
ROR2 Is Epigenetically Regulated in Endometrial Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
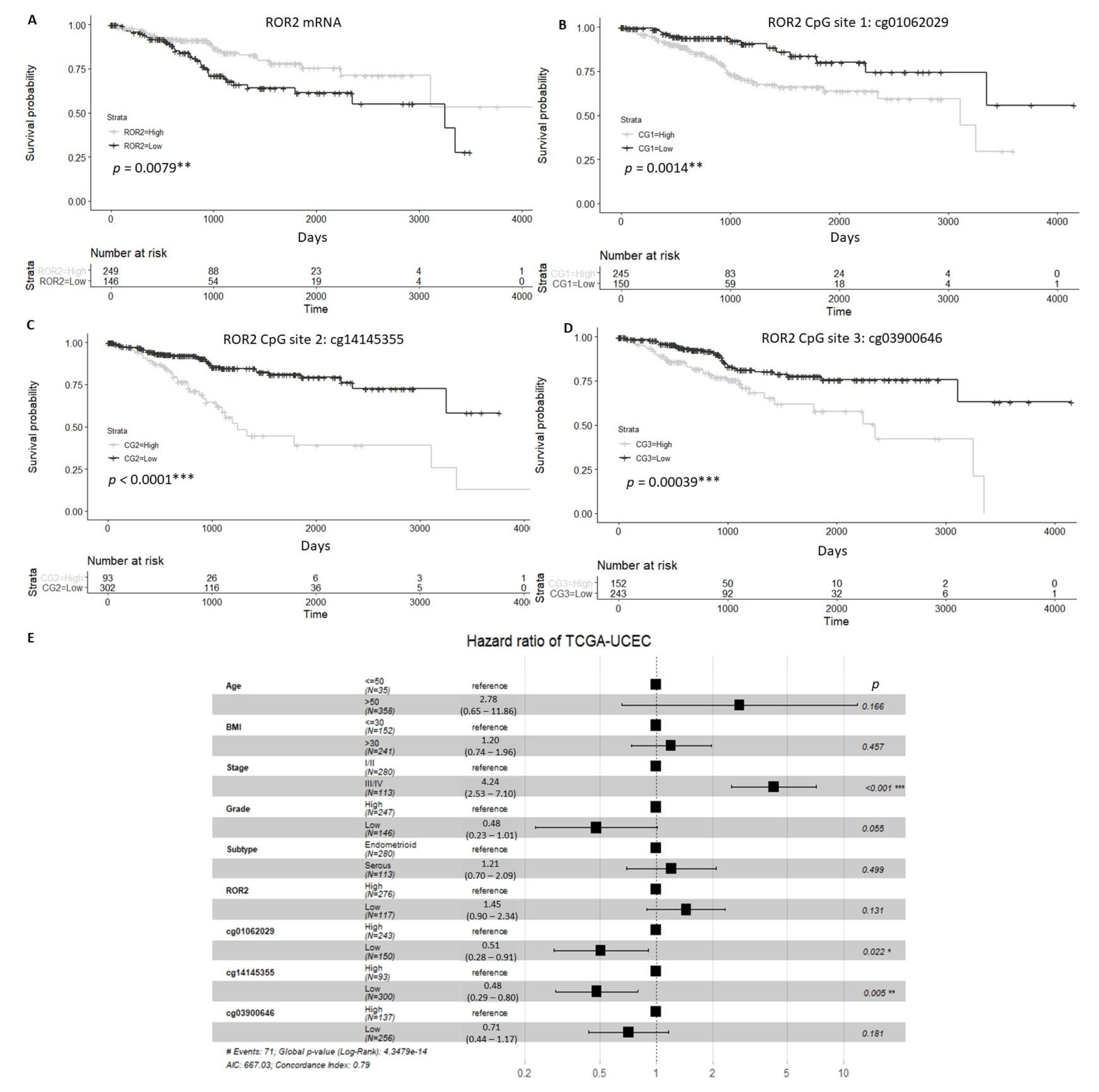
2.1. Expression and Methylation Status of ROR2 is Associated with Overall Survival in an EC Cohort
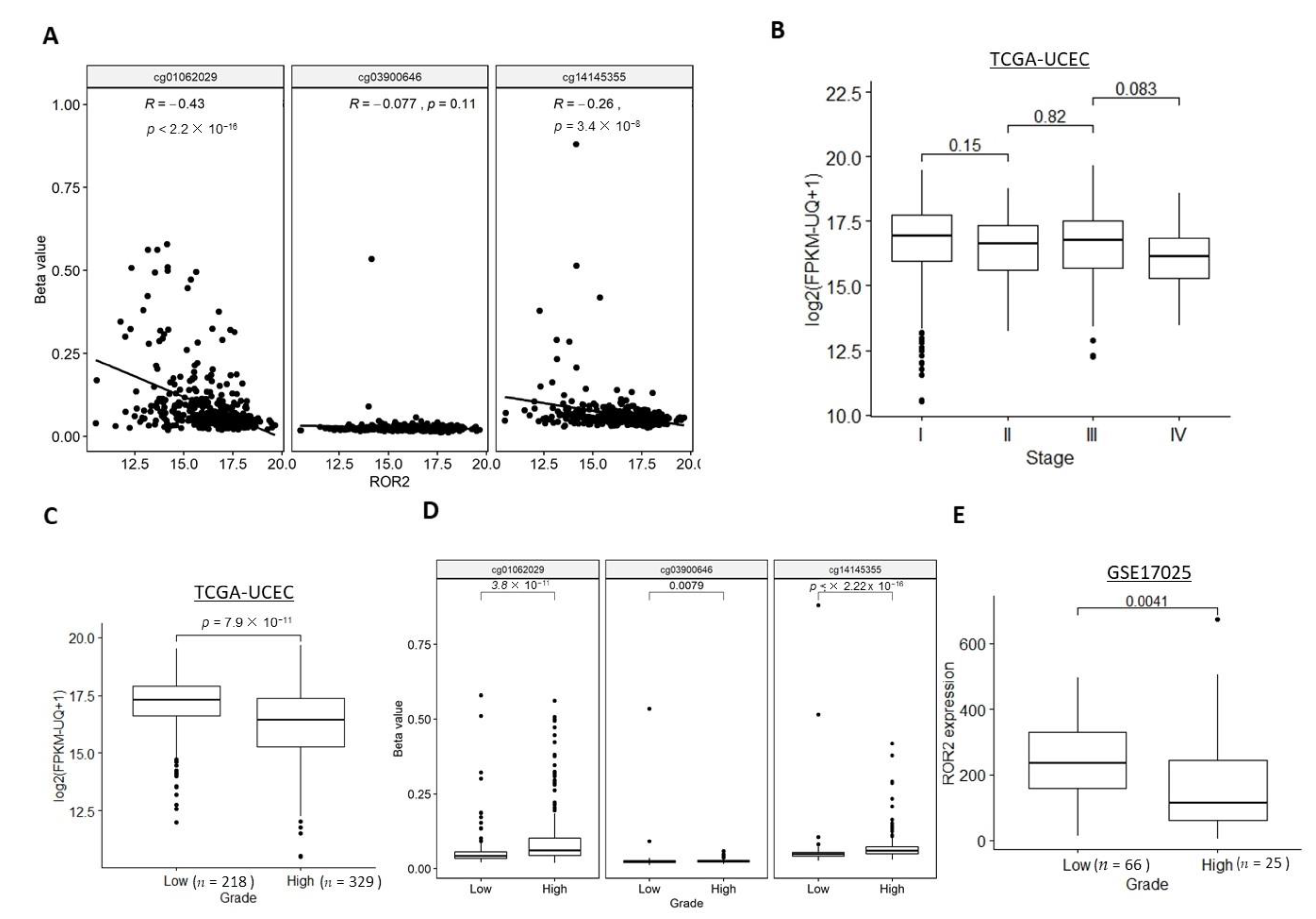
2.2. ROR2 Was Epigenetically Suppressed in High-Grade EC in Public Datasets
2.3. ROR2 Was Epigenetically Suppressed in Serous EC in Public Datasets
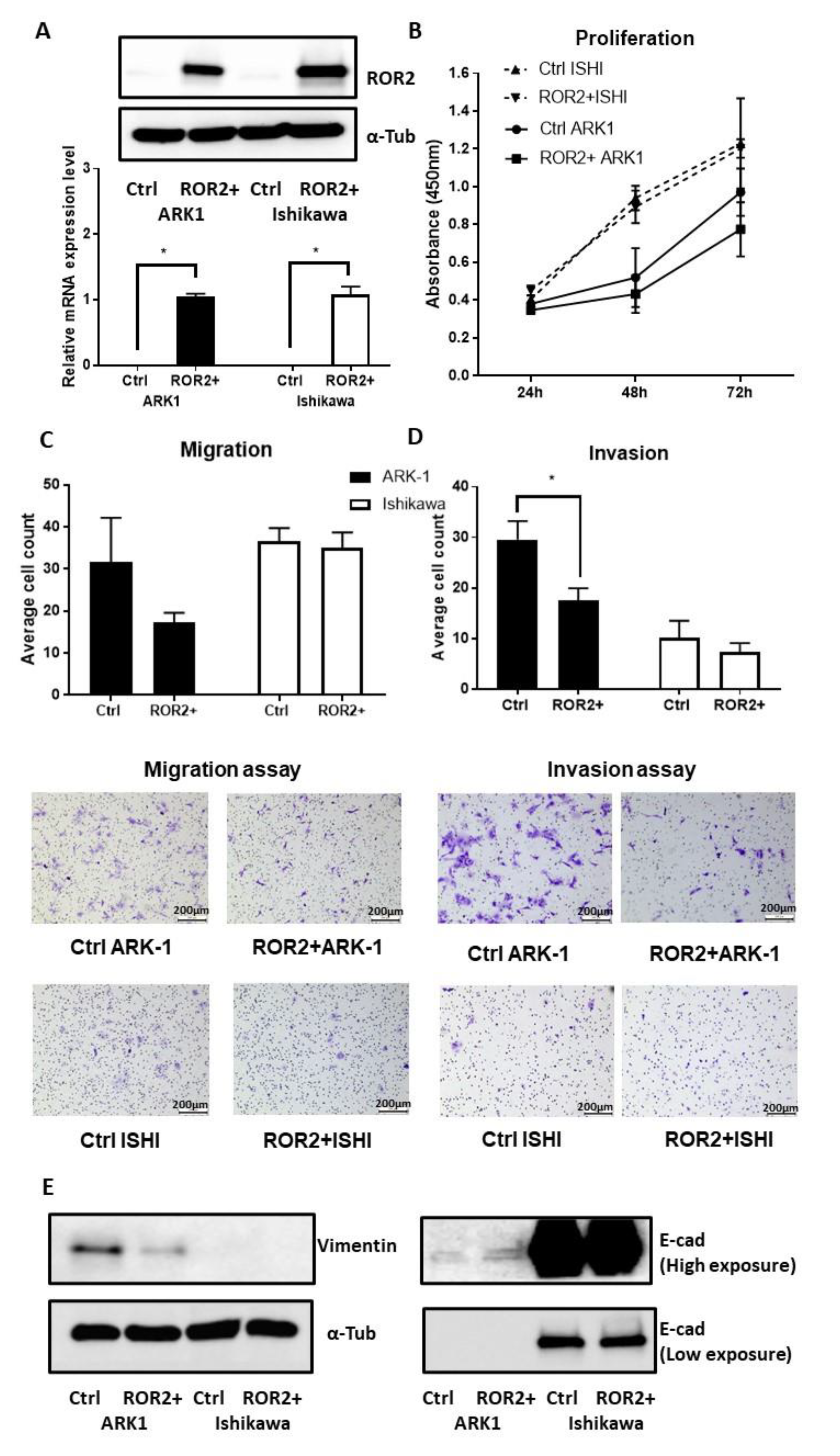
2.4. Expression of ROR2 was Regulated by Promoter Methylation in EC Cell Lines and Patient Samples
2.5. ROR2 Overexpression Inhibited Cell Invasion in ARK-1
3. Discussion
4. Materials and Methods
4.1. TCGA-UCEC Cohort
4.2. GEO Dataset
4.3. Cell Culture
4.4. Patient Samples
4.5. DNA and RNA Extraction
4.6. Combined Bisulphite Restriction Assay (COBRA)
4.7. Bisulphite Sequencing
4.8. qRTPCR
4.9. Western Blot
4.10. ROR2 Transfection
4.11. Proliferation Assay
4.12. Migration and Invasion Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Levine, D.A. Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Astec, T. Adjuvant external beam radiotherapy in the treatment of endometrial cancer (MRC ASTEC and NCIC CTG EN. 5 randomised trials): Pooled trial results, systematic review, and meta-analysis. Lancet 2009, 373, 137–146. [Google Scholar]
- León-Castillo, A.; De Boer, S.M.; Powell, M.E.; Mileshkin, L.R.; Mackay, H.J.; Leary, A.; Nijman, H.W.; Singh, N.; Pollock, P.M.; Bessette, P.; et al. Molecular Classification of the PORTEC-3 Trial for High-Risk Endometrial Cancer: Impact on Prognosis and Benefit From Adjuvant Therapy. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Noone, A.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D. SEER Cancer Statistics Review, 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2018. [Google Scholar]
- Hamilton, C.; Cheung, M.K.; Osann, K.; Chen, L.; Teng, N.N.; Longacre, T.; Powell, M.; Hendrickson, M.R.; Kapp, D.S.; Chan, J.K. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br. J. Cancer 2006, 94, 642–646. [Google Scholar] [CrossRef]
