
Histone Modifying Enzymes in Gynaecological Cancers

 , ,
, , 

Abstract
:Simple Summary
Abstract
1. Background
1.1. DNA Methylation
1.2. Histone Modifications
1.3. Enzymes Involved in Epigenetic Regulation
2. Epigenetic Modifiers in Ovarian Cancer
2.1. Histone Methyltransferases in Ovarian Cancer
2.2. Histone Demethylases in Ovarian Cancer
2.3. HDACs in Ovarian Cancer
3. Epigenetic Modifiers in Endometrial Cancer
3.1. Histone Methyltransferases in Endometrial Cancer
3.2. Histone Demethylases in Endometrial Cancer
3.3. HDACs in Endometrial Cancer
4. Epigenetic Modifiers in Cervical Cancer
5. Epigenetic Treatments in Gynaecological Cancers
5.1. HDAC Inhibitors
5.2. Histone Methyltransferase and Demethylase Inhibitors
5.3. Targeting “Readers” of Histone Modifications
5.4. Combination Therapy
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, Q.W.; Zhu, X.Y.; Li, Y.Y.; Meng, Z.Q. Epigenetic regulation and cancer (review). Oncol. Rep. 2014, 31, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lujambio, A.; Esteller, M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle 2007, 6, 1455–1459. [Google Scholar] [CrossRef]
- Kim, T.H.; Barrera, L.O.; Zheng, M.; Qu, C.; Singer, M.A.; Richmond, T.A.; Wu, Y.; Green, R.D.; Ren, B. A high-resolution map of active promoters in the human genome. Nature 2005, 436, 876–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, F.F. Epigenomics in cancer management. Cancer Manag. Res. 2010, 2, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J. Action at a distance: Epigenetic silencing of large chromosomal regions in carcinogenesis. Hum. Mol. Genet. 2007, 16, R88–R95. [Google Scholar] [CrossRef] [Green Version]
- Krusche, C.A.; Vloet, A.J.; Classen-Linke, I.; von Rango, U.; Beier, H.M.; Alfer, J. Class I histone deacetylase expression in the human cyclic endometrium and endometrial adenocarcinomas. Hum. Reprod. 2007, 22, 2956–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, H.; Zhao, J.; Vieth, E.; Nephew, K.P.; Matei, D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget 2016, 7, 84453. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, S.M.; Chen, M.W.; Chen, C.A.; Chien, M.H.; Hua, K.T.; Hsiao, M.; Kuo, M.L.; Wei, L.H. The H3K9 Methyltransferase G9a Represses E-cadherin and is Associated with Myometrial Invasion in Endometrial Cancer. Ann. Surg. Oncol. 2015, 22 (Suppl 3), S1556–S1565. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, H.; Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Suzuki, S.; Seino, S.; Seino, M.; Ohta, T.; Nagase, S.; et al. GSKJ4, A Selective Jumonji H3K27 Demethylase Inhibitor, Effectively Targets Ovarian Cancer Stem Cells. Anticancer Res. 2015, 35, 6607–6614. [Google Scholar]
- Hua, K.-T.; Wang, M.-Y.; Chen, M.-W.; Wei, L.-H.; Chen, C.-K.; Ko, C.-H.; Jeng, Y.-M.; Sung, P.-L.; Jan, Y.-H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef] [Green Version]
- Shao, G.; Wang, J.; Li, Y.; Liu, X.; Xie, X.; Wan, X.; Yan, M.; Jin, J.; Lin, Q.; Zhu, H.; et al. Lysine-specific demethylase 1 mediates epidermal growth factor signaling to promote cell migration in ovarian cancer cells. Sci. Rep. 2015, 5, 15344. [Google Scholar] [CrossRef] [Green Version]
- Theisen, E.R.; Gajiwala, S.; Bearss, J.; Sorna, V.; Sharma, S.; Janat-Amsbury, M. Reversible inhibition of lysine specific demethylase 1 is a novel anti-tumor strategy for poorly differentiated endometrial carcinoma. BMC Cancer 2014, 14, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.C.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.Z.; Luo, X.G.; Shen, J.; Zou, J.N.; Lu, Y.H.; Xi, T. Knockdown of SMYD3 by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep. 2008, 41, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Xu, B.; Jiang, D.; Huang, S.; Yu, H.; Wu, Z.; Wu, Q. Depletion of H3K79 methyltransferase Dot1L promotes cell invasion and cancer stem-like cell property in ovarian cancer. Am. J. Transl. Res. 2019, 11, 1145–1153. [Google Scholar] [PubMed]
