Dendritic Cells: Behind the Scenes of T-Cell Infiltration into the Tumor Microenvironment

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
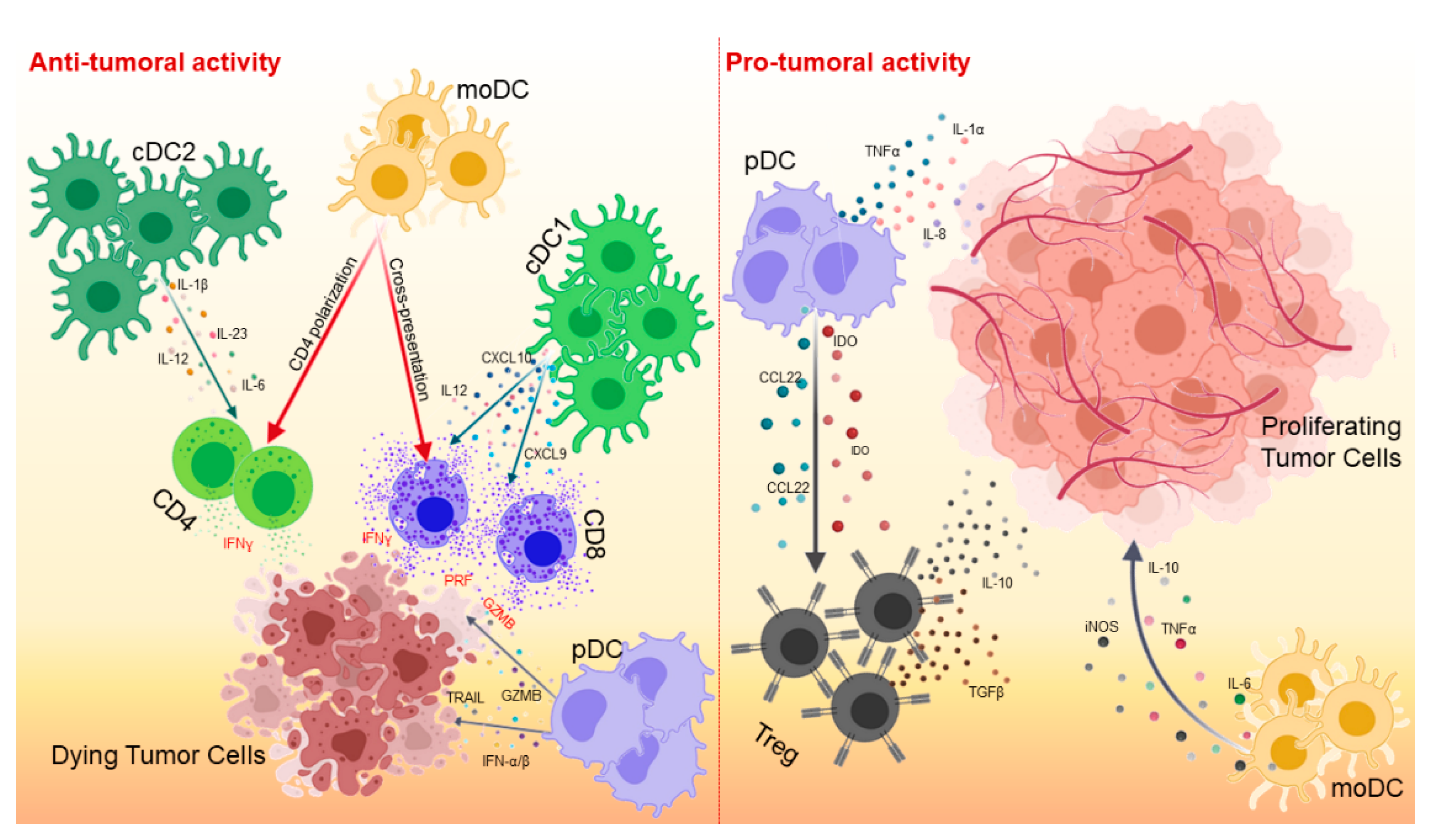
2. DC Cell Subsets in Cancer Immunology
2.1. cDCs
2.2. pDCs
2.3. MoDCs
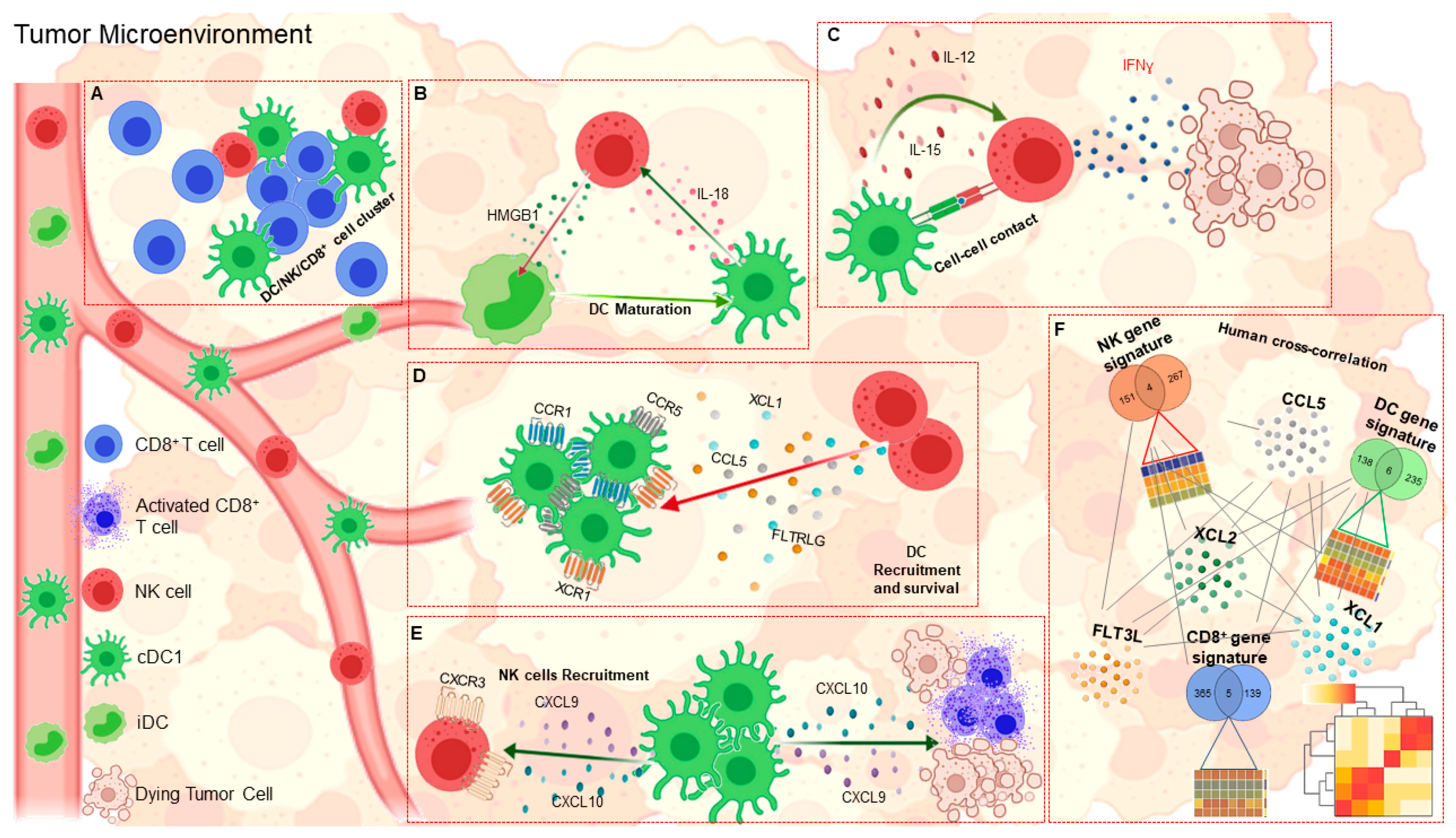
3. Chemokine Networks and DCs in the TME
4. DC-NK Cell Axis in Anti-Cancer Immunity
5. Prognostic Value of cDC1s in Solid Tumors
6. Clinical Trials Exploiting the Efficacy of Agents and Therapies That Promote the Immunogenic Functions of DCs in Cancer Immunotherapy
6.1. Agents Promoting the Immunogenic Functions of DCs
6.2. DCs and Chemotherapy
6.3. DCs and Radiation Therapy
6.4. DCs and Irreversible Electroporation
6.5. DCs and Cryoablation
6.6. PD-L1 on DCs and Immune Checkpoint Blockade Therapies
7. Ex Vivo Manipulation of cDC1s in Cancer Immunotherapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Monteagudo, C.; Martin, J.M.; Jorda, E.; Llombart-Bosch, A. CXCR3 chemokine receptor immunoreactivity in primary cutaneous malignant melanoma: Correlation with clinicopathological prognostic factors. J. Clin. Pathol. 2007, 60, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Dhodapkar, K.M.; Palucka, A.K. Interactions of tumor cells with dendritic cells: Balancing immunity and tolerance. Cell Death Differ. 2008, 15, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Idoyaga, J.; Fiorese, C.; Zbytnuik, L.; Lubkin, A.; Miller, J.; Malissen, B.; Mucida, D.; Merad, M.; Steinman, R.M. Specialized role of migratory dendritic cells in peripheral tolerance induction. J. Clin. Investig. 2013, 123, 844–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Audiger, C.; Rahman, M.J.; Yun, T.J.; Tarbell, K.V.; Lesage, S. The Importance of Dendritic Cells in Maintaining Immune Tolerance. J. Immunol. 2017, 198, 2223–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Cancer immunogenicity, danger signals, and DAMPs: What, when, and how? Biofactors 2013, 39, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Gallo, P.M.; Gallucci, S. The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front. Immunol. 2013, 4, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlitzer, A.; McGovern, N.; Ginhoux, F. Dendritic cells and monocyte-derived cells: Two complementary and integrated functional systems. Semin. Cell Dev. Biol. 2015, 41, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Bottcher, J.P.; e Sousa, C.R. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Chiang, M.C.; Tullett, K.M.; Lee, Y.S.; Idris, A.; Ding, Y.; McDonald, K.J.; Kassianos, A.; Leal Rojas, I.M.; Jeet, V.; Lahoud, M.H.; et al. Differential uptake and cross-presentation of soluble and necrotic cell antigen by human DC subsets. Eur. J. Immunol. 2016, 46, 329–339. [Google Scholar] [CrossRef]
- Balan, S.; Bhardwaj, N. Cross-Presentation of Tumor Antigens Is Ruled by Synaptic Transfer of Vesicles among Dendritic Cell Subsets. Cancer Cell 2020, 37, 751–753. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond cDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, S.; Saxena, M.; Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 2019, 348, 1–68. [Google Scholar] [CrossRef] [PubMed]
- Haniffa, M.; Shin, A.; Bigley, V.; McGovern, N.; Teo, P.; See, P.; Wasan, P.S.; Wang, X.N.; Malinarich, F.; Malleret, B.; et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 2012, 37, 60–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Alloatti, A.; Rookhuizen, D.C.; Joannas, L.; Carpier, J.M.; Iborra, S.; Magalhaes, J.G.; Yatim, N.; Kozik, P.; Sancho, D.; Albert, M.L.; et al. Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. J. Exp. Med. 2017, 214, 2231–2241. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- De Mingo Pulido, A.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74.e66. [Google Scholar] [CrossRef] [Green Version]
