14) |
| pT2 | 5 (38.4) | 16 (25) |
| pT3 | 2 (15.4) | 36 (56.3) |
| pT4 | 1 (7.8) | 3 (4.7) |
| N stage | ||
| N0/x | 11 (84.6) | 58 (90.6) |
| N1 | 2 (15.4) | 6 (9.4) |
| Perirenal fat invasion | 3 (23.1) | 40 (62.5) |
| Vascular invasion | 2 (15.4) | 10 (15.6) |
| Necrosis | 1 (7.8) | 24 (37.5) |
| SSIGN score | ||
| Intermediate risk | 7 (53.8) | 39 (60.9) |
| High risk | 6 (46.2) | 25 (39.1) |
| Genes | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| p | 95% CI | HR | p | 95% CI | HR | |
| DUOX1 | <0.001 | 1.404–2.751 | 1.965 | |||
| HS6ST2 | 0.001 | 2.084–14.157 | 5.432 | <0.001 | 2.710–14.880 | 6.35 |
| KRT20 | <0.001 | 1.419–2.926 | 2.037 | |||
| RCOR2 | <0.001 | 2.417–16.627 | 6.340 | |||
| SFN | <0.001 | 1.806–5.647 | 3.194 | |||
| SSC4D | 0.001 | 1.982–14.831 | 5.422 | |||
| WNT9A | 0.002 | 1.600–7.475 | 3.459 | |||
| pT Stage | <0.001 | 1.610–5.362 | 2.939 | 0.016 | 1.150–4.090 | 2.17 |
| Tumor size | <0.001 | 1.068–1.246 | 1.154 | 0.018 | 1.020–1.230 | 1.12 |
| ISUP | 0.045 | 1.010–2.533 | 1.599 | 0.021 | 1.100–3.370 | 1.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roldán, F.L.; Lozano, J.J.; Ingelmo-Torres, M.; Carrasco, R.; Díaz, E.; Ramirez-Backhaus, M.; Rubio, J.; Reig, O.; Alcaraz, A.; Mengual, L.; et al. Clinicopathological and Molecular Prognostic Classifier for Intermediate/High-Risk Clear Cell Renal Cell Carcinoma. Cancers 2021, 13, 6338. https://doi.org/10.3390/cancers13246338
Roldán FL, Lozano JJ, Ingelmo-Torres M, Carrasco R, Díaz E, Ramirez-Backhaus M, Rubio J, Reig O, Alcaraz A, Mengual L, et al. Clinicopathological and Molecular Prognostic Classifier for Intermediate/High-Risk Clear Cell Renal Cell Carcinoma. Cancers. 2021; 13(24):6338. https://doi.org/10.3390/cancers13246338
Chicago/Turabian StyleRoldán, Fiorella L., Juan J. Lozano, Mercedes Ingelmo-Torres, Raquel Carrasco, Esther Díaz, Miguel Ramirez-Backhaus, José Rubio, Oscar Reig, Antonio Alcaraz, Lourdes Mengual, and et al. 2021. "Clinicopathological and Molecular Prognostic Classifier for Intermediate/High-Risk Clear Cell Renal Cell Carcinoma" Cancers 13, no. 24: 6338. https://doi.org/10.3390/cancers13246338
APA StyleRoldán, F. L., Lozano, J. J., Ingelmo-Torres, M., Carrasco, R., Díaz, E., Ramirez-Backhaus, M., Rubio, J., Reig, O., Alcaraz, A., Mengual, L., & Izquierdo, L. (2021). Clinicopathological and Molecular Prognostic Classifier for Intermediate/High-Risk Clear Cell Renal Cell Carcinoma. Cancers, 13(24), 6338. https://doi.org/10.3390/cancers13246338