- Clarke, M.A.; Devesa, S.S.; Harvey, S.V.; Wentzensen, N. Hysterectomy-Corrected Uterine Corpus Cancer Incidence Trends and Differences in Relative Survival Reveal Racial Disparities and Rising Rates of Nonendometrioid Cancers. J. Clin. Oncol. 2019, 37, 1895–1908. [Google Scholar] [CrossRef]
- Wang, Y.; Hanifi-Moghaddam, P.; Hanekamp, E.E.; Kloosterboer, H.J.; Franken, P.; Veldscholte, J.; Van Doorn, H.C.; Ewing, P.C.; Kim, J.J.; Grootegoed, J.A.; et al. Progesterone Inhibition of Wnt/β-Catenin Signaling in Normal Endometrium and Endometrial Cancer. Clin. Cancer Res. 2009, 15, 5784–5793. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Mak, C.S.; Leung, T.H.; Chan, K.K.L.; Ngan, H.Y.-S. Down-regulation of Sox7 is associated with aberrant activation of Wnt/β-catenin signaling in endometrial cancer. Oncotarget 2012, 3, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Van Der Zee, M.; Jia, Y.; Wang, Y.; Heijmans-Antonissen, C.; Ewing, P.C.; Franken, P.; DeMayo, F.J.; Lydon, J.P.; Burger, C.W.; Fodde, R.; et al. Alterations in Wnt-β -catenin and Pten signalling play distinct roles in endometrial cancer initiation and progression. J. Pathol. 2013, 230, 48–58. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Hardisson, D.; Sanchez, C.; Sarrio, D.; Cassia, R.; García-Rostán, G.; Prat, J.; Guo, M.; Herman, J.G.; Matías-Guiu, X. Abnormalities of the APC/β-catenin pathway in endometrial cancer. Oncogene 2002, 21, 7981–7990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Yoshinaga, K.; Semba, S.; Kondo, E.; Ohmori, H.; Horii, A. Mutational analysis of the CTNNB1 (beta-catenin) gene in human endometrial cancer: Frequent mutations at codon 34 that cause nuclear accumulation. Oncol. Rep. 2000, 7, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, T.; Sakamoto, M.; Tsuda, H.; Maruyama, K.; Nozawa, S.; Hirohashi, S. β-Catenin mutation in carcinoma of the uterine endometrium. Cancer Res. 1998, 58, 3526–3528. [Google Scholar] [PubMed]
