
Printing the Pathway Forward in Bone Metastatic Cancer Research: Applications of 3D Engineered Models and Bioprinted Scaffolds to Recapitulate the Bone–Tumor Niche



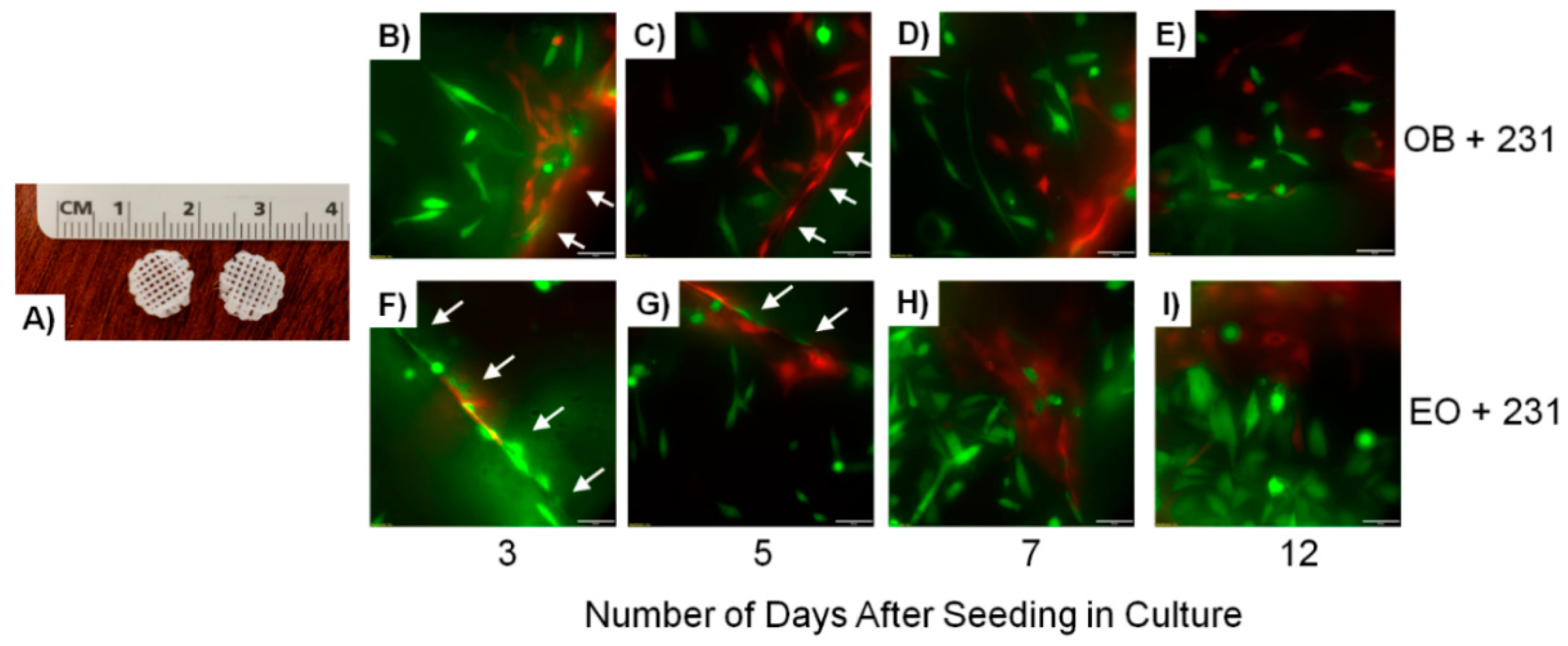{kind=link}
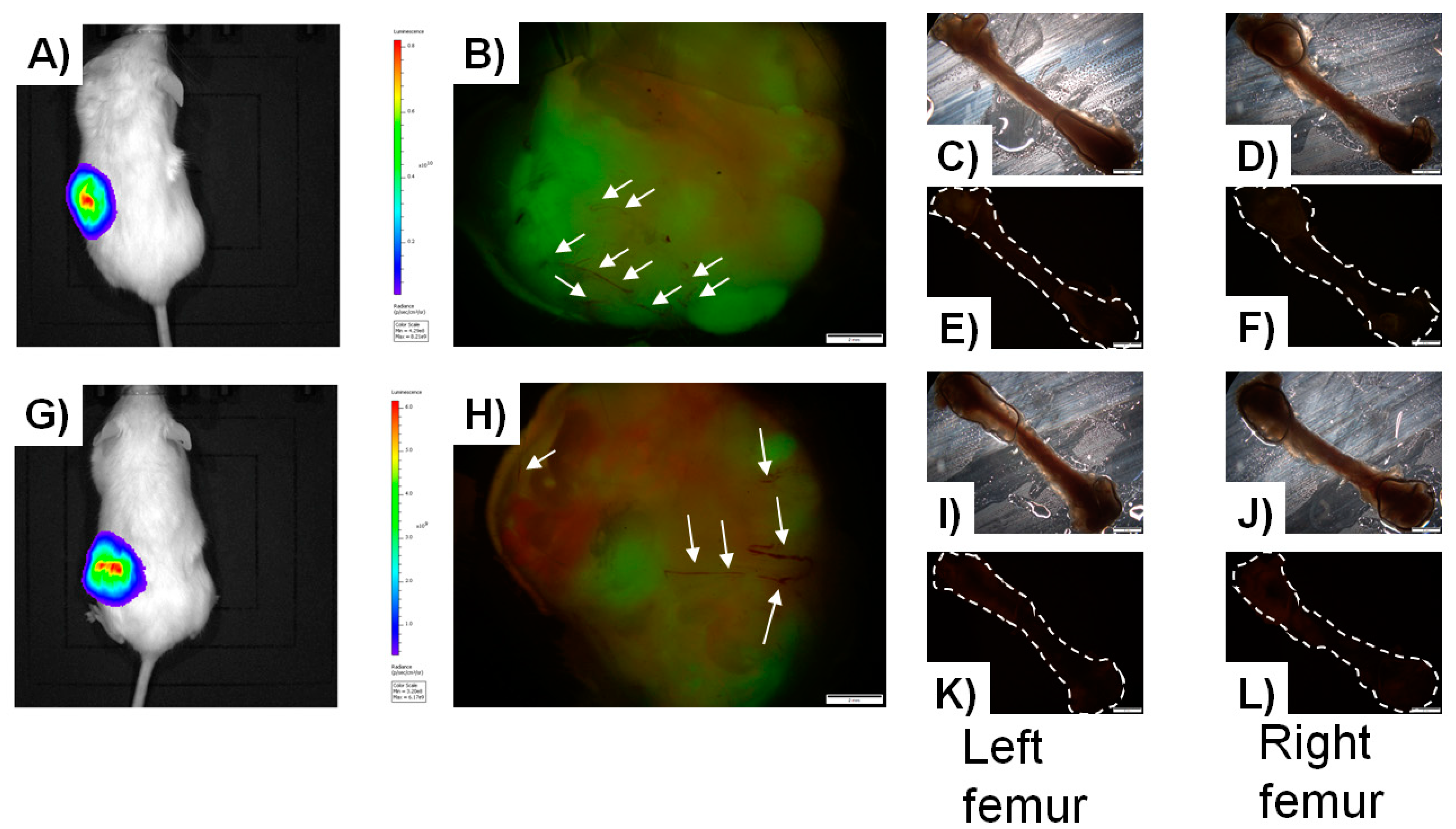{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Gross Anatomy of Bone and Bone Physiology
3. Bone Remodeling during Disease
4. Bone Is a Favored Site of Cancer Metastasis
4.1. Breast Cancer Cells Preferentially Metastasize to Bone
4.2. Multiple Myeloma Metastases to the Bone
4.3. Prostate Cancer Colonization of the Skeleton
4.4. Lung Cancer Metastasizes to the Skeleton
5. Models to Investigate Tumor-Bone Cell Interactions
5.1. Model Systems for Early Cancer Cell Dissemination to Bone
5.2. Models to Investigate Late-Stage Metastatic Cancer Cell Invasion, Colonization, and Survival in Bone
5.2.1. In-Vivo Models of Cancer Metastasis to Bone
5.2.2. Three-Dimensional Tissue-Engineered Models to Study Cancer–Bone Interactions during Disease Progression
Hydrogel Tissue Constructs
Bone-Like Scaffolds
Bioreactor-Based Engineered Tissue
