
Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes on



Abstract
:Simple Summary
Abstract
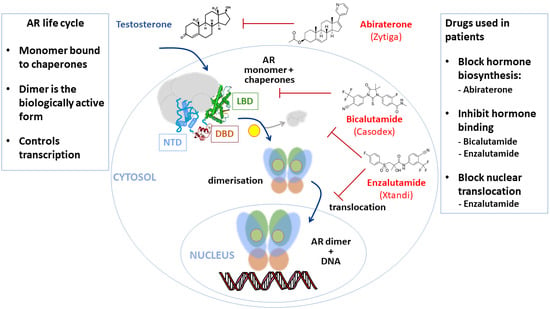
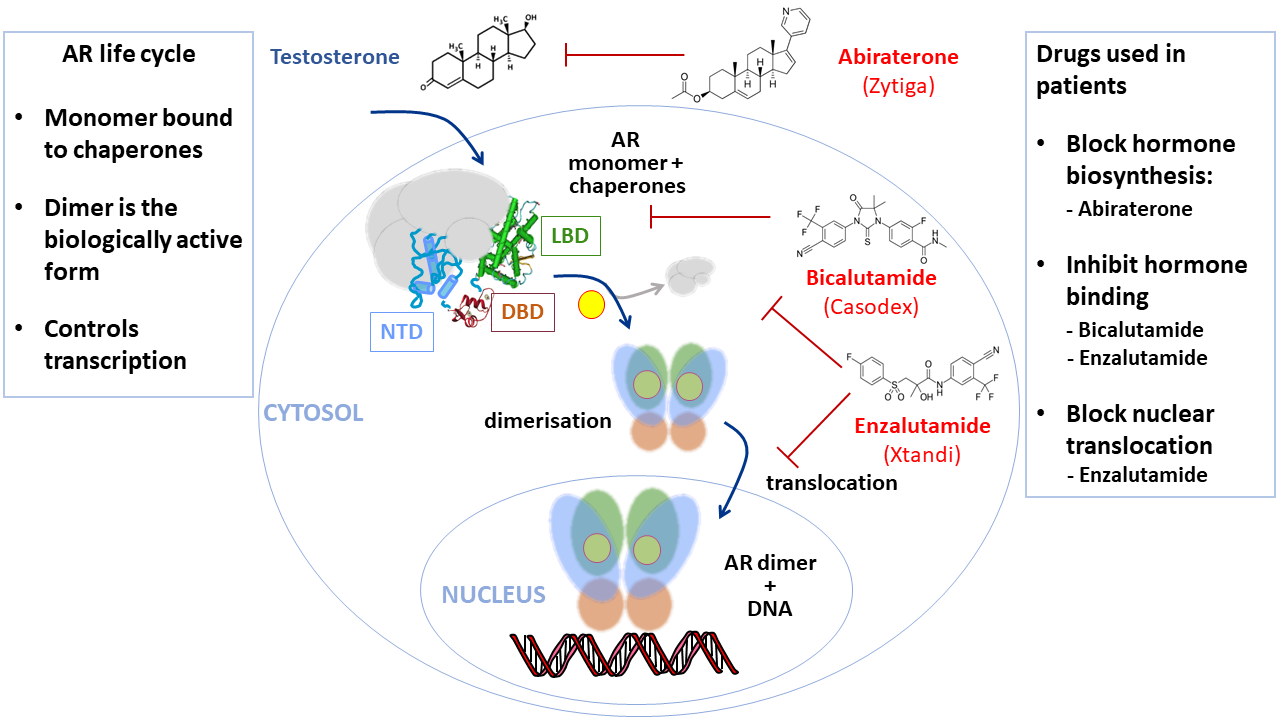{kind=link}
{kind=link}
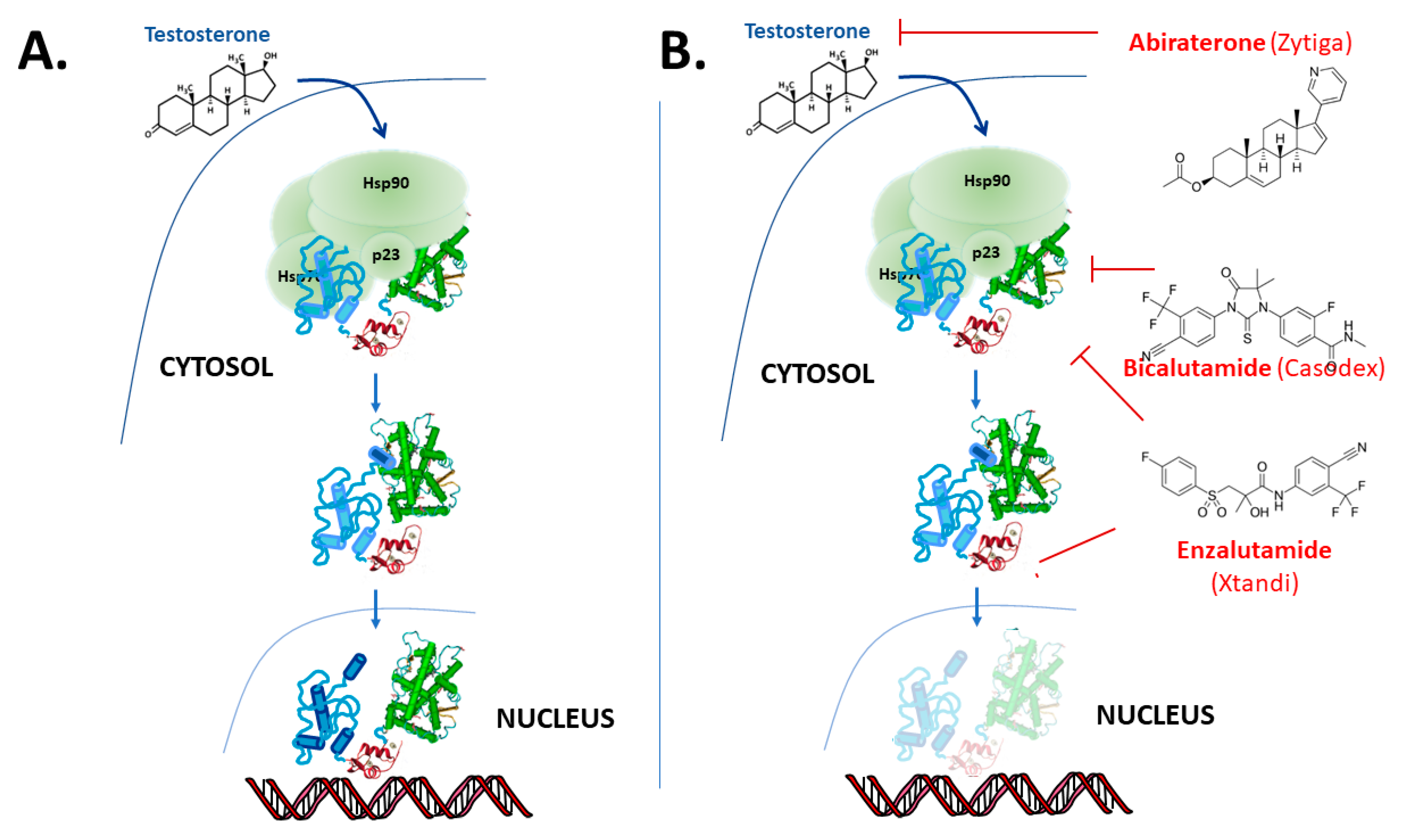{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Androgen Receptor—A Key Driver of PCa and Drug Target
3. Androgen Receptor Structure and Function
3.1. The N-Terminal Domain
3.2. The DNA Binding Domain
3.3. The Ligand Binding Domain
4. Targeting the Hormone-Binding Function of the Androgen Receptor
5. New Approaches to AR Inhibition in CRPC and Associated Clinical Trials
New Approaches to Existing Antiandrogens
6. Selective Androgen Receptor Degraders (SARDs)
7. Looking Outside the Ligand-Binding Pocket
7.1. The Amino-Terminal Domain
7.2. BF3 Surface Pocket
7.3. The DNA Binding Domain
8. Combination of Antiandrogen and Chemotherapy
9. The Future of Anti-Androgen Therapy
10. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Noone, A.M. Cancer Statistics Review 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2017.