- Henry, C.; Llamosas, E.; Knipprath-Meszaros, A.; Schoetzau, A.; Obermann, E.; Fuenfschilling, M.; Caduff, R.; Fink, D.; Hacker, N.; Ward, R.; et al. Targeting the ROR1 and ROR2 receptors in epithelial ovarian cancer inhibits cell migration and invasion. Oncotarget 2015, 6, 40310–40326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.; Llamosas, E.; Daniels, B.; Coopes, A.; Tang, K.; Ford, C. ROR1 and ROR2 play distinct and opposing roles in endometrial cancer. Gynecol. Oncol. 2018, 148, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Schambony, A.; Wedlich, D. Wnt-5A/Ror2 Regulate Expression of XPAPC through an Alternative Noncanonical Signaling Pathway. Dev. Cell 2007, 12, 779–792. [Google Scholar] [CrossRef] [Green Version]
- Nomachi, A.; Nishita, M.; Inaba, D.; Enomoto, M.; Hamasaki, M.; Minami, Y. Receptor Tyrosine Kinase Ror2 Mediates Wnt5a-induced Polarized Cell Migration by Activating c-Jun N-terminal Kinase via Actin-binding Protein Filamin A. J. Biol. Chem. 2008, 283, 27973–27981. [Google Scholar] [CrossRef] [Green Version]
- Mikels, A.; Minami, Y.; Nusse, R. Ror2 Receptor Requires Tyrosine Kinase Activity to Mediate Wnt5A Signaling. J. Biol. Chem. 2009, 284, 30167–30176. [Google Scholar] [CrossRef] [Green Version]
- Billiard, J.; Way, D.S.; Seestaller-Wehr, L.M.; Moran, R.A.; Mangine, A.; Bodine, P.V.N. The Orphan Receptor Tyrosine Kinase Ror2 Modulates Canonical Wnt Signaling in Osteoblastic Cells. Mol. Endocrinol. 2005, 19, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Kani, S.; Oishi, I.; Yamamoto, H.; Yoda, A.; Suzuki, H.; Nomachi, A.; Iozumi, K.; Nishita, M.; Kikuchi, A.; Takumi, T.; et al. The Receptor Tyrosine Kinase Ror2 Associates with and Is Activated by Casein Kinase Iϵ. J. Biol. Chem. 2004, 279, 50102–50109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Ford, C.; Ma, S.S.Q.; Quadir, A.; Ward, R.L. The dual role of the novel Wnt receptor tyrosine kinase, ROR2, in human carcinogenesis. Int. J. Cancer 2013, 133, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Ye, X.; Lin, L.; Shen, M.; Jiang, T. Up-regulation of ROR2 is associated with unfavorable prognosis and tumor progression in cervical cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 856–861. [Google Scholar] [PubMed]