- Oba, T.; Long, M.D.; Keler, T.; Marsh, H.C.; Minderman, H.; Abrams, S.I.; Liu, S.; Ito, F. Overcoming primary and acquired resistance to anti-PD-L1 therapy by induction and activation of tumor-residing cDC1s. Nat. Commun. 2020, 11, 5415. [Google Scholar] [CrossRef]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e289. [Google Scholar] [CrossRef]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.P.; Bern, M.D.; Davidson, J.T.t.; Bagadia, P.; Liu, T.; Briseno, C.G.; et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 2020, 584, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Leal Rojas, I.M.; Mok, W.H.; Pearson, F.E.; Minoda, Y.; Kenna, T.J.; Barnard, R.T.; Radford, K.J. Human Blood CD1c(+) Dendritic Cells Promote Th1 and Th17 Effector Function in Memory CD4(+) T Cells. Front. Immunol. 2017, 8, 971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Koucky, V.; Boucek, J.; Fialova, A. Immunology of Plasmacytoid Dendritic Cells in Solid Tumors: A Brief Review. Cancers 2019, 11, 470. [Google Scholar] [CrossRef] [Green Version]
- Villadangos, J.A.; Young, L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity 2008, 29, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Salvi, V.; Vermi, W.; Cavani, A.; Lonardi, S.; Carbone, T.; Facchetti, F.; Bosisio, D.; Sozzani, S. IL-21 May Promote Granzyme B-Dependent NK/Plasmacytoid Dendritic Cell Functional Interaction in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1493–1500. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, S.; Yang, Y.; Zhu, S.; Zhang, M.; Qiao, Y.; Liu, Y.J.; Chen, J. TLR-activated plasmacytoid dendritic cells inhibit breast cancer cell growth in vitro and in vivo. Oncotarget 2017, 8, 11708–11718. [Google Scholar] [CrossRef] [Green Version]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza-Gonzalez, A.; Zhou, G.; Vargas-Mendez, E.; Boor, P.P.; Mancham, S.; Verhoef, C.; Polak, W.G.; Grunhagen, D.; Pan, Q.; Janssen, H.; et al. Tumor-infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. Oncoimmunology 2015, 4, e1008355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, R.; Terlizzi, M.; Di Crescenzo, V.G.; Popolo, A.; Pecoraro, M.; Perillo, G.; Galderisi, A.; Pinto, A. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1alpha in an AIM2 inflammasome-dependent manner. Am. J. Pathol. 2015, 185, 3115–3124. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J.; Cheng, P.; Mottram, P.; Alvarez, X.; Moons, L.; Evdemon-Hogan, M.; Wei, S.; Zou, L.; Kryczek, I.; Hoyle, G.; et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004, 64, 5535–5538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verneau, J.; Sautes-Fridman, C.; Sun, C.M. Dendritic cells in the tumor microenvironment: Prognostic and theranostic impact. Semin. Immunol. 2020, 101410. [Google Scholar] [CrossRef]
- Kuhn, S.; Yang, J.; Ronchese, F. Monocyte-Derived Dendritic Cells Are Essential for CD8(+) T Cell Activation and Antitumor Responses After Local Immunotherapy. Front. Immunol. 2015, 6, 584. [Google Scholar] [CrossRef] [Green Version]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef] [Green Version]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [Green Version]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Hartung, E.; Guttler, S.; Mora, A.; Zhou, X.; Hegemann, A.; Plantinga, M.; Mazzini, E.; Stoitzner, P.; Gurka, S.; et al. Expression of XCR1 Characterizes the Batf3-Dependent Lineage of Dendritic Cells Capable of Antigen Cross-Presentation. Front. Immunol. 2012, 3, 214. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Kitahata, K.; Kawabata, F.; Kamei, M.; Hara, Y.; Takamura, S.; Oiso, N.; Kawada, A.; Yoshie, O.; Nakayama, T. A Highly Active Form of XCL1/Lymphotactin Functions as an Effective Adjuvant to Recruit Cross-Presenting Dendritic Cells for Induction of Effector and Memory CD8(+) T Cells. Front. Immunol. 2018, 9, 2775. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Zhao, J.; Winter, E.; Cattral, M.S. Recruitment and differentiation of conventional dendritic cell precursors in tumors. J. Immunol. 