Humanized Biomaterial Implants
6. Bioprinting
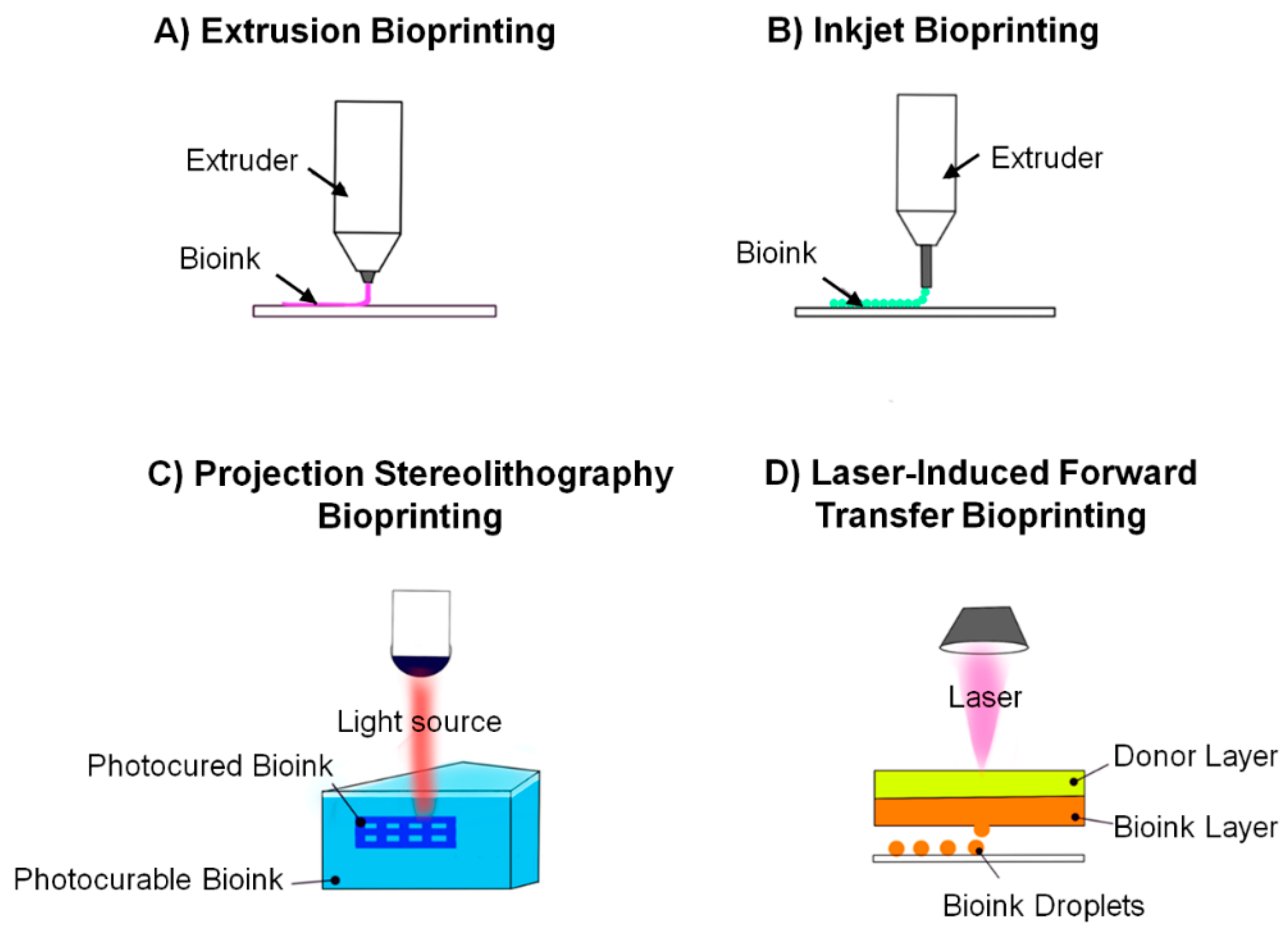
6.1. Basis of Bioprinting
6.2. Bioprinting Techniques
6.3. Bioinks
6.4. Applications of Bioprinting in Cancer
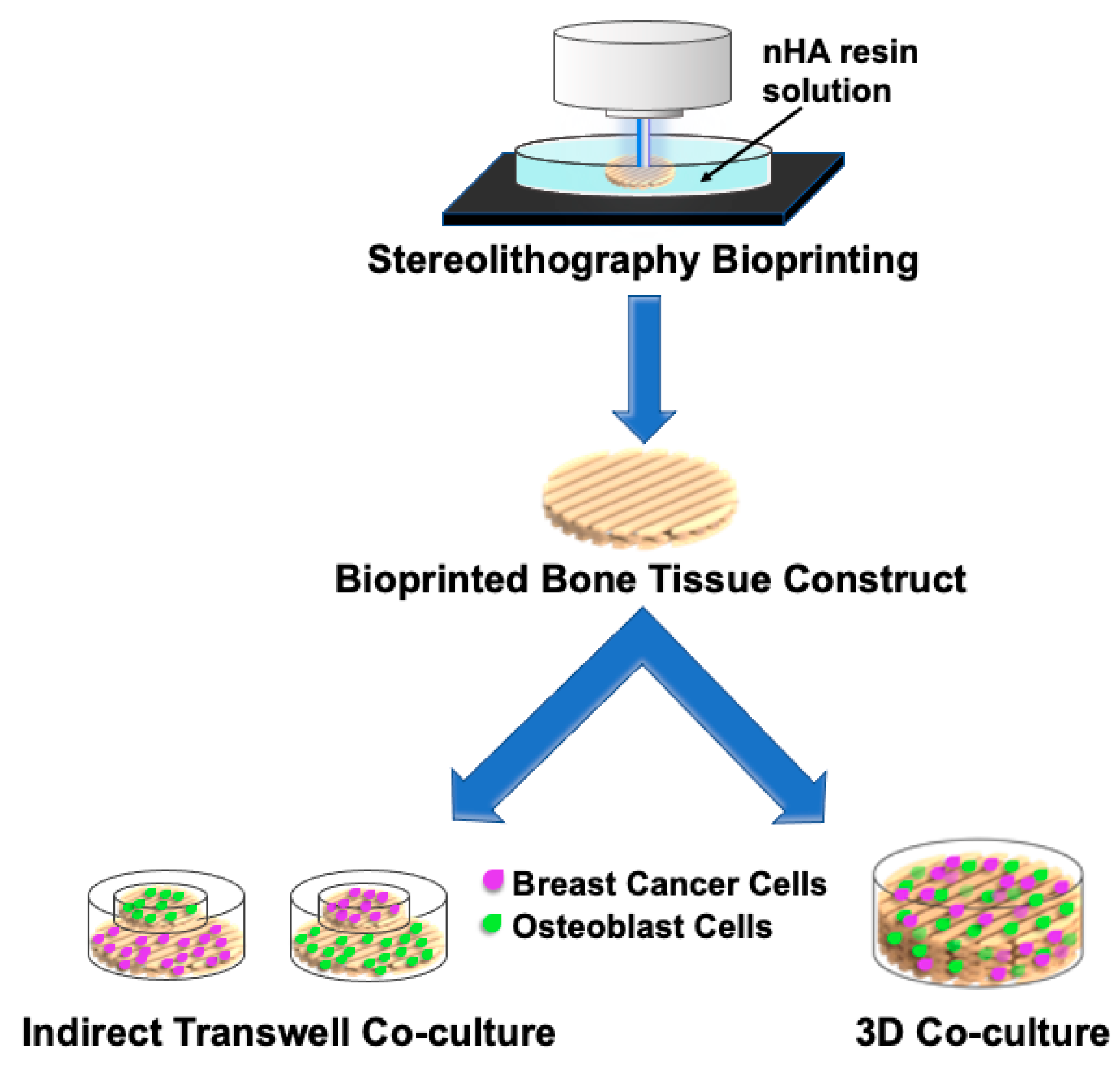
6.4.1. 3D Printed In-Vitro Models
6.4.2. Bioprinted Models for Breast Cancer Metastasis
7. Conclusions and Future Exploration
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.T. Mechanical properties of basement membrane in health and disease. Matrix Biol. 2017, 57–58, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Alford, A.I.; Kozloff, K.M.; Hankenson, K.D. Extracellular matrix networks in bone remodeling. Int. J. Biochem. Cell Biol. 2015, 65, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Gentili, C.; Cancedda, R. Cartilage and bone extracellular matrix. Curr. Pharm. Des. 2009, 15, 1334–1348. [Google Scholar] [CrossRef] [PubMed]
- Bone Metastasis: Symptoms and Diagnosis. Metastatic Breast Cancer Symptoms and Diagnosis. 2018. Available online: https://www.breastcancer.org/symptoms/types/recur_metast/metastic/bone (accessed on 11 December 2020).