- Lara, E.; Calvanese, V.; Huidobro, C.; Fernandez, A.F.; Moncada-Pazos, A.; Obaya, A.J.; Aguilera, O.; González-Sancho, J.M.; Sanchez, L.; Astudillo, A.; et al. Epigenetic repression of ROR2 has a Wnt-mediated, pro-tumourigenic role in colon cancer. Mol. Cancer 2010, 9, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.S.Q.; Srivastava, S.; Llamosas, E.; Hawkins, N.J.; Hesson, L.B.; Ward, R.L.; Ford, C.E. ROR2 is epigenetically inactivated in the early stages of colorectal neoplasia and is associated with proliferation and migration. BMC Cancer 2016, 16, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ying, J.; Tong, X.; Zhong, L.; Su, X.; Xiang, T.; Shu, X.; Rong, R.; Xiong, L.; Li, H.; et al. Epigenetic identification of receptor tyrosine kinase-like orphan receptor 2 as a functional tumor suppressor inhibiting β-catenin and AKT signaling but frequently methylated in common carcinomas. Cell. Mol. Life Sci. 2013, 71, 2179–2192. [Google Scholar] [CrossRef]
- Ma, S.S.; Henry, C.; Llamosas, E.; Higgins, R.; Daniels, B.; Hesson, L.B.; Hawkins, N.J.; Ward, R.L.; E Ford, C. Validation of specificity of antibodies for immunohistochemistry: The case of ROR2. Virchows Arch. 2017, 470, 99–108. [Google Scholar] [CrossRef]
- Sheetz, J.B.; Mathea, S.; Karvonen, H.; Malhotra, K.; Chatterjee, D.; Niininen, W.; Perttilä, R.; Preuss, F.; Suresh, K.; Stayrook, S.E.; et al. Structural Insights into Pseudokinase Domains of Receptor Tyrosine Kinases. Mol. Cell 2020, 79, 390–405.e7. [Google Scholar] [CrossRef]
- Nishida, M.; Kasahara, K.; Kaneko, M.; Iwasaki, H.; Hayashi, K. Establishment of a new human endometrial adenocarcinoma cell line, Ishikawa cells, containing estrogen and progesterone receptors. Nihon Sanka Fujinka Gakkai Zasshi 1985, 37, 1103–1111. [Google Scholar]
- Nishida, M. The Ishikawa cells from birth to the present. Hum. Cell 2002, 15, 104–117. [Google Scholar] [CrossRef]
- Hackenberg, R.; Hawighorst, T.; Hild, F.; Schulz, K.-D. Establishment of new epithelial carcinoma cell lines by blocking monolayer formation. J. Cancer Res. Clin. Oncol. 1997, 123, 669–673. [Google Scholar] [CrossRef]
- Richardson, G.S.; Dickersin, G.; Atkins, L.; MacLaughlin, D.T.; Raam, S.; Merk, L.P.; Bradley, F.M. KLE: A cell line with defective estrogen receptor derived from undifferentiated endometrial cancer. Gynecol. Oncol. 1984, 17, 213–230. [Google Scholar] [CrossRef]