- Dickson, K.A.; Cole, A.J.; Gill, A.J.; Clarkson, A.; Gard, G.B.; Chou, A.; Kennedy, C.J.; Henderson, B.R.; Fereday, S.; Traficante, N.; et al. The RING finger domain E3 ubiquitin ligases BRCA1 and the RNF20/RNF40 complex in global loss of the chromatin mark histone H2B monoubiquitination (H2Bub1) in cell line models and primary high-grade serous ovarian cancer. Hum. Mol. Genet. 2016, 25, 5460–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarcic, O.; Pateras, I.S.; Cooks, T.; Shema, E.; Kanterman, J.; Ashkenazi, H.; Boocholez, H.; Hubert, A.; Rotkopf, R.; Baniyash, M.; et al. RNF20 Links Histone H2B Ubiquitylation with Inflammation and Inflammation-Associated Cancer. Cell Rep. 2016, 14, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Black, J.C.; Manning, A.L.; Van Rechem, C.; Kim, J.; Ladd, B.; Cho, J.; Pineda, C.M.; Murphy, N.; Daniels, D.L.; Montagna, C.; et al. KDM4A Lysine Demethylase Induces Site-Specific Copy Gain and Rereplication of Regions Amplified in Tumors. Cell 2013, 154, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Ramadoss, S.; Sen, S.; Ramachandran, I.; Roy, S.; Chaudhuri, G.; Farias-Eisner, R. Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene 2016, 36, 6508. [Google Scholar] [CrossRef] [Green Version]
- Sone, K.; Piao, L.; Nakakido, M.; Ueda, K.; Jenuwein, T.; Nakamura, Y.; Hamamoto, R. Critical role of lysine 134 methylation on histone H2AX for γ-H2AX production and DNA repair. Nat. Commun. 2014, 5, 5691. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.-T.; Fan, Q.; Zhu, Z.; Kwan, S.-Y.; Chen, L.; Chen, J.-H.; Ying, Z.-L.; Zhou, Y.; Gu, W.; Wang, L.-H.; et al. KDM4B and KDM4A promote endometrial cancer progression by regulating androgen receptor, c-myc, and p27(kip1). Oncotarget 2015, 6, 31702–31720. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Zhang, J.; Cai, H.; Li, Y.; Zhang, Y.; Zhang, X.; Zhao, D.; Li, Z.; Ma, H.; Wang, J.; et al. HDAC as a therapeutic target for treatment of endometrial cancers. Curr. Pharm. Des. 2014, 20, 1847–1856. [Google Scholar] [CrossRef]
- Yano, M.; Yasuda, M.; Sakaki, M.; Nagata, K.; Fujino, T.; Arai, E.; Hasebe, T.; Miyazawa, M.; Miyazawa, M.; Ogane, N.; et al. Association of histone deacetylase expression with histology and prognosis of ovarian cancer. Oncol Lett 2018, 15, 3524–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Tian, G.; Murph, M.M. Molecular epigenetics in the management of ovarian cancer: Are we investigating a rational clinical promise? Front. Oncol. 2014, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Ma, P.; Jing, Y.; Yan, Y.; Cai, M.-C.; Zhang, M.; Zhang, S.; Peng, H.; Ji, Z.-L.; Di, W.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating FoxM1. Theranostics 2016, 6, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 2014, 53, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casciello, F.; Al-Ejeh, F.; Kelly, G.; Brennan, D.J.; Ngiow, S.F.; Young, A.; Stoll, T.; Windloch, K.; Hill, M.M.; Smyth, M.J.; et al. G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 7077–7082. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA A Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Bodelon, C.; Killian, J.K.; Sampson, J.N.; Anderson, W.F.; Matsuno, R.; Brinton, L.A.; Lissowska, J.; Anglesio, M.S.; Bowtell, D.D.L.; Doherty, J.A.; et al. Molecular Classification of Epithelial Ovarian Cancer Based on Methylation Profiling: Evidence for Survival Heterogeneity. Clin. Cancer Res. 2019, 25, 5937–5946. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489. [Google Scholar] [CrossRef]
- Fukasawa, K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005, 230, 6–19. [Google Scholar] [CrossRef]
- Saleemuddin, A.; Folkins, A.K.; Garrett, L.; Garber, J.; Muto, M.G.; Crum, C.P.; Tworoger, S. Risk factors for a serous cancer precursor ("p53 signature") in women with inherited BRCA mutations. Gynecol. Oncol. 2008, 111, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, J.F.; Gayther, S.A.; Russell, P.; Dearden, J.; Gore, M.; Blake, P.; Easton, D.; Ponder, B.A. Contribution of BRCA1 mutations to ovarian cancer. N. Engl. J. Med. 1997, 336, 1125–1130. [Google Scholar] [CrossRef]