- Miller, K.D.; Siegel, R.L.; Khan, R.; Jemal, A. Cancer statistics. Cancer Rehabil. 2018, 70, 7–30. [Google Scholar] [CrossRef]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell. Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Mastro, A.M.; Gay, C.V.; Welch, D. The skeleton as a unique environment for breast cancer cells. Clin. Exp. Metastasis 2003, 20, 275–284. [Google Scholar] [CrossRef]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef]
- Phadke, P.A.; Mercer, R.R.; Harms, J.F.; Jia, Y.; Frost, A.R.; Jewell, J.L.; Bussard, K.M.; Nelson, S.; Moore, C.; Kappes, J.C.; et al. Kinetics of metastatic breast cancer cell trafficking in bone. Clin. Cancer Res. 2006, 12, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2004, 20, 318–329. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Hernández, A.D.R. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Leonard, F.; Godin, B. 3D in vitro model for breast cancer research using magnetic levitation and bioprinting method. Toxic. Assess. 2016, 1406, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Reid, J.A.; Mollica, P.A.; Bruno, R.D.; Sachs, P.C. Consistent and reproducible cultures of large-scale 3D mammary epithelial structures using an accessible bioprinting platform. Breast Cancer Res. 2018, 20, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wergedal, J.E.; Mohan, S.; Lundy, M.; Baylink, D.J. Skeletal growth factor and other growth factors known to be present in bone matrix stimulate proliferation and protein synthesis in human bone cells. J. Bone Miner. Res. 1990, 5, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.P.; Cooke, M.M.; Christopherson, P.A.; Westfall, P.R.; McCarthy, G.M. Calcium hydroxyapatite promotes mitogenesis and matrix metalloproteinase expression in human breast cancer cell lines. Mol. Carcinog. 2001, 32, 111–117. [Google Scholar] [CrossRef]
- Petersen, O.W.; Rønnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064. [Google Scholar] [CrossRef] [Green Version]
- Dhurjati, R.; Krishnan, V.; Shuman, L.A.; Mastro, A.M.; Vogler, E.A. Metastatic breast cancer cells colonize and degrade three-dimensional osteoblastic tissue in vitro. Clin. Exp. Metastasis 2008, 25, 741–752. [Google Scholar] [CrossRef]
- Dhurjati, R.; Liu, X.; Gay, C.V.; Mastro, A.M.; Vogler, E.A. Extended-Term culture of bone cells in a compartmentalized bioreactor. Tissue Eng. 2006, 12, 3045–3054. [Google Scholar] [CrossRef]
- Krishnan, V.; Dhurjati, R.; Vogler, E.A.; Mastro, A.M. Osteogenesis in vitro: From pre-osteoblasts to osteocytes: A contribution from the Osteobiology Research Group, The Pennsylvania State University. In Vitro Cell. Dev. Biol. Anim. 2010, 46, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Shuman, L.A.; Sosnoski, D.M.; Dhurjati, R.; Vogler, E.A.; Mastro, A.M. Dynamic interaction between breast cancer cells and osteoblastic tissue: Comparison of Two- and Three-dimensional cultures. J. Cell. Physiol. 2011, 226, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Vogler, E.A.; Mastro, A.M. Three-Dimensional in vitro model to study osteobiology and osteopathology. J. Cell. Biochem. 2015, 116, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Vogler, E.A.; Sosnoski, D.M.; Mastro, A.M. In vitro mimics of bone remodeling and the vicious cycle of cancer in bone. J. Cell. Physiol. 2013, 229, 453–462. [Google Scholar] [CrossRef]
- Sosnoski, D.M.; Krishnan, V.; Kraemer, W.J.; Dunn-Lewis, C.; Mastro, A.M. Changes in cytokines of the bone microenvironment during breast cancer metastasis. Int. J. Breast Cancer 2012, 2012. [Google Scholar] [CrossRef]
- Lee, J.; Heckl, D.; Parekkadan, B. Multiple genetically engineered humanized microenvironments in a single mouse. Biomater. Res. 2016, 20, 19. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Li, M.; Milwid, J.; Dunham, J.; Vinegoni, C.; Gorbatov, R.; Iwamoto, Y.; Wang, F.; Shen, K.; Hatfield, K.; et al. Implantable microenvironments to attract hematopoietic stem/cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, 19638–19643. [Google Scholar] [CrossRef] [Green Version]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, 90036. [Google Scholar] [CrossRef]
- Chen, Y.; Jacamo, R.; Shi, Y.-X.; Wang, R.-Y.; Battula, V.L.; Konoplev, S.; Strunk, D.; Hofmann, N.A.; Reinisch, A.; Konopleva, M.; et al. Human extramedullary bone marrow in mice: A novel in vivo model of genetically controlled hematopoietic microenvironment. Blood 2012, 119, 4971–4980. [Google Scholar] [CrossRef] [Green Version]
- Marturano-Kruik, A.; Nava, M.M.; Yeager, K.; Chramiec, A.; Hao, L.; Robinson, S.; Guo, E.; Raimondi, M.T.; Vunjak-Novakovic, G. Human bone perivascular niche-on-a-chip for studying metastatic colonization. Proc. Natl. Acad. Sci. USA 2018, 115, 1256–1261. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Ha, L.; Cheng, G.; Wan, Y.; Xia, Y.; Sosnoski, D.M.; Mastro, A.M.; Zheng, S.-Y. A spontaneous 3D bone-on-a-chip for bone metastasis study of breast cancer cells. Small 2018, 14, e1702787. [Google Scholar] [CrossRef] [PubMed]
- Shupp, A.B.; Kolb, A.D.; Bussard, K.M. Novel Techniques to Study the Bone-Tumor Microenvironment, in Tumor Microenvironment: Advances in Experimental Medicine and Biology; Birbrair, A., Ed.; Springer: New York, NY, USA, 2020. [Google Scholar]
- Belgodere, J.A.; King, C.T.; Bursavich, J.B.; Burow, M.E.; Martin, E.C.; Jung, J.P. Engineering breast cancer microenvironments and 3D bioprinting. Front. Bioeng. Biotechnol. 2018, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Marks, S.C., Jr.; Odgren, P.R. Structure and Development of the Skeleton, in Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 3–16. [Google Scholar]
- Minguell, J.J.; Erices, A.; Conget, P. Mesenchymal stem cells. Exp. Biol. Med. 2001, 226, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Augat, P.; Schorlemmer, S. The role of cortical bone and its microstructure in bone strength. Age Ageing 2006, 35, ii27–ii31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florencio-Silva, R.; Sasso, G.R.d.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of bone tissue: Structure, function, and factors that influence bone cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mullender, M.M.; Van Der Meer, D.; Huiskes, H.R.; Lips, P. Osteocyte density changes in aging and osteoporosis. Bone 1996, 18, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Udagawa, N.; Takami, M.; Suda, T. Cells of bone: Osteoclast generation. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 109–126. [Google Scholar]
- Kanis, J.A.; McCloskey, E.V. Bone turnover and biochemical markers in malignancy. Cancer 1997, 80, 1538–1545. [Google Scholar] [CrossRef]
- Wilson, S.R.; Peters, C.; Saftig, P.; Brömme, D.; Cathepsin, K. Activity-Dependent regulation of osteoclast actin ring formation and bone resorption. J. Biol. Chem. 2009, 284, 2584–2592. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.C. How the osteoclast degrades bone. Bioessays 1998, 20, 837–846. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Therapeutic implications of suppressing osteoclast formation versus function. Rheumatology 2016, 55, ii61–ii63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenbeck, G. Formation and function of the ruffled border in osteoclasts. Semin. Cell Dev. Biol. 2002, 13, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Hauschka, P.V.; Mavrakos, A.E.; Iafrati, M.D.; Doleman, S.E.; Klagsbrun, M. Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-Sepharose. J. Biol. Chem. 1986, 261, 12665–12674. [Google Scholar] [CrossRef]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell … and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [Green Version]