2010, 184, 1261–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middel, P.; Brauneck, S.; Meyer, W.; Radzun, H.J. Chemokine-mediated distribution of dendritic cell subsets in renal cell carcinoma. BMC Cancer 2010, 10, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, J.; Di Domizio, J.; Salameire, D.; Bendriss-Vermare, N.; Aspord, C.; Muhammad, R.; Lefebvre, C.; Plumas, J.; Leccia, M.T.; Chaperot, L. Characterization of circulating dendritic cells in melanoma: Role of CCR6 in plasmacytoid dendritic cell recruitment to the tumor. J. Investig. Dermatol. 2010, 130, 1646–1656. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenmuller, W.; Brandes, M.; Wang, Z.; Herz, J.; Egen, J.G.; Germain, R.N. Peripheral prepositioning and local CXCL9 chemokine-mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity 2013, 38, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martinez-Cano, S.; Mejias-Perez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat. Commun. 2017, 8, 16073. [Google Scholar] [CrossRef]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e810. [Google Scholar] [CrossRef]
- Greyer, M.; Whitney, P.G.; Stock, A.T.; Davey, G.M.; Tebartz, C.; Bachem, A.; Mintern, J.D.; Strugnell, R.A.; Turner, S.J.; Gebhardt, T.; et al. T Cell Help Amplifies Innate Signals in CD8(+) DCs for Optimal CD8(+) T Cell Priming. Cell Rep. 2016, 14, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, K.; Shiraishi, N.; Sugita, K.; Mori, T.; Onoue, A.; Kobayashi, M.; Sakabe, J.; Yoshiki, R.; Tamamura, H.; Fujii, N.; et al. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am. J. Pathol. 2007, 171, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczek, I.; Lange, A.; Mottram, P.; Alvarez, X.; Cheng, P.; Hogan, M.; Moons, L.; Wei, S.; Zou, L.; Machelon, V.; et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005, 65, 465–472. [Google Scholar] [PubMed]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Conejo-Garcia, J.R.; Benencia, F.; Courreges, M.C.; Kang, E.; Mohamed-Hadley, A.; Buckanovich, R.J.; Holtz, D.O.; Jenkins, A.; Na, H.; Zhang, L.; et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat. Med. 2004, 10, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Diao, J.; Gu, H.; Khatri, I.; Zhao, J.; Cattral, M.S. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep. 2015, 13, 2851–2864. [Google Scholar] [CrossRef] [Green Version]
- Vulcano, M.; Albanesi, C.; Stoppacciaro, A.; Bagnati, R.; D’Amico, G.; Struyf, S.; Transidico, P.; Bonecchi, R.; Del Prete, A.; Allavena, P.; et al. Dendritic cells as a major source of macrophage-derived chemokine/CCL22 in vitro and in vivo. Eur. J. Immunol. 2001, 31, 812–822. [Google Scholar] [CrossRef]
- Rohrle, N.; Knott, M.M.L.; Anz, D. CCL22 Signaling in the Tumor Environment. Adv. Exp. Med. Biol. 2020, 1231, 79–96. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.C.; Lozier, A.; Flament, C.; Ricciardi-Castagnoli, P.; Bellet, D.; Suter, M.; Perricaudet, M.; Tursz, T.; Maraskovsky, E.; Zitvogel, L. Dendritic cells directly trigger NK cell functions: Cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 1999, 5, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Borg, C.; Jalil, A.; Laderach, D.; Maruyama, K.; Wakasugi, H.; Charrier, S.; Ryffel, B.; Cambi, A.; Figdor, C.; Vainchenker, W.; et al. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization in DCs. Blood 2004, 104, 3267–3275. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103(+) Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallandre, J.R.; Krzewski, K.; Bedel, R.; Ryffel, B.; Caignard, A.; Rohrlich, P.S.; Pivot, X.; Tiberghien, P.; Zitvogel, L.; Strominger, J.L.; et al. Dendritic cell and natural killer cell cross-talk: A pivotal role of CX3CL1 in NK cytoskeleton organization and activation. Blood 2008, 112, 4420–4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguille, S.; Van Acker, H.H.; Van den Bergh, J.; Willemen, Y.; Goossens, H.; Van Tendeloo, V.F.; Smits, E.L.; Berneman, Z.N.; Lion, E. Interleukin-15 Dendritic Cells Harness NK Cell Cytotoxic Effector Function in a Contact- and IL-15-Dependent Manner. PLoS ONE 2015, 10, e0123340. [Google Scholar] [CrossRef]