- Van Nyen, T.; Moiola, C.P.; Colas, E.; Annibali, D.; Amant, F. Modeling Endometrial Cancer: Past, Present, and Future. Int. J. Mol. Sci. 2018, 19, 2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Jordan, V.C.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, W.; Zhao, Y.; Wang, X.; Qi, Y.; Zhou, C.; Hua, Y.; Hou, J.; Jiang, S.-W. (Albert) Culture characters, genetic background, estrogen/progesterone receptor expression, and tumorigenic activities of frequently used sixteen endometrial cancer cell lines. Clin. Chim. Acta 2019, 489, 225–232. [Google Scholar] [CrossRef]
- Santin, A.; Rose, G.; Hiserodt, J.; Fruehauf, J.; Eck, L.; Garcia, R.; Schranz, V.; Disaia, P.; Pecorelli, S.; Granger, G. Effects of tumor necrosis factor-a plus interferon-y combined with high dose y irradiation on the expression of major histocompatibility complex molecules and intercellular adhesion molecule-1 in human ovarian cancers. Int. J. Cancer 1996, 65, 688–694. [Google Scholar] [CrossRef]
- McConechy, M.K.; Hoang, L.N.; Chui, M.H.; Senz, J.; Yang, W.; Rozenberg, N.; MacKenzie, R.; McAlpine, J.N.; Huntsman, D.G.; Clarke, B.A.; et al. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J. Pathol. Clin. Res. 2015, 1, 173–185. [Google Scholar] [CrossRef]
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R. In-tegrated molecular characterization of uterine carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Gotzmann, J.; Mikula, M.; Eger, A.; Schulte-Hermann, R.; Foisner, R.; Beug, H.; Mikulits, W. Molecular aspects of epithelial cell plasticity: Implications for local tumor invasion and metastasis. Mutat. Res. Mutat. Res. 2004, 566, 9–20. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Enomoto, M.; Hayakawa, S.; Itsukushima, S.; Ren, D.Y.; Matsuo, M.; Tamada, K.; Oneyama, C.; Okada, M.; Takumi, T.; Nishita, M.; et al. Autonomous regulation of osteosarcoma cell invasiveness by Wnt5a/Ror2 signaling. Oncogene 2009, 28, 3197–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.-L.; Su, P.-H.; Liao, Y.-P.; Wu, T.-I.; Hsu, Y.-T.; Lin, W.-Y.; Wang, H.-C.; Weng, Y.-C.; Ou, Y.-C.; Huang, T.H.