- Sottnik, J.L.; Dai, J.; Zhang, H.; Campbell, B.; Keller, E.T. Tumor-Induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res. 2015, 75, 2151–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, C.; Haugh, M.; Schaffler, M.; Majeska, R.; McNamara, L.M. Osteocyte differentiation is regulated by extracellular matrix stiffness and intercellular separation. J. Mech. Behav. Biomed. Mater. 2013, 28, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Robling, A.G.; Turner, C.H. Mechanical signaling for bone modeling and remodeling. Crit. Rev. Eukaryot. Gene Expr. 2009, 19, 319–338. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Wolff, J. Das Gesetz der Transformation der Knochen; Hirschwald: Berlin, Germany, 1892. [Google Scholar]
- Klein-Nulend, J.; Bacabac, R.G.; Bakker, A.D. Mechanical loading and how it affects bone cells: The role of the osteocyte cytoskeleton in maintaining our skeleton. Eur. Cells Mater. 2012, 24, 278–291. [Google Scholar] [CrossRef]
- Klein-Nulend, J.; Van Der Plas, A.; Semeins, C.M.; Ajubi, N.E.; Erangos, J.A.; Nijweide, P.J.; Burger, E. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. 1995, 9, 441–445. [Google Scholar] [CrossRef]
- Mastro, A.M.; Gay, C.V.; Welch, D.; Donahue, H.J.; Jewell, J.; Mercer, R.; DiGirolamo, D.; Chislock, E.M.; Guttridge, K. Breast cancer cells induce osteoblast apoptosis: A possible contributor to bone degradation. J. Cell. Biochem. 2004, 91, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Josse, J.; Velard, F.; Gangloff, S. Staphylococcus aureus vs. osteoblast: Relationship and consequences in osteomyelitis. Front. Cell. Infect. Microbiol. 2015, 5, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, C.; DeBerg, M.A.; Bellahcène, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheum. 2008, 58, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Mazzucchelli, G.; Lambert, C.; Comblain, F.; Depauw, E.; Henrotin, Y. Comparison of secretome from osteoblasts derived from sclerotic versus non-sclerotic subchondral bone in OA: A pilot study. PLoS ONE 2018, 13, e0194591. [Google Scholar] [CrossRef]
- Yoneda, T. Mechanisms of preferential metastasis of breast cancer to bone—(Review). Int. J. Oncol. 1996, 9, 103–109. [Google Scholar] [CrossRef]
- Lentino, J.R. Prosthetic joint infections: Bane of orthopedists, challenge for infectious disease specialists. Clin. Infect. Dis. 2003, 36, 1157–1161. [Google Scholar] [CrossRef]
- Berendt, T.; Byren, I. Bone and joint infection. Clin. Med. 2004, 4, 510–518. [Google Scholar] [CrossRef]
- Tillander, J.; Hagberg, K.; Berlin, Ö.; Hagberg, L.; Brånemark, R. Osteomyelitis risk in patients with transfemoral amputations treated with osseointegration prostheses. Clin. Orthop. Relat. Res. 2017, 475, 3100–3108. [Google Scholar] [CrossRef] [Green Version]
- Kellesarian, S.V.; Javed, F.; Romanos, G.E. Osteomyelitis arising around osseointegrated dental implants: A systematic review. Implant Dent. 2018, 27, 226–235. [Google Scholar] [CrossRef]
- Semel, G.; Wolff, A.; Shilo, D.; Akrish, S.; Emodi, O.; Rachmiel, A. Mandibular osteomyelitis associated with dental implants. A case series. Eur. J. Oral Implantol. 2016, 9, 435–442. [Google Scholar]
- Bosse, M.J.; Gruber, H.E.; Ramp, W.K. Internalization of bacteria by osteoblasts in a patient with recurrent, long-term osteomyelitis: A case report. JBJS Case Connect. 2005, 87, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Dapunt, U.; Maurer, S.; Giese, T.; Gaida, M.M.; Hänsch, G.M. The macrophage inflammatory proteins MIP1α(CCL3) and MIP2α(CXCL2) in implant-associated osteomyelitis: Linking inflammation to bone degradation. Mediat. Inflamm. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bost, K.L.; Bento, J.L.; Ellington, J.K.; Marriott, I.; Hudson, M.C. Induction of colony-stimulating factor expression following staphylococcus or salmonellainteraction with mouse or human osteoblasts. Infect. Immun. 2000, 68, 4834–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, K.M.; Friedland, J.S. Regulation of chemokine gene expression and secretion in Staphylococcus aureus-infected osteoblasts. Microbes Infect. 2004, 6, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Ning, R.; Zhang, X.; Guo, X.; Li, Q. Staphylococcus aureus regulates secretion of interleukin-6 and monocyte chemoattractant protein-1 through activation of nuclear factor kappaB signaling pathway in human osteoblasts. Braz. J. Infect. Dis. 2011, 15, 189–194. [Google Scholar]
- Gasper, N.A.; Petty, C.C.; Schrum, L.W.; Marriott, I.; Bost, K.L. Bacterium-Induced CXCL10 secretion by osteoblasts can be mediated in part through toll-like receptor 4. Infect. Immun. 2002, 70, 4075–4082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somayaji, S.N.; Ritchie, S.; Sahraei, M.; Marriott, I.; Hudson, M.C. Staphylococcus aureus induces expression of receptor activator of NF-kappaB ligand and prostaglandin E2 in infected murine osteoblasts. Infect. Immun. 2008, 76, 5120–5126. [Google Scholar] [CrossRef] [Green Version]
- Widaa, A.; Claro, T.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein a plays a critical role in mediating bone destruction and bone loss in osteomyelitis. PLoS ONE 2012, 7, e40586. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Pesesse, L.; Gabay, O.; Delcour, J.-P.; Msika, P.; Baudouin, C.; Henrotin, Y. Regulation of subchondral bone osteoblast metabolism by cyclic compression. Arthritis Rheum. 2012, 64, 1193–1203. [Google Scholar] [CrossRef]
- Bianco, D.; Todorov, A.; Cengic, T.; Pagenstert, G.; Schären, S.; Netzer, C.; Hügle, T.; Geurts, J. Alterations of subchondral bone progenitor cells in human knee and hip osteoarthritis lead to a bone sclerosis phenotype. Int. J. Mol. Sci. 2018, 19, 475. [Google Scholar] [CrossRef]
- Martineau, X.; Abed, É.; Martel-Pelletier, J.; Pelletier, J.-P.; Lajeunesse, D. Alteration of Wnt5a expression and of the non-canonical Wnt/PCP and Wnt/PKC-Ca2+ pathways in human osteoarthritis osteoblasts. PLoS ONE 2017, 12, e0180711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinder, M.; Chislock, E.M.; Bussard, K.M.; Shuman, L.; Mastro, A.M. Metastatic breast cancer induces an osteoblast inflammatory response. Exp. Cell Res. 2008, 314, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2007, 27, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, C.; Morris, M.J.; Den, R.; Coleman, R.E. Current perspectives on bone metastases in castrate-resistant prostate cancer. Cancer Metastasis Rev. 2018, 37, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logothetis, C.J.; Lin, S.-H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer 2005, 5, 21–28. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res. 2019, 21, 31. [Google Scholar] [CrossRef] [Green Version]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Khoo, W.H.; Terry, R.L.; Down, J.M.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef] [Green Version]
- Mundy, G.R. Bone Remodeling and Its Disorders; Martin Dunitz Ltd.: London, UK, 1999. [Google Scholar]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Liede, A.; Jerzak, K.J.; Hernandez, R.K.; Wade, S.W.; Sun, P.; Narod, S.A. The incidence of bone metastasis after early-stage breast cancer in Canada. Breast Cancer Res. Treat. 2016, 156, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.M.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Uzzo, R.; Amato, R.J.; Ellis, G.K.; Hakimian, B.; Roodman, G.D.; Smith, M.R. The science and practice of bone health in oncology: Managing bone loss and metastasis in patients with solid tumors. J. Natl. Compr. Cancer Netw. 2009, 7 (Suppl. S7), S-1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manders, K.; Van De Poll-Franse, L.V.; Creemers, G.-J.; Vreugdenhil, G.; Van Der Sangen, M.J.C.; Nieuwenhuijzen, G.A.P.; Roumen, R.M.H.; Voogd, A.C. Clinical management of women with metastatic breast cancer: A descriptive study according to age group. BMC Cancer 2006, 6, 179. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Guise, T.A.; Mundy, G.R. Cancer and bone. Endocr. Rev. 1998, 19, 18–54. [Google Scholar]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases: Fig. 1. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [Green Version]