- Ruscito, I.; Dimitrova, D.; Vasconcelos, I.; Gellhaus, K.; Schwachula, T.; Bellati, F.; Zeillinger, R.; Benedetti-Panici, P.; Vergote, I.; Mahner, S.; et al. BRCA1 gene promoter methylation status in high-grade serous ovarian cancer patients—A study of the tumour Bank ovarian cancer (TOC) and ovarian cancer diagnosis consortium (OVCAD). Eur. J. Cancer 2014, 50, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L.; Nemeth, E.; Tran, H.; Shvartsman, H.; Cass, I.; Narod, S.; Karlan, B.Y. BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res. 2000, 60, 5329–5333. [Google Scholar]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef]
- Yi, M.; Dong, B.; Qin, S.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, R.W.; Coleman, R.L.; Brown, J.; Moore, K.N. Phase III trials in ovarian cancer: The evolving landscape of front line therapy. Gynecol. Oncol. 2019, 153, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Ruscito, I.; Bellati, F.; Ray-Coquard, I.; Mirza, M.R.; du Bois, A.; Gasparri, M.L.; Costanzi, F.; De Marco, M.P.; Nuti, M.; Caserta, D.; et al. Incorporating Parp-inhibitors in Primary and Recurrent Ovarian Cancer: A Meta-analysis of 12 phase II/III randomized controlled trials. Cancer Treat. Rev. 2020, 87, 102040. [Google Scholar] [CrossRef]
- Shash, E.; Peccatori, F.A.; Azim, H.A., Jr. Optimizing the use of epidermal growth factor receptor inhibitors in advanced non-small-lung cancer (NSCLC). J. Thorac. Dis. 2011, 3, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Cai, Q.; Godwin, A.K.; Zhang, R. Enhancer of Zeste Homolog 2 Promotes the Proliferation and Invasion of Epithelial Ovarian Cancer Cells. Mol. Cancer Res. 2010, 8, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zeng, X.; Chen, S.; Ding, L.; Zhong, J.; Zhao, J.C.; Wang, L.; Sarver, A.; Koller, A.; Zhi, J.; et al. BRCA1 is a negative modulator of the PRC2 complex. EMBO J. 2013, 32, 1584–1597. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.Y.; Granowicz, E.M.; Maillard, I.; Thomas, D.; Hess, J.L. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood 2011, 117, 4759–4768. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Jiang, Q.; Lemieux, M.; Jeannotte, L.; Su, L.; Zhang, Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 2006, 8, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.J.; Wu, H.; Achille, N.J.; Reisenauer, M.R.; Chou, C.W.; Zeleznik-Le, N.J.; Hemenway, C.S.; Zhang, W. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 2010, 70, 10234–10242. [Google Scholar] [CrossRef] [Green Version]
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERα gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, eaav5590. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhang, X.-X.; Li, M.-C.; Cao, C.-H.; Wan, D.-Y.; Xi, B.-X.; Tan, J.-H.; Wang, J.; Yang, Z.-Y.; Feng, X.-X.; et al. C/EBPβ enhances platinum resistance of ovarian cancer cells by reprogramming H3K79 methylation. Nat. Commun. 2018, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, D.; Li, M.; Cao, C.; Wan, D.; Xi, B.; Li, W.; Tan, J.; Wang, J.; Wu, Z.; et al. Prognostic and therapeutic value of disruptor of telomeric silencing-1-like (DOT1L) expression in patients with ovarian cancer. J. Hematol. Oncol. 2017, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Kang, D.; Suzuki, T.; Masuda, A.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. The Histone Methyltransferase SMYD2 Methylates PARP1 and Promotes Poly(ADP-ribosyl)ation Activity in Cancer Cells. Neoplasia 2014, 16, 257–264.e252. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Allali-Hassani, A.; Antonysamy, S.; Chang, S.; Chen, L.H.; Curtis, C.; Emtage, S.; Fan, L.; Gheyi, T.; Li, F.; et al. LLY-507, a Cell-active, Potent, and Selective Inhibitor of Protein-lysine Methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Hsieh, C.Y.; Huang, K.E.; Chang, C.; Kang, H.Y. Cryptotanshinone down-regulates androgen receptor signaling by modulating lysine-specific demethylase 1 function. Int. J. Cancer 2012, 131, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer 2010, 127, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.L.; Pak, J.H.; Park, J.Y.; Choi, W.H.; Lee, J.Y.; Kim, J.H.; Nam, J.H. Expression profile of histone deacetylases 1, 2 and 3 in ovarian cancer tissues. J. Gynecol. Oncol. 2008, 19, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Denkert, C.; Noske, A.; Darb-Esfahani, S.; Dietel, M.; Kalloger, S.E.; Huntsman, D.G.; Köbel, M. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia 2008, 10, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Khabele, D.; Son, D.S.; Parl, A.K.; Goldberg, G.L.; Augenlicht, L.H.; Mariadason, J.M.; Rice, V.M. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: Implications for therapy. Cancer Biol. Ther. 2007, 6, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Garrett, L.A.; Growdon, W.B.; Rueda, B.R.; Foster, R. Influence of a novel histone deacetylase inhibitor panobinostat (LBH589) on the growth of ovarian cancer. J. Ovarian Res. 2016, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Langdon, S.P.; Tse, M.; Mullen, P.; Um, I.H.; Faratian, D.; Harrison, D.J. The role of HDAC2 in chromatin remodelling and response to chemotherapy in ovarian cancer. Oncotarget 2016, 7, 4695–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 Modulate RGS10 Expression and Decrease Ovarian Cancer Chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, G.; Wolff, J.E.; Aldoss, I.; Hu, B.; Dinh, M.; Torres, A.; Chen, X.; Rizzieri, D.; Sood, A.; Odenike, O.; et al. First-in-human study of ABBV-075 (mivebresib), a pan-inhibitor of bromodomain and extra terminal (BET) proteins, in patients (pts) with relapsed/refractory (RR) acute myeloid leukemia (AML): Preliminary data. J. Clin. Oncol. 2018, 36, 7019. [Google Scholar] [CrossRef]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2016, 36, 1707–1720. [Google Scholar] [CrossRef]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011, 31, 2723–2732. [Google Scholar]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.S.; et al. Confirmation of ProMisE: A simple, genomics-based clinical classifier for endometrial cancer. Cancer 2017, 123, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Y.; Li, J.; Cragun, J.; Hatch, K.; Chambers, S.K.; Zheng, W. Lynch syndrome related endometrial cancer: Clinical significance beyond the endometrium. J. Hematol. Oncol. 2013, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.T.; Dovey, O.M.; Lezina, L.; Luo, J.L.; Gant, T.W.; Barlev, N.; Bradley, A.; Cowley, S.M. Lysine-Specific Demethylase 1 Regulates the Embryonic Transcriptome and CoREST Stability. Mol. Cell. Biol. 2010, 30, 4851–4863. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Franco, E.L.; Duarte-Franco, E.; Ferenczy, A. Cervical cancer: Epidemiology, prevention and the role of human papillomavirus infection. CMAJ 2001, 164, 1017–1025. [Google Scholar]
- Bodily, J.M.; Mehta, K.P.M.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1 mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.S.; Rajasekaran, N.; Ju, W.; Shin, Y.K. Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer. J. Clin. Med. 2015, 4, 1126–1155. [Google Scholar] [CrossRef] [Green Version]
- Sartor, M.A.; Dolinoy, D.C.; Jones, T.R.; Colacino, J.A.; Prince, M.E.P.; Carey, T.E.; Rozek, L.S. Genome-wide methylation and expression differences in HPV(+) and HPV(−) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 2011, 6, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Salvati, A.; Gigantino, V.; Nassa, G.; Giurato, G.; Alexandrova, E.; Rizzo, F.; Tarallo, R.; Weisz, A. The Histone Methyltransferase DOT1L Is a Functional Component of Estrogen Receptor Alpha Signaling in Ovarian Cancer Cells. Cancers 2019, 11, 1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.; Atiya, H.I.; Wang, Y.; Pisanic, T.R.; Wang, T.H.; Shih, I.M.; Foy, K.K.; Frisbie, L.; Buckanovich, R.J.; Chomiak, A.A.; et al. Epigenomic Reprogramming toward Mesenchymal-Epithelial Transition in Ovarian-Cancer-Associated Mesenchymal Stem Cells Drives Metastasis. Cell Rep. 2020, 33, 108473. [Google Scholar] [CrossRef] [PubMed]