-M.; et al. Integrated Epigenomics Analysis Reveals a DNA Methylation Panel for Endometrial Cancer Detection Using Cervical Scrapings. Clin. Cancer Res. 2017, 23, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, Y.J.; Verset, G.; Schats, K.; Van Dam, P.-J.; Seremet, T.; Kockx, M.; Van Laethem, J.-L.B.; Neyns, B. Phase I clinical trial of decitabine (5-aza-2’-deoxycytidine) administered by hepatic arterial infusion in patients with unresectable liver-predominant metastases. ESMO Open 2019, 4, e000464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinardo, C.D.; Stein, A.S.; Stein, E.M.; Fathi, A.T.; Schuh, A.C.; Fernández, P.M.; Odenike, O.; Kantarjian, H.; Stone, R.M.; Collins, R.; et al. Mutant IDH (mIDH) inhibitors, ivosidenib or enasidenib, with azacitidine (AZA) in patients with acute myeloid leukemia (AML). J. Clin. Oncol. 2018, 36, 7042. [Google Scholar] [CrossRef]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Cortes, J.; Ravandi, F.; Jabbour, E.; Hendrickson, S.; Brandt, M.; Pierce, S.; Gordon, T.; et al. Phase IB/II study of nivolumab with azacytidine (AZA) in patients (pts) with relapsed AML. J. Clin. Oncol. 2017, 35, 7026. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Fram, R.J.; Hua, Z.; Ades, L. Phase 3 Study of First Line Pevonedistat (PEV)+ Azacitidine (AZA) Versus Single-Agent AZA in Patients with Higher-Risk Myelodysplastic Syndromes (HR MDS), Chronic Myelomonocytic Leukemia (CMML) or low-Blast Acute Mye-Logenous Leukemia (AML); American Society of Clinical Oncology: Alexandria, VA, USA, 2018. [Google Scholar]
- Liu, D.; Gunther, K.; Enriquez, L.A.; Daniels, B.; O’Mara, T.A.; Tang, K.; Spurdle, A.B.; Ford, C.E. ROR1 is upregulated in endometrial cancer and represents a novel therapeutic target. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Kondo, E.; Furukawa, T.; Yoshinaga, K.; Kijima, H.; Semba, S.; Yatsuoka, T.; Yokoyama, T.; Fukushige, S.; Horii, A. Not hMSH2 but hMLH1 is frequently silenced by hypermethylation in endometrial cancer but rarely silenced in pancreatic cancer with microsatellite instability. Int. J. Oncol. 2000, 17, 535–576. [Google Scholar] [CrossRef]