- Guise, T.A. Molecular mechanisms of osteolytic bone metastases. Cancer 2000, 88, 2892–2898. [Google Scholar] [CrossRef]
- Marathe, D.D.; Marathe, A.; Mager, D.E. Integrated model for denosumab and ibandronate pharmacodynamics in postmenopausal women. Biopharm. Drug Dispos. 2011, 32, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Anagnostis, P.; Vakalopoulou, S.; Christoulas, D.; Paschou, S.A.; Papatheodorou, A.; Garipidou, V.; Kokkoris, P.; Terpos, E. The role of sclerostin/dickkopf-1 and receptor activator of nuclear factor kB ligand/osteoprotegerin signalling pathways in the development of osteoporosis in patients with haemophilia A and B: A cross-sectional study. Haemophilia 2017, 24, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.-J.; Liang, Q.; Zhong, J.-H.; Zhu, M.; Meng, F.-Y.; Wu, N.; Liang, R.; Yuan, B.-Y. Ibandronate to treat skeletal-related events and bone pain in metastatic bone disease or multiple myeloma: A meta-analysis of randomised clinical trials. BMJ Open 2015, 5, e007258. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.N.; Lie, J.D.; Wan, C.K.V.; Cameron, M.; Austel, A.G.; Nguyen, J.K.; Van, K.; Hyun, D. Osteoporosis: A review of treatment options. Pharm. Ther. 2018, 43, 92–104. [Google Scholar]
- Greenberg, A.J.; Rajkumar, S.V.; Therneau, T.M.; Singh, P.; Dispenzieri, A.; Kumar, S.K. Relationship between initial clinical presentation and the molecular cytogenetic classification of myeloma. Leukemia 2014, 28, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, S.; Roodman, G.D. Multiple myeloma and bone: The fatal interaction. Cold Spring Harb. Perspect. Med. 2018, 8, a031286. [Google Scholar] [CrossRef]
- Bataille, R.; Chappard, D.; Marcelli, C.; Dessauw, P.; Sany, J.; Baldet, P.; Alexandre, C. Mechanisms of bone destruction in multiple myeloma: The importance of an unbalanced process in determining the severity of lytic bone disease. J. Clin. Oncol. 1989, 7, 1909–1914. [Google Scholar] [CrossRef]
- Ehrlich, L.A.; Chung, H.Y.; Ghobrial, I.; Choi, S.J.; Morandi, F.; Colla, S.; Rizzoli, V.; Roodman, G.D.; Giuliani, N. IL-3 is a potential inhibitor of osteoblast differentiation in multiple myeloma. Blood 2005, 106, 1407–1414. [Google Scholar] [CrossRef]
- Silbermann, R.; Bolzoni, M.; Storti, P.; Guasco, D.; Bonomini, S.; Zhou, D.; Wu, J.; Anderson, J.L.; Windle, J.J.; Aversa, F.; et al. Bone marrow monocyte-/macrophage-derived activin A mediates the osteoclastogenic effect of IL-3 in multiple myeloma. Leukemia 2014, 28, 951–954. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694. [Google Scholar] [CrossRef]
- Waning, D.L.; Mohammad, S.K.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef]
- Nyman, J.S.; Merkel, A.R.; Uppuganti, S.; Nayak, B.; Rowland, B.; Makowski, A.J.; Oyajobi, B.O.; Sterling, J.A. Combined treatment with a transforming growth factor beta inhibitor (1D11) and bortezomib improves bone architecture in a mouse model of myeloma-induced bone disease. Bone 2016, 91, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, S.; Del Prete, D.; Jin, S.; Sun, Q.; Huston, A.J.; Kostov, F.E.; Sammut, B.; Hong, C.-S.; Anderson, J.L.; Patrene, K.D.; et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 2011, 118, 6871–6880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, S.; Fulciniti, M.; Yan, H.; Vallet, S.; Eda, H.; Patel, K.; Santo, L.; Cirstea, D.; Hideshima, T.; Schirtzinge, L.; et al. In vivo and in vitro effects of a novel anti-Dkk1 neutralizing antibody in multiple myeloma. Bone 2013, 53, 487–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.-T.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.J.; Scher, H.I. Clinical approaches to osseous metastases in prostate cancer. Oncologist 2003, 8, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Docherty, F.E.; Brown, H.K.; Reeves, K.J.; Fowles, A.C.M.; Ottewell, P.D.; Dear, T.N.; Holen, I.; Croucher, P.I.; Eaton, C.L. Prostate cancer cells preferentially home to osteoblast-rich areas in the early stages of bone metastasis: Evidence from in vivo models. J. Bone Miner. Res. 2014, 29, 2688–2696. [Google Scholar] [CrossRef] [Green Version]
- Roudier, M.P.; Morrissey, C.; True, L.D.; Higano, C.S.; Vessella, R.L.; Ott, S.M. Histopathological assessment of prostate cancer bone osteoblastic metastases. J. Urol. 2008, 180, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Eastham, J.A. Bone health in men receiving androgen deprivation therapy for prostate cancer. J. Urol. 2007, 177, 17–24. [Google Scholar] [CrossRef]
- Sekita, A.; Matsugaki, A.; Nakano, T. Disruption of collagen/apatite alignment impairs bone mechanical function in osteoblastic metastasis induced by prostate cancer. Bone 2017, 97, 83–93. [Google Scholar] [CrossRef]
- Matsugaki, A.; Aramoto, G.; Ninomiya, T.; Sawada, H.; Hata, S.; Nakano, T. Abnormal arrangement of a collagen/apatite extracellular matrix orthogonal to osteoblast alignment is constructed by a nanoscale periodic surface structure. Biomaterials 2015, 37, 134–143. [Google Scholar] [CrossRef]
- Charhon, S.A.; Chapuy, M.C.; Delvin, E.E.; Valentin-Opran, A.; Edouard, C.M.; Meunier, P.J. Histomorphometric analysis of sclerotic bone metastases from prostatic carcinoma with special reference to osteomalacia. Cancer 1983, 51, 918–924. [Google Scholar] [CrossRef]
- Clarke, N.W.; McClure, J.; George, N.J.R. Morphometric evidence for bone resorption and replacement in prostate cancer. BJU Int. 1991, 68, 74–80. [Google Scholar] [CrossRef]
- Wan, X.; Corn, P.G.; Yang, J.; Palanisamy, N.; Starbuck, M.W.; Efstathiou, E.; Li-Ning-Tapia, E.M.; Zurita, A.J.; Aparicio, A.; Ravoori, M.K.; et al. Prostate cancer cell–stromal cell crosstalk via FGFR1 mediates antitumor activity of dovitinib in bone metastases. Sci. Transl. Med. 2014, 6, 252ra122. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Yang, J.; Peleg, S.; Sikes, C.R.; Kreimann, E.L.; Daliani, D.; Olive, M.; Raymond, K.A.; Janus, T.J.; Logothetis, C.; et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clin. Cancer Res. 2003, 9, 2587–2597. [Google Scholar] [PubMed]
- Oberneder, R.; Riesenberg, R.; Kriegmair, M.; Bitzer, U.; Klammert, R.; Schneede, P.; Hofstetter, A.; Pantel, K. Immunocytochemical detection and phenotypic characterization of micrometastatic tumour cells in bone marrow of patients with prostate cancer. Urol. Res. 1994, 22, 3–8. [Google Scholar] [CrossRef]
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carducci, M.A.; Nelson, J.B.; Bowling, M.K.; Rogers, T.; Eisenberger, M.A.; Sinibaldi, V.; Donehower, R.; Leahy, T.L.; Carr, R.A.; Isaacson, J.D.; et al. Atrasentan, an endothelin-receptor antagonist for refractory adenocarcinomas: Safety and pharmacokinetics. J. Clin. Oncol. 2002, 20, 2171–2180. [Google Scholar] [CrossRef]
- Carducci, M.A.; Padley, R.J.; Breul, J.; Vogelzang, N.J.; Zonnenberg, B.A.; Daliani, D.D.; Schulman, C.C.; Nabulsi, A.A.; Humerickhouse, R.A.; Weinberg, M.A.; et al. Effect of endothelin-a receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: A randomized, phase ii, placebo-controlled trial. J. Clin. Oncol. 2003, 21, 679–689. [Google Scholar] [CrossRef]