- Oki, S.; Sone, K.; Oda, K.; Hamamoto, R.; Ikemura, M.; Maeda, D.; Takeuchi, M.; Tanikawa, M.; Mori-Uchino, M.; Nagasaka, K.; et al. Oncogenic histone methyltransferase EZH2: A novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget 2017, 8, 40402–40411. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Wang, Z.; Liu, D. Interference with endogenous EZH2 reverses the chemotherapy drug resistance in cervical cancer cells partly by up-regulating Dicer expression. Tumour Biol. 2016, 37, 6359–6369. [Google Scholar] [CrossRef]
- Chen, R.J.; Shun, C.T.; Yen, M.L.; Chou, C.H.; Lin, M.C. Methyltransferase G9a promotes cervical cancer angiogenesis and decreases patient survival. Oncotarget 2017, 8, 62081–62098. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Shin, S.H.; Yoon, H.; Huh, J.; Shin, H.W.; Chun, Y.S.; Park, J.W. FIH Is an Oxygen Sensor in Ovarian Cancer for G9a/GLP-Driven Epigenetic Regulation of Metastasis-Related Genes. Cancer Res. 2018, 78, 1184–1199. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, L.; Wei, L.; Hou, Z.; Ye, W.; Huang, S. Chaetocin induces caspase-dependent apoptosis in ovarian cancer cells via the generation of reactive oxygen species. Oncol. Lett. 2019, 18, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. Human Papillomavirus E7 Oncoprotein Subverts Host Innate Immunity via SUV39H1-Mediated Epigenetic Silencing of Immune Sensor Genes. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Qiu, L.; Hong, Y.; Karnik, T.; Tadros, G.; Mau, B.; Ma, T.; Mu, Y.; New, J.; Louie, R.J.; et al. The histone demethylase KDM4B regulates peritoneal seeding of ovarian cancer. Oncogene 2017, 36, 2565–2576. [Google Scholar] [CrossRef] [Green Version]
- Soldi, R.; Ghosh Halder, T.; Weston, A.; Thode, T.; Drenner, K.; Lewis, R.; Kaadige, M.R.; Srivastava, S.; Daniel Ampanattu, S.; Rodriguez Del Villar, R.; et al. The novel reversible LSD1 inhibitor SP-2577 promotes anti-tumor immunity in SWItch/Sucrose-NonFermentable (SWI/SNF) complex mutated ovarian cancer. PLoS ONE 2020, 15, e0235705. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Stimson, L.; Wood, V.; Khan, O.; Fotheringham, S.; La Thangue, N.B. HDAC inhibitor-based therapies and haematological malignancy. Ann. Oncol. 2009, 20, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P.; Gynecologic Oncology, G. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Matulonis, U.; Berlin, S.; Lee, H.; Whalen, C.; Obermayer, E.; Penson, R.; Liu, J.; Campos, S.; Krasner, C.; Horowitz, N. Phase I study of combination of vorinostat, carboplatin, and gemcitabine in women with recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother. Pharmacol. 2015, 76, 417–423. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Wilson, A.J.; Sarfo-Kantanka, K.; Barrack, T.; Steck, A.; Saskowski, J.; Crispens, M.A.; Khabele, D. Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol. Oncol. 2016, 143, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues Moita, A.J.; Bandolik, J.J.; Hansen, F.K.; Kurz, T.; Hamacher, A.; Kassack, M.U. Priming with HDAC Inhibitors Sensitizes Ovarian Cancer Cells to Treatment with Cisplatin and HSP90 Inhibitors. Int. J. Mol. Sci 2020, 21, 8300. [Google Scholar] [CrossRef]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.A.; Tsao, M.S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; Disilvestro, P.A.; Fader, A.N. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Wasim, L.; Chopra, M. Panobinostat induces apoptosis via production of reactive oxygen species and synergizes with topoisomerase inhibitors in cervical cancer cells. Biomed. Pharmacother. 