- Gerosa, F.; Baldani-Guerra, B.; Nisii, C.; Marchesini, V.; Carra, G.; Trinchieri, G. Reciprocal activating interaction between natural killer cells and dendritic cells. J. Exp. Med. 2002, 195, 327–333. [Google Scholar] [CrossRef]
- Semino, C.; Angelini, G.; Poggi, A.; Rubartelli, A. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood 2005, 106, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Piccioli, D.; Sbrana, S.; Melandri, E.; Valiante, N.M. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J. Exp. Med. 2002, 195, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Morandi, B.; Mortara, L.; Chiossone, L.; Accolla, R.S.; Mingari, M.C.; Moretta, L.; Moretta, A.; Ferlazzo, G. Dendritic cell editing by activated natural killer cells results in a more protective cancer-specific immune response. PLoS ONE 2012, 7, e39170. [Google Scholar] [CrossRef] [Green Version]
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008, 68, 8437–8445. [Google Scholar] [CrossRef] [Green Version]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e1310. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melaiu, O.; Chierici, M.; Lucarini, V.; Jurman, G.; Conti, L.A.; De Vito, R.; Boldrini, R.; Cifaldi, L.; Castellano, A.; Furlanello, C.; et al. Cellular and gene signatures of tumor-infiltrating dendritic cells and natural-killer cells predict prognosis of neuroblastoma. Nat. Commun. 2020, 11, 5992. [Google Scholar] [CrossRef]
- Belounis, A.; Ayoub, M.; Cordeiro, P.; Lemieux, W.; Teira, P.; Haddad, E.; Herblot, S.; Duval, M. Patients’ NK cell stimulation with activated plasmacytoid dendritic cells increases dinutuximab-induced neuroblastoma killing. Cancer Immunol. Immunother. 2020, 69, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Lucarini, V.; Giovannoni, R.; Fruci, D.; Gemignani, F. News on immune checkpoint inhibitors as immunotherapy strategies in adult and pediatric solid tumors. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Michea, P.; Noel, F.; Zakine, E.; Czerwinska, U.; Sirven, P.; Abouzid, O.; Goudot, C.; Scholer-Dahirel, A.; Vincent-Salomon, A.; Reyal, F.; et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat. Immunol. 2018, 19, 885–897. [Google Scholar] [CrossRef]
- Hubert, M.; Gobbini, E.; Couillault, C.; Manh, T.V.; Doffin, A.C.; Berthet, J.; Rodriguez, C.; Ollion, V.; Kielbassa, J.; Sajous, C.; et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Ladanyi, A.; Kiss, J.; Somlai, B.; Gilde, K.; Fejos, Z.; Mohos, A.; Gaudi, I.; Timar, J. Density of DC-LAMP(+) mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol. Immunother. 2007, 56, 1459–1469. [Google Scholar] [CrossRef]
- Truxova, I.; Kasikova, L.; Hensler, M.; Skapa, P.; Laco, J.; Pecen, L.; Belicova, L.; Praznovec, I.; Halaska, M.J.; Brtnicky, T.; et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J. Immunother. Cancer 2018, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Goc, J.; Germain, C.; Vo-Bourgais, T.K.; Lupo, A.; Klein, C.; Knockaert, S.; de Chaisemartin, L.; Ouakrim, H.; Becht, E.; Alifano, M.; et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 2014, 74, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mina, M.; Boldrini, R.; Citti, A.; Romania, P.; D’Alicandro, V.; De Ioris, M.; Castellano, A.; Furlanello, C.; Locatelli, F.; Fruci, D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015, 4, e1019981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melaiu, O.; Mina, M.; Chierici, M.; Boldrini, R.; Jurman, G.; Romania, P.; D’Alicandro, V.; Benedetti, M.C.; Castellano, A.; Liu, T.; et al. PD-L1 Is a Therapeutic Target of the Bromodomain Inhibitor JQ1 and, Combined with HLA Class I, a Promising Prognostic Biomarker in Neuroblastoma. Clin. Cancer Res. 2017, 23, 4462–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Turbocharging vaccines: Emerging adjuvants for dendritic cell based therapeutic cancer vaccines. Curr. Opin. Immunol. 2017, 47, 35–43. [Google Scholar] [CrossRef]