- Suominen, M.I.; Fagerlund, K.M.; Rissanen, J.P.; Konkol, Y.M.; Morko, J.P.; Peng, Z.; Alhoniemi, E.J.; Laine, S.K.; Corey, E.; Mumberg, D.; et al. Radium-223 inhibits osseous prostate cancer growth by dual targeting of cancer cells and bone microenvironment in mouse models. Clin. Cancer Res. 2017, 23, 4335–4346. [Google Scholar] [CrossRef] [Green Version]
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.; Falconer, A.M.D.; Wilkinson, D.J.; Europe-Finner, G.N.; Litherland, G.J.; Rowan, A.D. Protein kinase D3 modulates MMP1 and MMP13 expression in human chondrocytes. PLoS ONE 2018, 13, e0195864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.-H.; Tan, T.-W.; Fu, W.-M.; Yang, R.-S. Involvement of matrix metalloproteinase-9 in stromal cell-derived factor-1/CXCR4 pathway of lung cancer metastasis. Carcinogenesis 2007, 29, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Yamada, K.; Sugiura, T.; Hida, T.; Mitsudomi, T. Predictors of surivival in patients with bone metastasis of lung cancer. Clin. Ortho. Relat. Res. 2008, 466, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Hill, C.A. Bronchioloalveolar carcinoma: A review. Radiology 1984, 150, 15–20. [Google Scholar] [CrossRef]
- Gordon, J.A.R.; Tye, C.E.; Sampaio, A.V.; Underhill, T.M.; Hunter, G.K.; Goldberg, H.A. Bone sialoprotein expression enhances osteoblast differentiation and matrix mineralization in vitro. Bone 2007, 41, 462–473. [Google Scholar] [CrossRef]
- Kang, E.J.; Lee, S.Y.; Kim, H.J.; Min, K.H.; Hur, G.-Y.; Shim, J.J.; Kang, K.H.; Oh, S.C.; Seo, J.H.; Lee, S.; et al. Prognostic factors and skeletal-related events in patients with small cell lung cancer with bone metastases at the time of diagnosis. Oncologist 2016, 90, 103–111. [Google Scholar] [CrossRef]
- Lang, J.; Zhao, Q.; He, Y.; Yu, X. Bone turnover markers and novel biomarkers in lung cancer bone metastases. Biomarkers 2018, 23, 518–526. [Google Scholar] [CrossRef]
- Papotti, M.; Kalebic, T.; Volante, M.; Chiusa, L.; Bacillo, E.; Cappia, S.; Lausi, P.O.; Novello, S.; Borasio, P.; Scagliotti, G.V. Bone sialoprotein is predictive of bone metastases in resectable non–small-cell lung cancer: A retrospective case-control study. J. Clin. Oncol. 2006, 24, 4818–4824. [Google Scholar] [CrossRef]
- Brown, J.E.; Cook, R.J.; Major, P.; Lipton, A.; Saad, F.; Smith, M.R.; Lee, K.-A.; Zheng, M.; Hei, Y.-J.; Coleman, R.E. Bone turnover markers as predictors of skeletal complications in prostate cancer, lung cancer, and other solid tumors. J. Natl. Cancer Inst. 2005, 97, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.; Demers, L.M.; Gouveia-Oliveira, A.; Schaller, J.; Costa, E.B.; De Moura, M.C.; Lipton, A. Prospective evaluation of the peptide-bound collagen type I cross-links N-telopeptide and C-telopeptide in predicting bone metastases status. J. Clin. Oncol. 2002, 20, 850–856. [Google Scholar] [PubMed]
- Liu, B.; Zhao, Y.; Yuan, J.; Zeng, L.; Sun, R.; Meng, X.; Yang, S. Elevated N-telopeptide as a potential diagnostic marker for bone metastasis in lung cancer: A meta-analysis. PLoS ONE 2017, 12, e0187860. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yan, S.; Su, Y.; Chen, H.; Wang, S.; Sun, B.; Zhang, L. Serum cross-linked N-telopeptide of type I collagen as a biomarker of bone metastases for patients with lung cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2018, 11, 12864–12869. [Google Scholar]
- Hu, Z.; Lin, D.; Yuan, J.; Xiao, T.; Zhang, H.; Sun, W.; Han, N.; Ma, Y.; Di, X.; Gao, M.; et al. Overexpression of osteopontin is associated with more aggressive phenotypes in human non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 4646–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothari, A.N.; Arffa, M.; Chang, V.; Blackwell, R.H.; Syn, W.-K.; Zhang, J.; Mi, Z.; Kuo, P.C. Osteopontin—A master regulator of epithelial-mesenchymal transition. J. Clin. Med. 2016, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A. Models, mechanisms, and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Braun, S.; Naume, B. Circulating and disseminated tumor cells. J. Clin. Oncol. 2005, 23, 1623–1626. [Google Scholar] [CrossRef]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef]
- Quayle, L.; Ottewell, P.D.; Holen, I. Bone metastasis: Molecular mechanisms implicated in tumour cell dormancy in breast and prostate cancer. Curr. Cancer Drug Targets 2015, 15, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Scimeca, M.; Antonacci, C.; Toschi, N.; Giannini, E.; Bonfiglio, R.; Buonomo, C.O.; Pistolese, C.A.; Tarantino, U.; Bonanno, E. Breast osteoblast-like cells: A reliable early marker for bone metastases from breast cancer. Clin. Breast Cancer 2018, 18, e659–e669. [Google Scholar] [CrossRef]
- Bodenstine, T.M.; Beck, B.H.; Cao, X.; Cook, L.M.; Ismail, A.; Powers, S.J.K.; Powers, J.K.; Mastro, A.M.; Welch, D. Pre-osteoblastic MC3T3-E1 cells promote breast cancer growth in bone in a murine xenograft model. Chin. J. Cancer 2011, 30, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Ghajar, C.M.; Bissell, M.J. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv. Drug Deliv. Rev. 2014, 69-70, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse models of metastasis: Progress and prospects. Dis. Model. Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Christofori, G. Rebuilding cancer metastasis in the mouse. Mol. Oncol. 2013, 7, 283–296. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; De Almeida, D.L.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Naumov, G.N.; Macdonald, I.C.; Weinmeister, P.M.; Kerkvliet, N.; Nadkarni, K.V.; Wilson, S.M.; Morris, V.L.; Groom, A.C.; Chambers, A.F. Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res. 2002, 62, 2162–2168. [Google Scholar]
- Chen, Y.-C.; Mastro, A.M.; Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Vogler, E.A. Abstract 4891: Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Tumor Biol. 2014, 32, 335–344. [Google Scholar] [CrossRef]
- Angeloni, V.; Negrini, N.C.; De Marco, C.; Bertoldi, S.; Tanzi, M.C.; Daidone, M.G.; Farè, S. Polyurethane foam scaffold as in vitro model for breast cancer bone metastasis. Acta Biomater. 2017, 63, 306–316. [Google Scholar] [CrossRef]
- Khanna, C.; Hunter, K. Modeling metastasis in vivo. Carcinogenesis 2005, 26, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, R.B. Human breast cancer cell line xenografts as models of breast cancer—The immunobiologies of recipient mice and the characteristics of several tumorigenic cell lines. Breast Cancer Res. Treat. 1996, 39, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.E.; Ottewell, P.D.; Rucci, N.; Peyruchaud, O.; Pagnotti, G.M.; Chiechi, A.; Buijs, J.T.; Sterling, J.A. Murine models of breast cancer bone metastasis. BoneKEy Rep. 2016, 5, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shupp, A.B.; Kolb, A.D.; Bussard, K.M. Novel techniques to study the bone-tumor microenvironment. Adv. Exp. Med. Biol. 2020, 1225, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, T.; Williams, P.J.; Hiraga, T.; Niewolna, M.; & Nishimura, R. A bone-seeking clone exhibits different biological properties from the MDA-MD-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner Res. 2001, 16, 1486–1495. [Google Scholar] [CrossRef]
- Cassereau, L.; Miroshnikova, Y.A.; Ou, G.; Lakins, J.; Weaver, V.M. A 3D tension bioreactor platform to study the interplay between ECM stiffness and tumor phenotype. J. Biotechnol. 2015, 193, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Matei, I.; Rampersaud, S.; Lyden, D. Engineered niches model the onset of metastasis. Nat. Biomed. Eng. 2018, 2, 885–887. [Google Scholar] [CrossRef]