2016, 84, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, J.; Ding, H.; Chen, L.; Zhang, Y.; Liu, R.; Xu, P.; Du, D.; Lu, W.; Liu, J.; et al. Identification of novel EZH2 inhibitors through pharmacophore-based virtual screening and biological assays. Bioorganic Med. Chem. Lett. 2016, 26, 3813–3817. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e408. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination–proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenguer-Daize, C.; Astorgues-Xerri, L.; Odore, E.; Cayol, M.; Cvitkovic, E.; Noel, K.; Bekradda, M.; MacKenzie, S.; Rezai, K.; Lokiec, F.; et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 2016, 139, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Soria, J.C.; Stathis, A.; Delord, J.P.; Awada, A.; Peters, S.; Lewin, J.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. A phase Ib trial with MK-8628/OTX015, a small molecule inhibitor of bromodomain (BRD) and extra-terminal (BET) proteins, in patients with selected advanced solid tumors. Eur. J. Cancer 2016, 69, S2–S3. [Google Scholar] [CrossRef]
- Liu, A.; Fan, D.; Wang, Y. The BET bromodomain inhibitor i-BET151 impairs ovarian cancer metastasis and improves antitumor immunity. Cell Tissue Res. 2018, 374, 577–585. [Google Scholar] [CrossRef]
- Wilson, A.J.; Hoover, A.; Harris, W.; Liu, E.; Khabele, D.; Yull, F. Abstract 626: Bromodomain inhibition in ovarian cancer and the tumor microenvironment to improve PARP inhibitor response. Cancer Res. 2018, 78, 626. [Google Scholar] [CrossRef]
- Yin, M.; Guo, Y.; Hu, R.; Cai, W.L.; Li, Y.; Pei, S.; Sun, H.; Peng, C.; Li, J.; Ye, R.; et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 2020, 11, 1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, G.Y.L.; Shaw, R.; Schaub, F.X.; Stork, I.N.; Gurley, K.E.; Bridgwater, C.; Diaz, R.L.; Rosati, R.; Swan, H.A.; Ince, T.A.; et al. BET, SRC, and BCL2 family inhibitors are synergistic drug combinations with PARP inhibitors in ovarian cancer. EBioMedicine 2020, 60, 102988. [Google Scholar] [CrossRef]
- Boshuizen, J.; Peeper, D.S. Rational Cancer Treatment Combinations: An Urgent Clinical Need. Mol. Cell 2020, 78, 1002–1018. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuco, V.; De Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS ONE 2011, 6, e29085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A.; Gynecologic Oncology, G. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.Y.; Liao, W.S.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]


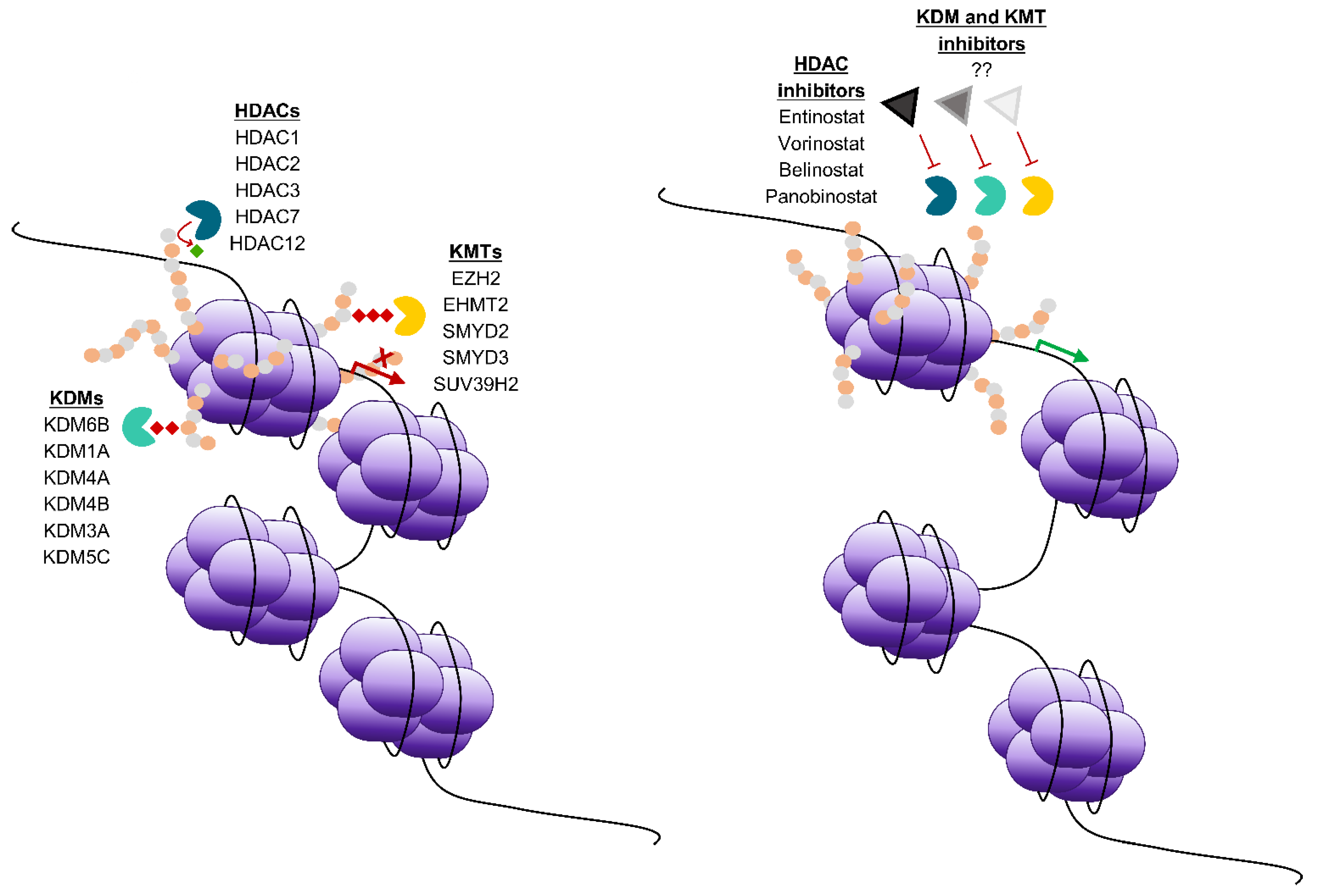{kind=link}
| Enzyme | Enzyme Type | Mechanism | Modification | Cancer | Expression | Reference(s) |
|---|---|---|---|---|---|---|
| EZH2 | Writer | Methyltransferase | H3K27me2, H3K27me3 | Ovarian, Endometrial | ↑ | [14,15] |
| KDM6B | Eraser | Demethylase | H3K27me2, H3K27me3 | Ovarian | ↑ | [16] |
| EHMT2 | Writer | Methyltransferase | H3K9me, H3K9me2 | Ovarian, Endometrial | ↑ | [15,17] |
| LSD1 | Eraser | Demethylase | H3K4me2, H3K4me | Ovarian, Endometrial | ↑ | [18] |
| LSD1 | Eraser | Demethylase | H3K9me | Endometrial | ↑ | [19] |
| EP400 | Writer | Acetyltransferase | HAc | Cervical | ↓ | [20] |
| KDM5C | Eraser | Demethylase | H3K4me, H3K4me2, H3K4me3 | Cervical | ↓ | [20] |
| SMYD2 | Writer | Methyltransferase | H3K4me3 | Ovarian | ↑ | [21] |
| SMYD3 | Writer | Methyltransferase | H3K4me3, H4K5me | Cervical | ↑ | [21] |
| DOT1L | Writer | Methyltransferase | H3K79me2 | Ovarian | ↑ | [22] |
| RNF20-RNF40 complex | Writer | Ubiquitinating enzyme | H2Bub | Ovarian | ↓ | [23,24] |
| KDM4A | Eraser | Demethylase | H3K9me3, H3K36 | Ovarian | ↑ | [25] |
| KDM3A | Eraser | Demethylase | H3K9me, H3K9me2 | Ovarian | ↑ | [26] |
| SUV39H2 | Writer | Methyltransferase | H3K9me3 | Cervical | ↑ | [27] |
| KDM4B | Eraser | Demethylase | H3K9me2, H3K9me3 | Endometrial | ↑ | [28] |
| HDAC1 | Eraser | Deacetylase | HAc | Ovarian, Endometrial | ↑ | [29,30] |