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Ghabili, K.; Tosoian, J.J.; Schaeffer, E.M.; Pavlovich, C.P.; Golzari, S.E.J.; Khajir, G.; Andreas, D.; Benzon, B.; Vuica-Ross, M.; Ross, A.E. The History of Prostate Cancer from Antiquity: Review of Paleopathological Studies. Urology 2016, 97, 8–12. [Google Scholar] [CrossRef]
- Minozzi, S.; Lunardini, A.; Caldarini, C.; Caramella, D.; Fornaciari, G.; Catalano, P.; Giuffra, V. Metastatic Prostate Carcinoma from Imperial Rome (1st to 2nd Centuries AD). Pathobiology 2018, 85, 289–299. [Google Scholar] [CrossRef]
- Prates, C.; Sousa, S.; Oliveira, C.; Ikram, S. Prostate metastatic bone cancer in an Egyptian Ptolemaic mummy, a proposed radiological diagnosis. Int. J. Paleopathol. 2011, 1, 98–103. [Google Scholar] [CrossRef]
- Schultz, M.; Parzinger, H.; Posdnjakov, D.V.; Chikisheva, T.A.; Schmidt-Schultz, T.H. Oldest known case of metastasizing prostate carcinoma diagnosed in the skeleton of a 2,700-year-old Scythian king from Arzhan (Siberia, Russia). Int. J. Cancer 2007, 121, 2591–2595. [Google Scholar] [CrossRef]
- Sriprasad, S.; Feneley, M.R.; Thompson, P.M. History of prostate cancer treatment. Surg. Oncol. 2009, 18, 185–191. [Google Scholar] [CrossRef]
- Guillaumier, S.; Peters, M.; Arya, M.; Afzal, N.; Charman, S.; Dudderidge, T.; Hosking-Jervis, F.; Hindley, R.G.; Lewi, H.; McCartan, N.; et al. A Multicentre Study of 5-year Outcomes Following Focal Therapy in Treating Clinically Significant Nonmetastatic Prostate Cancer. Eur. Urol. 2018, 74, 422–429. [Google Scholar] [CrossRef]
- Fukami, M.; Homma, K.; Hasegawa, T.; Ogata, T. Backdoor pathway for dihydrotestosterone biosynthesis: Implications for normal and abnormal human sex development. Dev. Dyn. 2012, 242, 320–329. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Antignac, J.P.; Le Bizec, B.; Morvan, M.-L.; Svechnikov, K.; Söder, O.; Savchuk, I.; Monteiro, A.; Soffientini, U.; Johnston, Z.C.; et al. Alternative (backdoor) androgen production and masculinization in the human fetus. PLoS Biol. 2019, 17, e3000002. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.D.; Griffin, J.E.; Russell, D.W. Steroid 5 Alpha-Reductase 2 Deficiency. Endocr. Rev. 1993, 14, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer: I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. J. Urol. 2002, 168, 9–12. [Google Scholar] [CrossRef]
- He, B.; Gampe, R.T.; Kole, A.J.; Hnat, A.T.; Stanley, T.B.; An, G.; Stewart, E.L.; Kalman, R.I.; Minges, J.T.; Wilson, E.M. Structural Basis for Androgen Receptor Interdomain and Coactivator Interactions Suggests a Transition in Nuclear Receptor Activation Function Dominance. Mol. Cell 2004, 16, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, M.E.; Cunha, S.M.; Brink, M.C.; Mattern, K.A.; Nigg, A.L.; Dubbink, H.J.; Verschure, P.J.; Trapman, J.; Houtsmuller, A.B. Compartmentalization of androgen receptor protein–protein interactions in living cells. J. Cell Biol. 2007, 177, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutress, M.L.; Whitaker, H.C.; Mills, I.G.; Stewart, M.; Neal, D.E. Structural Basis for the Nuclear Import of the Human An-drogen Receptor. J. Cell. Sci. 2008, 121, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Jänne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A Hierarchical Network of Transcription Factors Governs Androgen Receptor-Dependent Prostate Cancer Growth. Mol. Cell 2007, 27, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O’Malley, B.W. Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 2020, 79, 812–823.e4. [Google Scholar]