- Vanderburgh, J.P.; Guelcher, S.A.; Sterling, J.A. 3D bone models to study the complex physical and cellular interactions between tumor and the bone microenvironment. J. Cell. Biochem. 2018, 119, 5053–5059. [Google Scholar] [CrossRef]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Page, J.M.; Merkel, A.R.; Ruppender, N.S.; Guo, R.; Dadwal, U.C.; Cannonier, S.; Basu, S.; Guelcher, S.A.; Sterling, J.A. Matrix rigidity regulates the transition of tumor cells to a bone-destructive phenotype through integrin B3 and TGF-B receptor type II. Biomaterials 2015, 64, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Matsugaki, A.; Sekita, A.; Nakano, T. Alteration of osteoblast arrangement via direct attack by cancer cells: New insights into bone metastasis. Sci. Rep. 2017, 7, 44824. [Google Scholar] [CrossRef] [Green Version]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, B.P.; Naved, B.A.; Nyberg, E.L.; Dias, M.; Holmes, C.A.; Elisseeff, J.H.; Dorafshar, A.H.; Grayson, W. Three-Dimensional printing of bone extracellular matrix for craniofacial regeneration. ACS Biomater. Sci. Eng. 2016, 2, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Temple, J.P.; Hutton, D.L.; Hung, B.P.; Huri, P.Y.; Cook, C.A.; Kondragunta, R.; Jia, X.; Grayson, W. Engineering anatomically shaped vascularized bone grafts with hASCs and 3D-printed PCL scaffolds. J. Biomed. Mater. Res. Part A 2014, 102, 4317–4325. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.A.; Kwak, J.-G.; Peyton, S.R.; Lee, J. Implantable pre-metastatic niches for the study of the microenvironmental regulation of disseminated human tumour cells. Nat. Biomed. Eng. 2018, 2, 915–929. [Google Scholar] [CrossRef]
- Patricio, T.; Domingos, M.; Gloria, A.; Bártolo, P. Characterisation of PCL and PCL/PLA scaffolds for tissue engineering. Procedia CIRP 2013, 5, 110–114. [Google Scholar] [CrossRef]
- Kolb, A.D.; Shupp, A.B.; Bussard, K.M. Unpublished Data; Thomas Jefferson University: Philadelphia, PA, USA, 2021. [Google Scholar]
- Dababneh, A.B.; Ozbolat, I.T. Bioprinting technology: A current state-of-the-art review. J. Manuf. Sci. Eng. 2014, 136, 061016. [Google Scholar] [CrossRef]
- Cleversey, C.; Robinson, M.; Willerth, S.M. 3D printing breast tissue models: A review of past work and directions for future work. Micromachines 2019, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Klebe, R.J. Cytoscribing: A method for micropositioning cells and the construction of two- and three-dimensional synthetic tissues. Exp. Cell Res. 1988, 179, 362–373. [Google Scholar] [CrossRef]
- Ashammakhi, N.; Hasan, A.; Kaarela, O.; Byambaa, B.; Sheikhi, A.; Gaharwar, A.K.; Khademhosseini, A. Advancing frontiers in bone bioprinting. Adv. Healthc. Mater. 2019, 8, e1801048. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Hubbell, K.; Schilling, A.F.; Dai, G.; Cui, X. Bioprinting cartilage tissue from mesenchymal stem cells and PEG hydrogel. Toxic. Assess. 2017, 1612, 391–398. [Google Scholar] [CrossRef]
- Gao, G.; Schilling, A.F.; Yonezawa, T.; Wang, J.; Dai, G.; Cui, X. Bioactive nanoparticles stimulate bone tissue formation in bioprinted three-dimensional scaffold and human mesenchymal stem cells. Biotechnol. J. 2014, 9, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.; Huang, G.; Liu, J.; Zhang, X.; Ma, Y.; Lu, T.; Xu, F. Bioprinting-Based high-throughput fabrication of three-dimensional MCF-7 human breast cancer cellular spheroids. Engineering 2015, 1, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Mirani, B.; Pagan, E.; Shojaei, S.; Duchscherer, J.; Toyota, B.D.; Ghavami, S.; Akbari, M. A 3D bioprinted hydrogel mesh loaded with all-trans retinoic acid for treatment of glioblastoma. Eur. J. Pharmacol. 2019, 854, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liu, C.; Wang, Z.; Luo, Y. 3D printed core-shell hydrogel fiber scaffolds with NIR-triggered drug release for localized therapy of breast cancer. Int. J. Pharm. 2020, 580, 119219. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Y.; Ozbolat, I.T. A hybrid bioprinting approach for scale-up tissue fabrication. J. Manuf. Sci. Eng. 2014, 136, 061013. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Cui, X. Three-Dimensional bioprinting in tissue engineering and regenerative medicine. Biotechnol. Lett. 2015, 38, 203–211. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar]
- Gudapati, H.; Dey, M.; Ozbolat, I.T. A comprehensive review on droplet-based bioprinting: Past, present and future. Biomaterials 2016, 102, 20–42. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Guvendiren, M. Recent advances in bioink design for 3D bioprinting of tissues and organs. Front. Bioeng. Biotechnol. 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placone, J.K.; Engler, A.J. Recent advances in extrusion-based 3D printing for biomedical applications. Adv. Healthc. Mater. 2017, 7, e1701161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colosi, C.; Shin, S.R.; Manoharan, V.; Massa, S.; Costantini, M.; Barbetta, A.; Dokmeci, M.R.; Dentini, M.; Khademhosseini, A. Microfluidic bioprinting of heterogeneous 3D tissue constructs using low-viscosity bioink. Adv. Mater. 2016, 28, 677–684. [Google Scholar] [CrossRef]
- Ding, S.; Feng, L.; Wu, J.; Zhu, F.; Yao, R.; Tan, Z. Bioprinting of stem cells: Interplay of bioprinting process, bioinks, and stem cell properties. ACS Biomater. Sci. Eng. 2018, 4, 3108–3124. [Google Scholar] [CrossRef] [PubMed]
- Skoog, S.A.; Goering, P.L.; Narayan, R.J. Stereolithography in tissue engineering. J. Mater. Sci. Mater. Med. 2014, 25, 845–856. [Google Scholar] [CrossRef]
- Li, J.; Wu, C.; Chu, P.K.; Gelinsky, M. 3D printing of hydrogels: Rational design strategies and emerging biomedical applications. Mater. Sci. Eng. Rep. 2020, 140, 100543. [Google Scholar] [CrossRef]
- Lee, A.; Hudson, A.; Shiwarski, D.J.; Tashman, J.W.; Hinton, T.J.; Yerneni, S.S.; Bliley, J.M.; Campbell, P.G.; Feinberg, A.W. 3D bioprinting of collagen to rebuild components of the human heart. Science 2019, 365, 482–487. [Google Scholar] [CrossRef]
- Mirdamadi, E.; Tashman, J.W.; Shiwarski, D.J.; Palchesko, R.N.; Feinberg, A.W. Fresh 3D bioprinting a full-size model of the human heart. ACS Biomater. Sci. Eng. 2020, 6, 6453–6459. [Google Scholar] [CrossRef]
- Kačarević, Z.P.; Rider, P.; Alkildani, S.; Retnasingh, S.; Smeets, R.; Jung, O.; Ivanišević, Z.; Barbeck, M. An introduction to 3D bioprinting: Possibilities, challenges and future aspects. Materials 2018, 11, 2199. [Google Scholar] [CrossRef] [Green Version]
- Fedorovich, N.E.; Alblas, J.; De Wijn, J.R.; Hennink, W.E.; Verbout, A.J.; Dhert, W.J. Hydrogels as extracellular matrices for skeletal tissue engineering: State-of-the-Art and novel application in organ printing. Tissue Eng. 2007, 13, 1905–1925. [Google Scholar] [CrossRef]
- Guvendiren, M.; Burdick, J.A. Engineering synthetic hydrogel microenvironments to instruct stem cells. Curr. Opin. Biotechnol. 2013, 24, 841–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzobo, K.; Motaung, K.S.C.M.; Adesida, A. Recent trends in decellularized extracellular matrix bioinks for 3D printing: An updated review. Int. J. Mol. Sci. 2019, 20, 4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabirian, F.; Mozafari, M. Decellularized ECM-derived bioinks: Prospects for the future. Methods 2020, 171, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.Y.; Park, S.-H. ECM-Based Bioink for Tissue Mimetic 3D Printing, in Biomimetic Medical Materials: From Nanotechnology to 3D Printing; Noh, I., Ed.; Springer: Singapore, 2018; pp. 335–354. [Google Scholar]