- Salvesen, H.B.; Macdonald, N.; Ryan, A.; Jacobs, I.J.; Lynch, E.D.; Akslen, L.A.; Das, S. PTEN methylation is associated with advanced stage and microsatellite instability in endometrial carcinoma. Int. J. Cancer 2000, 91, 22–26. [Google Scholar] [CrossRef]
- Xiong, Y.; Dowdy, S.C.; Xue, A.; Shujuan, J.; Eberhardt, N.L.; Podratz, K.C.; Jiang, S.-W. Opposite alterations of DNA methyl-transferase gene expression in endometrioid and serous endometrial cancers. Gynecol. Oncol. 2005, 96, 601–609. [Google Scholar] [CrossRef]
- Plumb, J.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2’-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044. [Google Scholar]
- Goldman, M.; Craft, B.; Brooks, A.; Zhu, J.; Haussler, D. The UCSC Xena Platform for cancer genomics data visualization and interpretation. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Day, R.S.; McDade, K.K.; Chandran, U.R.; Lisovich, A.; Conrads, T.P.; Hood, B.L.; Kolli, V.K.; Kirchner, D.; Litzi, T.; Maxwell, G.L. Identifier mapping performance for integrating transcriptomics and proteomics experimental results. BMC Bioinform. 2011, 12, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Histo Type | Stage | Grade |
|---|---|---|---|
| HSA2405 | Serous | III | 3 |
| HSA3180 | Endometrioid | IIIA | 1 |
| HSA0105 | Endometrioid | IIIA | 2 |
| HSA0131 | MMMT * | IB | 3 |
| HSA0423 | Endometrioid | IV | 1 |
| HSA3081 | Endometrioid | IA | 3 |
| HSA1571 | Endometrioid | IA | 3 |
| HSA0114 | Endometrioid | IA | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Enriquez, L.; Ford, C.E. ROR2 Is Epigenetically Regulated in Endometrial Cancer. Cancers 2021, 13, 383. https://doi.org/10.3390/cancers13030383
Liu D, Enriquez L, Ford CE. ROR2 Is Epigenetically Regulated in Endometrial Cancer. Cancers. 2021; 13(3):383. https://doi.org/10.3390/cancers13030383
Chicago/Turabian StyleLiu, Dongli, Luis Enriquez, and Caroline E. Ford. 2021. "ROR2 Is Epigenetically Regulated in Endometrial Cancer" Cancers 13, no. 3: 383. https://doi.org/10.3390/cancers13030383
APA StyleLiu, D., Enriquez, L., & Ford, C. E. (2021). ROR2 Is Epigenetically Regulated in Endometrial Cancer. Cancers, 13(3), 383. https://doi.org/10.3390/cancers13030383