- Chi, H.; Li, C.; Zhao, F.S.; Zhang, L.; Ng, T.B.; Jin, G.; Sha, O. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Front. Pharmacol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Martins, K.A.; Bavari, S.; Salazar, A.M. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev. Vaccines 2015, 14, 447–459. [Google Scholar] [CrossRef]
- Molenkamp, B.G.; Sluijter, B.J.; van Leeuwen, P.A.; Santegoets, S.J.; Meijer, S.; Wijnands, P.G.; Haanen, J.B.; van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8+ T-cell reactivity in melanoma patients. Clin. Cancer Res. 2008, 14, 4532–4542. [Google Scholar] [CrossRef] [Green Version]
- Anandasabapathy, N.; Breton, G.; Hurley, A.; Caskey, M.; Trumpfheller, C.; Sarma, P.; Pring, J.; Pack, M.; Buckley, N.; Matei, I.; et al. Efficacy and safety of CDX-301, recombinant human Flt3L, at expanding dendritic cells and hematopoietic stem cells in healthy human volunteers. Bone Marrow Transpl. 2015, 50, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.D.; Limagne, E.; Thibaudin, M.; Ghiringhelli, F. Immunogenic Cell Death and Elimination of Immunosuppressive Cells: A Double-Edged Sword of Chemotherapy. Cancers 2020, 12, 2637. [Google Scholar] [CrossRef] [PubMed]
- Asadzadeh, Z.; Safarzadeh, E.; Safaei, S.; Baradaran, A.; Mohammadi, A.; Hajiasgharzadeh, K.; Derakhshani, A.; Argentiero, A.; Silvestris, N.; Baradaran, B. Current Approaches for Combination Therapy of Cancer: The Role of Immunogenic Cell Death. Cancers 2020, 12, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Daguenet, E.; Louati, S.; Wozny, A.S.; Vial, N.; Gras, M.; Guy, J.B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magne, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 2020, 9, 1777624. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Davalos, R.V.; Bischof, J.C. A review of basic to clinical studies of irreversible electroporation therapy. IEEE Trans. Biomed. Eng. 2015, 62, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wang, Z.; Lei, K.; Liao, J.; Peng, Z.; Lin, M.; Liang, P.; Yu, J.; Peng, S.; Chen, S.; et al. Irreversible electroporation induces CD8(+) T cell immune response against post-ablation hepatocellular carcinoma growth. Cancer Lett. 2021. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Huang, X.; Zhang, Y.; Lin, X.; Li, S. T-cell activation and immune memory enhancement induced by irreversible electroporation in pancreatic cancer. Clin. Transl. Med. 2020, 10, e39. [Google Scholar] [CrossRef]
- Zhao, J.; Wen, X.; Tian, L.; Li, T.; Xu, C.; Wen, X.; Melancon, M.P.; Gupta, S.; Shen, B.; Peng, W.; et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat. Commun. 2019, 10, 899. [Google Scholar] [CrossRef] [Green Version]
- Aarts, B.M.; Klompenhouwer, E.G.; Rice, S.L.; Imani, F.; Baetens, T.; Bex, A.; Horenblas, S.; Kok, M.; Haanen, J.; Beets-Tan, R.G.H.; et al. Cryoablation and immunotherapy: An overview of evidence on its synergy. Insights Imaging 2019, 10, 53. [Google Scholar] [CrossRef]
- Erinjeri, J.P.; Thomas, C.T.; Samoilia, A.; Fleisher, M.; Gonen, M.; Sofocleous, C.T.; Thornton, R.H.; Siegelbaum, R.H.; Covey, A.M.; Brody, L.A.; et al. Image-guided thermal ablation of tumors increases the plasma level of interleukin-6 and interleukin-10. J. Vasc. Interv. Radiol. 2013, 24, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Liang, S.Z.; Wang, X.H.; Liang, Y.Q.; Zhang, M.J.; Niu, L.Z.; Chen, J.B.; Li, H.B.; Xu, K.C. Clinical efficacy of percutaneous cryoablation combined with allogenic NK cell immunotherapy for advanced non-small cell lung cancer. Immunol. Res. 2017, 65, 880–887. [Google Scholar] [CrossRef]