| HDAC2 | Eraser | Deacetylase | HAc | Ovarian, Endometrial | ↑ | [29,30] |
| HDAC3 | Eraser | Deacetylase | HAc | Ovarian, Endometrial | ↑ | [29,30] |
| HDAC7 | Eraser | Deacetylase | HAc | Ovarian | ↑ | [31] |
| HDAC12 | Eraser | Deacetylase | HAc | Ovarian | ↑ | [31] |
| BRD4 | Reader | BET protein | - | Ovarian | ↑ | [32] |
| Enzyme | Epigenetic Inhibitor | Combination Agent(s) | Cancer Type | Model/Cell Lines Tested | Inhibitor Dose and Treatment Duration | Outcome | Reference(s) |
|---|---|---|---|---|---|---|---|
| KMT4/DOT1L | EPZ004777 | None | Ovarian | PEO1 and PEO4 cell lines | 0.1 µM, 72 h | Growth arrest | [96] |
| EZH2 | GSK126 | Cisplatin | Ovarian | OVCAR3 and CA-MSC orthotopic mouse model | Various concentrations | Cell viability of OVCAR3 cells unaffected, but decreased ability of OVCAR3 cells to metastasise | [97] |
| EZH2 | GSK126 | 5-AZA dC | Ovarian | NSG model | 30 mg/kg, 3 times a week for 2 weeks | Increased efficacy of adoptive T-cell therapy in vivo | [58] |
| EZH2 | GSK126 | None, Cisplatin or Doxorubicin | Endometrial | Various cancer cell lines | 0.025–20 µM, 8 days | Decreased cell proliferation and induction of apoptosis. Additive effects with cisplatin or doxorubicin | [98] |
| EZH2 | DZNep | None | Cervical | HeLa and HeLa/DDP cells | Various concentrations, 72 h | Reversal of cisplatin resistance observed in the HeLa/DDP cell line | [99] |
| G9a (EHMT2) | BIX01294 | None | Cervical | CaSki, HeLa and SiHa cell lines | 5 µM, 72 h | Cell migration and invasion attenuated in BIX01294-treated cells | [100] |
| G9a (EHMT2) | BIX01294 | None | Cervical | Subcutaneous SiHa cell line xenograft cervical cancer tumor model | 5 mg/kg and 10 mg/kg, 39 days | Xenograft tumour growth significantly attenuated from day 29 at a dose of 10 mg/kg compared to control | [100] |
| G9a (EHMT2) | UNC0638 | None | Ovarian | SKOV-3, ES-2, and PA-1 cell lines | 2 µM, 48 h | Increase in metastasis suppressor genes such as CDH10 | [101] |
| G9a (EHMT2) | UNC0638 | None | Ovarian | SKOV-3 cell line | 2 µM, 48 h | Decreased metastasis-related signaling | [17] |
| SUV39H1/SUV39H2 | Chaetocin | None | Ovarian | OVCAR3 cell line | IC50–60.66 nM, 24 h | Inhibited proliferation, induced ROS accumulation and resulted in caspase-induced cell death in OVCAR-3 cells | [102] |
| SUV39H1/SUV39H2 | Chaetocin | None | Cervical | HeLa and CaSki cell lines | 150 nM, 24 h | Restored the innate immune response to exogenous DNA | [103] |
| KDM6A/6B | GSK-J4 | None | Cervical | SiHa and HeLa cell lines | 25–100 µM, 72 h | Decreased cell viability in SiHa cell line, no effect in HeLa cell line | [104] |
| KDM6A/6B | GSK-J4 | None | Cervical | CaSki cell line | 0–30 µM, 72 h | Decreased cell viability | [104] |
| KDM6A/6B | GSK-J4 | None | Ovarian | A2780 cancer stem cell like cells | 0.5–10 µM, 72 h | Decreased cell viability | [16] |
| LSD1 | SP-2577 | None | Ovarian | SWI/SNF-mutated cell lines | 0.01–1.1 µM, 72 h | Affects cell viability, and induces expression of inflammatory cytokines in organoids | [105] |
| LSD1 | HCI2509 | None | Endometrial | AN3CA and KLE cell lines | IC50–500 nM, 96 h | Apoptotic cell death in cell lines, tumour regression in orthotopic xenografts | [19] |
| BRD4 | JQ1, I-BET151 | None | Ovarian | Various cell lines | 0.01–10 µM | Cell cycle arrest in all subtypes of ovarian cancer cell lines tested | [32] |
| BRD4 | JQ1 | None | Ovarian | OVCAR-3 cell line xenograft and patient-derived xenograft model | 50 mg/kg | Decreased tumour volume | [32] |
| NCT Number | Trial | Inhibitor Name | Inhibitor Type | Combination Agent(s) | Epigenetic Target | Epigenetic Inhibitor Dose | Cancer Type * | Recruitment Status |
|---|---|---|---|---|---|---|---|---|
| NCT04651127 | Phase I/II | Chidamide | Class I HDAC inhibitor | Toripalimab | Class I HDACs | 30 mg/day orally, twice a week | Persistent, Recurrent, or Metastatic Cervical Cancer | Recruiting |