- Rickard, D.; Sullivan, T.; Shenker, B.; Leboy, P.; Kazhdan, I. Induction of rapid osteoblast differentiation in rat bone marrow stromal cell cultures by dexamethasone and BMP-2. Dev. Biol. 1994, 161, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Poldervaart, M.T.; Wang, H.; Van Der Stok, J.; Weinans, H.; Leeuwenburgh, S.C.G.; Öner, F.C.; Dhert, W.J.A.; Alblas, J. Sustained release of BMP-2 in bioprinted alginate for osteogenicity in mice and rats. PLoS ONE 2013, 8, e72610. [Google Scholar] [CrossRef] [PubMed]
- Balasundaram, G.; Sato, M.; Webster, T.J. Using hydroxyapatite nanoparticles and decreased crystallinity to promote osteoblast adhesion similar to functionalizing with RGD. Biomaterials 2006, 27, 2798–2805. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-S.; Park, M.S.; Jeon, O.; Choi, C.Y.; Kim, B.-S. Poly(lactide-co-glycolide)/hydroxyapatite composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 1399–1409. [Google Scholar] [CrossRef]
- Zhou, X.; Zhu, W.; Nowicki, M.; Miao, S.; Cui, H.; Holmes, B.; Glazer, R.I.; Zhang, L.G. 3D bioprinting a cell-laden bone matrix for breast cancer metastasis study. ACS Appl. Mater. Interfaces 2016, 8, 30017–30026. [Google Scholar] [CrossRef]
- Jakus, A.E.; Rutz, A.L.; Jordan, S.W.; Kannan, A.; Mitchell, S.M.; Yun, C.; Koube, K.D.; Yoo, S.C.; Whiteley, H.E.; Richter, C.-P.; et al. Hyperelastic “bone”: A highly versatile, growth factor–free, osteoregenerative, scalable, and surgically friendly biomaterial. Sci. Transl. Med. 2016, 8, 358ra127. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-H.; Jakus, A.E.; Jordan, S.W.; Dumanian, Z.; Parker, K.; Zhao, L.; Patel, P.K.; Shah, R.N. Three-Dimensionally printed hyperelastic bone scaffolds accelerate bone regeneration in critical-size calvarial bone defects. Plast. Reconstr. Surg. 2019, 143, 1397–1407. [Google Scholar] [CrossRef]
- Alluri, R.; Jakus, A.; Bougioukli, S.; Pannell, W.; Sugiyama, O.; Tang, A.; Shah, R.; Lieberman, J.R. 3D printed hyperelastic “bone” scaffolds and regional gene therapy: A novel approach to bone healing. J. Biomed. Mater. Res. Part A 2018, 106, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Plantz, M.A.; Hsu, W.K. Recent research advances in biologic bone graft materials for spine surgery. Curr. Rev. Musculoskelet. Med. 2020, 13, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Lewicki, J.; Bergman, J.; Kerins, C.; Hermanson, O. Optimization of 3D bioprinting of human neuroblastoma cells using sodium alginate hydrogel. Bioprinting 2019, 16, 00053. [Google Scholar] [CrossRef]
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Decellularized extracellular matrix for bioengineering physiomimetic 3D in vitro tumor models. Trends Biotechnol. 2020, 38, 1397–1414. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yao, R.; Ouyang, L.; Ding, H.; Zhang, T.; Zhang, K.; Cheng, S.; Sun, W. Three-Dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication 2014, 6, 035001. [Google Scholar] [CrossRef]
- Zhao, S.; Zuo, W.-J.; Shao, Z.-M.; Jiang, Y.-Z. Molecular subtypes and precision treatment of triple-negative breast cancer. Ann. Transl. Med. 2020, 8, 499. [Google Scholar] [CrossRef]
- Tang, M.; Xie, Q.; Gimple, R.C.; Prager, B.C.; Qiu, Z.; Schimelman, J.; Wang, P.; Lee, D.; Yu, A.; Miller, T.E.; et al. Abstract 320: 3D-bioprinting of biomimetic multicellular glioblastoma tissues enable modeling of tumor-immune interactions. Mol. Cell. Biol. Genet. 2020, 80, 320. [Google Scholar] [CrossRef]
- Hakobyan, D.; Médina, C.; Dusserre, N.; Stachowicz, M.L.; Handschin, C.; Fricain, J.C.; Guillermet-Guibert, J.; Oliveira, H. Laser-Assisted 3D bioprinting of exocrine pancreas spheroid models for cancer initiation study. Biofabrication 2020, 12, 035001. [Google Scholar] [CrossRef]
- Radhakrishnan, J.; Varadaraj, S.; Dash, S.K.; Sharma, A.; Verma, R.S. Organotypic cancer tissue models for drug screening: 3D constructs, bioprinting and microfluidic chips. Drug Discov. Today 2020, 25, 879–890. [Google Scholar] [CrossRef]
- Zhu, W.; Castro, N.J.; Cui, H.; Zhou, X.; Boualam, B.; McGrane, R.; Glazer, R.I.; Zhang, L.G. A 3D printed nano bone matrix for characterization of breast cancer cell and osteoblast interactions. Nanotechnology 2016, 27, 315103. [Google Scholar] [CrossRef]
- Kolb, A.D.; Bussard, K.M. The bone extracellular matrix as an ideal milieu for cancer cell metastases. Cancers 2019, 11, 1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Bahraminasab, M. Challenges on optimization of 3D-printed bone scaffolds. Biomed. Eng. Online 2020, 19, 1–33. [Google Scholar] [CrossRef]
- Wang, L.; Xu, M.; Luo, L.; Zhou, Y.; Si, P. Iterative feedback bio-printing-derived cell-laden hydrogel scaffolds with optimal geometrical fidelity and cellular controllability. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Heikal, L.; Ferns, G.A.; Ghezzi, P.; Nokhodchi, A.; Maniruzzaman, M. 3D bioprinting of novel biocompatible scaffolds for endothelial cell repair. Polymers 2019, 11, 1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koons, G.L.; Mikos, A.G. Progress in three-dimensional printing with growth factors. J. Control. Release 2019, 295, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Foresti, R.; Rossi, S.; Pinelli, S.; Alinovi, R.; Barozzi, M.; Sciancalepore, C.; Galetti, M.; Caffarra, C.; Lagonegro, P.; Scavia, G.; et al. Highly-Defined bioprinting of long-term vascularized scaffolds with Bio-Trap: Complex geometry functionalization and process parameters with computer aided tissue engineering. Materialia 2020, 9, 100560. [Google Scholar] [CrossRef]
- Hanumantharao, S.N.; Que, C.A.; Vogl, B.J.; Rao, S. Engineered Three-Dimensional Scaffolds Modulating Fate of Breast Cancer Cells Using Stiffness and Morphology Related Cell Adhesion. IEEE Open J. Eng. Med. Biol. 2020, 1, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Freeman, F.E.; Pitacco, P.; Van Dommelen, L.H.A.; Nulty, J.; Browe, D.C.; Shin, J.-Y.; Alsberg, E.; Kelly, D.J. 3D bioprinting spatiotemporally defined patterns of growth factors to tightly control tissue regeneration. Sci. Adv. 2020, 6, eabb5093. [Google Scholar] [CrossRef]
- Sun, B.; Lian, M.; Han, Y.; Mo, X.; Jiang, W.; Qiao, Z.; Dai, K. A 3D-Bioprinted dual growth factor-releasing intervertebral disc scaffold induces nucleus pulposus and annulus fibrosus reconstruction. Bioact. Mater. 2021, 6. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, A.M.; Kolb, A.D.; Shupp, A.B.; Shine, K.M.; Bussard, K.M. Printing the Pathway Forward in Bone Metastatic Cancer Research: Applications of 3D Engineered Models and Bioprinted Scaffolds to Recapitulate the Bone–Tumor Niche. Cancers 2021, 13, 507. https://doi.org/10.3390/cancers13030507
Hughes AM, Kolb AD, Shupp AB, Shine KM, Bussard KM. Printing the Pathway Forward in Bone Metastatic Cancer Research: Applications of 3D Engineered Models and Bioprinted Scaffolds to Recapitulate the Bone–Tumor Niche. Cancers. 2021; 13(3):507. https://doi.org/10.3390/cancers13030507
Chicago/Turabian StyleHughes, Anne M., Alexus D. Kolb, Alison B. Shupp, Kristy M. Shine, and Karen M. Bussard. 2021. "Printing the Pathway Forward in Bone Metastatic Cancer Research: Applications of 3D Engineered Models and Bioprinted Scaffolds to Recapitulate the Bone–Tumor Niche" Cancers 13, no. 3: 507. https://doi.org/10.3390/cancers13030507
APA StyleHughes, A. M., Kolb, A. D., Shupp, A. B., Shine, K. M., & Bussard, K. M. (2021). Printing the Pathway Forward in Bone Metastatic Cancer Research: Applications of 3D Engineered Models and Bioprinted Scaffolds to Recapitulate the Bone–Tumor Niche. Cancers, 13(3), 507. https://doi.org/10.3390/cancers13030507