- Yuanying, Y.; Lizhi, N.; Feng, M.; Xiaohua, W.; Jianying, Z.; Fei, Y.; Feng, J.; Lihua, H.; Jibing, C.; Jialiang, L.; et al. Therapeutic outcomes of combining cryotherapy, chemotherapy and DC-CIK immunotherapy in the treatment of metastatic non-small cell lung cancer. Cryobiology 2013, 67, 235–240. [Google Scholar] [CrossRef]
- Den Brok, M.H.; Sutmuller, R.P.; Nierkens, S.; Bennink, E.J.; Frielink, C.; Toonen, L.W.; Boerman, O.C.; Figdor, C.G.; Ruers, T.J.; Adema, G.J. Efficient loading of dendritic cells following cryo and radiofrequency ablation in combination with immune modulation induces anti-tumour immunity. Br. J. Cancer 2006, 95, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakkala, C.; Denys, A.; Kandalaft, L.; Duran, R. Cryoablation and immunotherapy of cancer. Curr. Opin. Biotechnol. 2020, 65, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Yakkala, C.; Chiang, C.L.; Kandalaft, L.; Denys, A.; Duran, R. Cryoablation and Immunotherapy: An Enthralling Synergy to Confront the Tumors. Front. Immunol. 2019, 10, 2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.J.; Anyaegbu, C.C.; Lee-Pullen, T.F.; Spalding, L.J.; Platell, C.F.; McCoy, M.J. PD-L1+ dendritic cells in the tumor microenvironment correlate with good prognosis and CD8+ T cell infiltration in colon cancer. Cancer Sci. 2020. [Google Scholar] [CrossRef]
- Oh, S.A.; Wu, D.C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 1, 681–691. [Google Scholar] [CrossRef]
- Zhao, Y.; Harrison, D.L.; Song, Y.; Ji, J.; Huang, J.; Hui, E. Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in cis to Attenuate PD-1 Signaling in T Cells. Cell Rep. 2018, 24, 379–390.e376. [Google Scholar] [CrossRef] [Green Version]
- Chaudhri, A.; Xiao, Y.; Klee, A.N.; Wang, X.; Zhu, B.; Freeman, G.J. PD-L1 Binds to B7-1 Only In Cis on the Same Cell Surface. Cancer Immunol. Res. 2018, 6, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, C.K.; Lin, C.H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e1059. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Scheuerpflug, A.; Ahmetlic, F.; Bauer, V.; Riedel, T.; Rocken, M.; Mocikat, R. The role of dendritic cells for therapy of B-cell lymphoma with immune checkpoint inhibitors. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Ollion, V.; Colletti, N.; Chelbi, R.; Montanana-Sanchis, F.; Liu, H.; Vu Manh, T.P.; Sanchez, C.; Savoret, J.; Perrot, I.; et al. Human XCR1+ dendritic cells derived in vitro from CD34+ progenitors closely resemble blood dendritic cells, including their adjuvant responsiveness, contrary to monocyte-derived dendritic cells. J. Immunol. 2014, 193, 1622–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, S.; Arnold-Schrauf, C.; Abbas, A.; Couespel, N.; Savoret, J.; Imperatore, F.; Villani, A.C.; Vu Manh, T.P.; Bhardwaj, N.; Dalod, M. Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell Rep. 2018, 24, 1902–1915.e1906. [Google Scholar] [CrossRef] [Green Version]
- Balan, S.; Dalod, M. In Vitro Generation of Human XCR1(+) Dendritic Cells from CD34(+) Hematopoietic Progenitors. Methods Mol. Biol. 2016, 1423, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Thordardottir, S.; Hangalapura, B.N.; Hutten, T.; Cossu, M.; Spanholtz, J.; Schaap, N.; Radstake, T.R.; van der Voort, R.; Dolstra, H. The aryl hydrocarbon receptor antagonist StemRegenin 1 promotes human plasmacytoid and myeloid dendritic cell development from CD34+ hematopoietic progenitor cells. Stem Cells Dev. 2014, 23, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Watanabe, E.; Shimizu, M.; Negishi, Y.; Kondo, Y.; Takahashi, H. Induction of tumor-specific CD8(+) cytotoxic T lymphocytes from naive human T cells by using Mycobacterium-derived mycolic acid and lipoarabinomannan-stimulated dendritic cells. Cancer Immunol. Immunother. 2019, 68, 1605–1619. [Google Scholar] [CrossRef] [Green Version]