| NCT02728492 | Phase I | Quisinostat | HDAC inhibitor | Gemcitabine, Carboplatin, Paclitaxel | HDACs | 8 mg every other day | Non-small Cell Lung Cancer, Epithelial Ovarian Cancer | Completed |
| NCT02948075 | Phase II | Quisinostat | HDAC inhibitor | Carboplatin, Paclitaxel | HDACs | 12 mg every other day | Ovarian Cancer | Completed |
| NCT02915523 | Phase I/II | Entinostat | HDAC inhibitor | Avelumab | HDACs | 5 mg weekly, 3 months | Advanced Epithelial Ovarian Cancer | Unknown |
| NCT00772798 | Phase II | Vorinostat | HDAC inhibitor | Paclitaxel, Carboplatin | HDACs | 400 mg once daily orally, days 4–10 of a 25 day cycle | Recurrent Ovarian Cancer | Unknown |
| NCT03345485 | Phase I/II | Tinostamustine | alkylating HDAC inhibitor | - | HDACs | 60 mg/m2 up to 100 mg/m2, day 1 and 15 of a 28 day cycle | Advanced Solid Tumors | Recruiting |
| NCT00020579 | Phase I | Entinostat | HDAC inhibitor | - | HDACs | Dose escalation study | Advanced Solid Tumors or Lymphoma | Completed |
| NCT00421889 | Phase I/II | Belinostat | HDAC inhibitor | Carboplatin, Paclitaxel | HDACs | 1000 mg/m2, days 1–5 of a 21 day cycle | Ovarian Cancer in Need of Relapse Treatment | Completed |
| NCT00976183 | Phase I/II | Vorinostat | HDAC inhibitor | Carboplatin, Paclitaxel | HDACs | 200 mg once a day | Advanced Stage Ovarian Carcinoma | Terminated |
| NCT04315233 | Phase I | Belinostat | HDAC inhibitor | Ribociclib | HDACs | 600 mg/m2, days 1–5 of a 28 day cycle | Metastatic Triple Neg Breast Cancer & Recurrent Ovarian Cancer | Recruiting |
| NCT02601937 | Phase I | Tazemetostat | KMT inhibitor | - | EZH2 | Dose escalation study | Pediatric Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma | Recruiting |
| NCT00301756 | Phase II | Belinostat | HDAC inhibitor | - | HDACs | - | Recurrent or Persistent Ovarian Epithelial or Primary Peritoneal Cavity Cancer | Completed |
| NCT04703920 | Phase I | Belinostat | HDAC inhibitor | Talazoparib | HDACs | Up to 1000 mg/m2 IV once daily on days 1– 5 of a 21-day cycle | Metastatic Castration Resistant Prostate Cancer, and Metastatic Ovarian Cancer | Not yet recruiting |
| NCT03018249 | Phase I | Entinostat | HDAC inhibitor | Medroxyprogesterone Acetate | HDACs | - | Endometrial cancer | Active, not recruiting |
| NCT00132067 | Phase II | Vorinostat | HDAC inhibitor | - | HDACs | - | Recurrent or Persistent Ovarian Epithelial or Primary Peritoneal Cavity Cancer | Completed |
| NCT02661815 | Phase I | Ricolinostat | HDAC inhibitor | Paclitaxel | HDACs | 80 mg/m2 per week (3 out of 4 weeks). | Gynecologic cancers | Terminated |
| NCT04357873 | Phase II | Vorinostat | HDAC inhibitor | Pembrolizumab | HDACs | 400 mg once daily, until progression | metastatic squamous cell carcinoma (head and neck, lung, cervix, vulva, anus and penis) | Recruiting |
| NCT04498520 | Phase I | Abexinostat | HDAC inhibitor | Palbociclib, Fulvestrant | HDACs | - | Breast and Gynecologic cancers | Not yet recruiting |
| NCT00413322 | Phase I | Belinostat | HDAC inhibitor | 5-Fluorouracil | HDACs | 300, 600, or 1000 mg/m2 belinostat for 5 days every 21 days starting with cycle 1 | Advanced Solid Tumors | Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramarao-Milne, P.; Kondrashova, O.; Barry, S.; Hooper, J.D.; Lee, J.S.; Waddell, N. Histone Modifying Enzymes in Gynaecological Cancers. Cancers 2021, 13, 816. https://doi.org/10.3390/cancers13040816
Ramarao-Milne P, Kondrashova O, Barry S, Hooper JD, Lee JS, Waddell N. Histone Modifying Enzymes in Gynaecological Cancers. Cancers. 2021; 13(4):816. https://doi.org/10.3390/cancers13040816
Chicago/Turabian StyleRamarao-Milne, Priya, Olga Kondrashova, Sinead Barry, John D. Hooper, Jason S. Lee, and Nicola Waddell. 2021. "Histone Modifying Enzymes in Gynaecological Cancers" Cancers 13, no. 4: 816. https://doi.org/10.3390/cancers13040816
APA StyleRamarao-Milne, P., Kondrashova, O., Barry, S., Hooper, J. D., Lee, J. S., & Waddell, N. (2021). Histone Modifying Enzymes in Gynaecological Cancers. Cancers, 13(4), 816. https://doi.org/10.3390/cancers13040816