- Silk, K.M.; Silk, J.D.; Ichiryu, N.; Davies, T.J.; Nolan, K.F.; Leishman, A.J.; Carpenter, L.; Watt, S.M.; Cerundolo, V.; Fairchild, P.J. Cross-presentation of tumour antigens by human induced pluripotent stem cell-derived CD141(+)XCR1+ dendritic cells. Gene Ther. 2012, 19, 1035–1040. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, R.F.; Haight, A.E.; Hirschmann-Jax, C.; Yvon, E.S.; Rill, D.R.; Mei, Z.; Smith, S.C.; Inman, S.; Cooper, K.; Alcoser, P.; et al. Local and systemic effects of an allogeneic tumor cell vaccine combining transgenic human lymphotactin with interleukin-2 in patients with advanced or refractory neuroblastoma. Blood 2003, 101, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Russell, H.V.; Strother, D.; Mei, Z.; Rill, D.; Popek, E.; Biagi, E.; Yvon, E.; Brenner, M.; Rousseau, R. Phase I trial of vaccination with autologous neuroblastoma tumor cells genetically modified to secrete IL-2 and lymphotactin. J. Immunother. 2007, 30, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| DC Subset | Mouse Surface Markers | Human Surface Markers |
|---|---|---|
| cDC1 | CD11c+ | CD11c low |
| MHC-II+ | HLA-DR+ | |
| CD103+ | CD141+ | |
| XCR1+ | XCR1+ | |
| CLEC9A+ | CLEC9A+ | |
| DEC205+ | DEC205+ | |
| CD8α+ | ||
| cDC2 | CD11c+ | CD11c+ |
| MHC-II+ | HLA-DR+ | |
| CD172a+ | CD172a+ | |
| CD11b+ | CD1a+ | |
| CD1c+ | ||
| pDC | CD11c low | CD11c− |
| MHC-II low | HLA-DR low | |
| CXCR3+ | CXCR3+ | |
| CD317+ | CD123+ | |
| SIGLEC-H+ | CD303+ | |
| B220+ | CD304+ | |
| moDC | CD11c+ | CD11c+ |
| MHC-II+ | HLA-DR+ | |
| CD11b+ | CD11b+ | |
| CD14+ | CD14+ | |
| CD64+ | CD64+ | |
| CD206+ | CD206+ | |
| CD209+ | CD209+ | |
| CCR2+ | CCR2+ | |
| Ly6C+ | CD1a+ | |
| CD1c+ |
| Agonist | Cancer Type(s) | Phase(s) | Interventions | Trials | |
|---|---|---|---|---|---|
| FLT3L | CDX-301 | Metastatic Breast Cancer, Head and Neck Squamous Cell Carcinoma | I/II | Radiation, Poly ICLC, Pembrolizumab | NCT03789097 |
| CDX-301 | Non-Small Cell Lung Cancer | II | Radiation | NCT02839265 | |
| CDX-301 | Colorectal Cancer, Metastatic Cancer | I | - | NCT00003431 | |
| Ad-hCMV-TK and Ad-hCMV-Flt3L | Malignant Glioma, Glioblastoma Multiforme | I | - | NCT01811992 | |
| CDX-301 | Non-Small Cell Lung Cancer Lung Cancer | I/II | Anti-CD40 Agonist Antibody, SBRT | NCT04491084 | |
| CDX-301 | Stage IV Melanoma, Stage IV Renal Cell Cancer, Recurrent Renal Cell Cancer, Recurrent Melanoma | II | gp100, MART-1, Montanide ISA-51 tyrosinase peptide | NCT00019396 | |
| CDX-301 | Kidney Cancer, Melanoma (Skin) | I | Recombinant CD40-ligand | NCT00020540 | |
| CDX-301 | Cutaneous, Mucosal and Ocular Melanoma | II | DEC-205/NY-ESO-1, Fusion Protein CDX-1401, Neoantigen-based, Melanoma-Poly-ICLC Vaccine | NCT02129075 | |
| CDX-301 | Melanoma, Non Small Cell Lung Cancer and others | I | CDX-1140, Pembrolizumab, Chemotherapy | NCT03329950 | |
| CDX-301 | Breast Cancer | I | Anti-CD40 Agonist, Poly ICLC, Radiation | NCT04616248 | |
| CDX-301 | Breast Cancer | I/II | Filgrastim, Thrombopoietin, Interleukin-3 | NCT00006225 | |
| TLR2 | CBLB612 | Breast Cancer | II | - | NCT02778763 |
| TLR4 | GLA-SE | Colorectal Cancer Metastatic | I | FOLFOX, Nivolumab, Ipilimumab | NCT03982121 |
| GSK1795091 | Neoplasms | I | GSK3174998, GSK3359609, Pembrolizumab | NCT03447314 | |
| GSK1795091 | Neoplasms | I | - | NCT02798978 | |
| GLA-SE | Melanoma | I | MART-1 Antigen | NCT02320305 | |
| GLA-SE | Soft Tissue Sarcoma | I | Radiation | NCT02180698 | |
| GLA-SE | Merkel Cell Carcinoma | I | - | NCT02035657 | |
| OM-174 | Neoplasms | I | - | NCT01800812 | |
| TLR3 | Hiltonol | Melanoma | I/II | NY-ESO-1 protein, Montanide | NCT01079741 |
| Hiltonol | Head and Neck Squamous Cell Carcinoma, Breast and others | I/II | Durvalumab, Tremelimumab | NCT02643303 | |
| Hiltonol | Ovarian cancer and others | I | OC-L, Montanide | NCT02452775 | |
| Hiltonol | Ovarian cancer | I | Oregovomab | NCT03162562 | |
| Ampligen | Ovarian cancer | I/II | OC-L, Montanide, Prevnar | NCT01312389 | |
| Hiltonol | Glioma | II | Autologous tumor lysate-pulsed DC vaccination, Tumor lysate-pulsed DC vaccination+0.2% resiquimod | NCT01204684 | |
| Hiltonol | Metastatic colon cancer Neoplasms | I/II | Pembrolizumab | NCT02834052 | |
| Hiltonol | Glioma | II | Bevacizumab, Peptide Vaccine, Poly-ICLC as immune adjuvant, Keyhole limpet hemocyanin | NCT02754362 | |
| TLR7 | RO7119929 | Hepatocellular Carcinoma, Biliary Tract Cancer, Secondary Liver Cancer, Liver Metastases | I | Tocilizumab | NCT04338685 |
| SHR2150 | Neoplasms | I/II | Chemotherapy, PD1 Ab, CD47 Ab | NCT04588324 | |
| Imiquimod (R837) | Breast Cancer | II | - | NCT00899574 | |
| Melanoma and others | I | PD-1 Antibody Blockade | NCT04116320 | ||
| Breast Cancer | I/II | Cyclophosphamide, Radiation | NCT01421017 | ||
| High Grade Cervical Intraepithelial Neoplasia | I | Topical Fluorouracil | NCT03196180 | ||
| DSP-0509 | Neoplasms | I/II | Pembrolizumab | NCT03416335 | |
| MEDI9197 | Neoplasms | I | Durvalumab | NCT02556463 | |
| Resiquimod | Neoplasms | I | NY-ESO-1, Montanide ISA®-51 VG | NCT00821652 | |
| 852A | Breast Cancer and others | II | - | NCT00319748 | |
| NJH395 | NON-breast HER2+ Cancers | I | - | NCT03696771 | |
| BNT411 | Neoplasms | I/II | Atezolizumab, Carboplatin, Etoposide | NCT04101357 | |
| TQ-A3334 | Non-Small Cell Lung Cancer | I/II | Anlotinib | NCT04273815 | |
| NKTR-262 | Melanoma and others | I/II | Bempegaldesleukin, Nivolumab | NCT03435640 | |
| BCDC-1001 | Breast Cancer, Gastric Cancer | I/II | Pembrolizumab | NCT04278144 | |
| LHC165 PDR001 | Neoplasms | I | - | NCT03301896 | |
| TLR9 | CpG | Pancreatic Cancer, Metastatic Pancreatic Cancer | I | Irreversible Electroporation, Nivolumab | NCT04612530 |
| CMP-001 | Melanoma | II | Nivolumab, [18F]F-AraG PET/CT | NCT04401995 | |
| CMP-001 | Locally Advanced Malignant Solid Neoplasm, Metastatic Pancreatic Adenocarcinoma | I/II | Agonistic Anti-OX40 | NCT04387071 | |
| Tilsotolimod | Advanced Neoplasms | I | Ipilimumab, Nivolumab | NCT04270864 | |
| Malignant Melanoma | II | - | NCT04126876 | ||
| SD-101 | Metastatic Pancreatic Adenocarcinoma, Refractory Pancreatic Adenocarcinoma, Stage IV Pancreatic Cancer AJCC | I | Nivolumab, Radiation | ||
| SD-101 | Advanced Malignant Solid Neoplasm, Extracranial Solid Neoplasm, Metastatic Malignant Solid Neoplasm | I | Anti-OX40 Antibody BMS 986178 | NCT03831295 | |
| CMP-001 | Melanoma | II | Nivolumab | NCT03618641 | |
| CMP-001 | Colorectal Neoplasms Malignant, Liver Metastases | I | Radiation, Nivolumab, Ipilimumab | NCT03507699 | |
| IMO-2125 | Metastatic Melanoma | III | Ipilimumab | NCT03445533 | |
| CMP-001 | Advanced Cancers | II | Avelumab, Utomilumab, PF-04518600, PD 0360324 | NCT02554812 | |
| EMD 1201081 | Head and Neck Squamous Cell Carcinoma | I | 5-FU, Cisplatin, Cetuximab | NCT01360827 | |
| EMD 1201081 | Head and Neck Squamous Cell Carcinoma | II | Cetuximab | NCT01040832 | |
| IMO-2055 | Colorectal Cancer | I | Cetuximab, FOLFIRI | NCT00719199 | |
| CpG-7909 | Esophageal Cancer | I/II | URLC10-177, TTK-567 | NCT00669292 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucarini, V.; Melaiu, O.; Tempora, P.; D’Amico, S.; Locatelli, F.; Fruci, D. Dendritic Cells: Behind the Scenes of T-Cell Infiltration into the Tumor Microenvironment. Cancers 2021, 13, 433. https://doi.org/10.3390/cancers13030433
Lucarini V, Melaiu O, Tempora P, D’Amico S, Locatelli F, Fruci D. Dendritic Cells: Behind the Scenes of T-Cell Infiltration into the Tumor Microenvironment. Cancers. 2021; 13(3):433. https://doi.org/10.3390/cancers13030433
Chicago/Turabian StyleLucarini, Valeria, Ombretta Melaiu, Patrizia Tempora, Silvia D’Amico, Franco Locatelli, and Doriana Fruci. 2021. "Dendritic Cells: Behind the Scenes of T-Cell Infiltration into the Tumor Microenvironment" Cancers 13, no. 3: 433. https://doi.org/10.3390/cancers13030433
APA StyleLucarini, V., Melaiu, O., Tempora, P., D’Amico, S., Locatelli, F., & Fruci, D. (2021). Dendritic Cells: Behind the Scenes of T-Cell Infiltration into the Tumor Microenvironment. Cancers, 13(3), 433. https://doi.org/10.3390/cancers13030433