- Powell, S.M.; Christiaens, V.; Voulgaraki, D.; Waxman, J.; Claessens, F.; Bevan, C.L. Mechanisms of Androgen Receptor Sig-nalling Via Steroid Receptor Coactivator-1 in Prostate. Endocr. Relat. Cancer 2004, 11, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Bevan, C.L.; Hoare, S.; Claessens, F.; Heery, D.M.; Parker, M.G. The AF1 and AF2 Domains of the Androgen Receptor Inter-act with Distinct Regions of SRC1. Mol. Cell. Biol. 1999, 19, 8383–8392. [Google Scholar] [CrossRef] [Green Version]
- De Mol, E.; Szulc, E.; di Sanza, C.; Marttnez-Cristtbal, P.; Bertoncini, C.W.; Fenwick, R.B.; Frigoll-Vivas, M.; Masín, M.; Hunter, I.; Buzzn, V.; et al. Regulation of Androgen Receptor Activity by Transient Interactions of Its Transactivation Domain with General Transcription Regulators. SSRN Electron. J. 2018, 26, 145. [Google Scholar] [CrossRef] [Green Version]
- De Moll, E.; Fenwick, R.B.; Phang, C.T.W.; Buzón, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; García, J.; Bertoncini, C.W.; Estébanez-Perpiñá, E.; et al. EPI-001, A Compound Active against Castration-Resistant Prostate Cancer, Targets Transactivation Unit 5 of the Androgen Receptor. ACS Chem. Biol. 2016, 11, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Cano, L.Q.; Lavery, D.N.; Bevan, C.L. Mini-review: Foldosome regulation of androgen receptor action in prostate cancer. Mol. Cell. Endocrinol. 2013, 369, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Reebye, V.; Querol-Cano, L.; Lavery, D.N.; Brooke, G.N.; Powell, S.M.; Chotai, D.; Walker, M.M.; Whitaker, H.C.; Wait, R.; Hurst, H.C.; et al. Role of the HSP90-Associated Cochaperone p23 in Enhancing Activity of the Androgen Receptor and Significance for Prostate Cancer. Mol. Endocrinol. 2012, 26, 1694–1706. [Google Scholar] [CrossRef] [Green Version]
- Shatkina, L.; Mink, S.; Rogatsch, H.; Klocker, H.; Langer, G.; Nestl, A.; Cato, A.C. The Cochaperone Bag-1L Enhances Androgen Receptor Action Via Interaction with the NH2-Terminal Region of the Receptor. Mol. Cell. Biol. 2003, 23, 7189–7197. [Google Scholar] [CrossRef] [Green Version]
- Yong, W.; Yang, Z.; Periyasamy, S.; Chen, H.; Yucel, S.; Li, W.; Lin, L.Y.; Wolf, I.M.; Cohn, M.J.; Baskin, L.S.; et al. Essential Role for Co-chaperone Fkbp52 but Not Fkbp51 in Androgen Receptor-mediated Signaling and Physiology. J. Biol. Chem. 2007, 282, 5026–5036. [Google Scholar] [CrossRef] [Green Version]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taulès, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388. [Google Scholar] [CrossRef]
- Presman, D.M.; Hager, G.L. More than meets the dimer: What is the quaternary structure of the glucocorticoid receptor? Transcription 2017, 8, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Presman, D.M.; Ganguly, S.; Schiltz, R.L.; Johnson, T.A.; Karpova, T.S.; Hager, G.L. DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc. Natl. Acad. Sci. USA 2016, 113, 8236–8241. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Panizo, A.; Perez, P.; Rojas, A.M.; Fuentes-Prior, P.; Estebanez-Perpina, E. Non-Canonical Dimerization of the An-drogen Receptor and Other Nuclear Receptors: Implications for Human Disease. Endocr. Relat. Cancer 2019, 26, R479–R497. [Google Scholar] [CrossRef] [Green Version]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; E Harding, A.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nat. Cell Biol. 1991, 352, 77–79. [Google Scholar] [CrossRef]
- Faber, P.W.; van Rooij, H.C.; van der Korput, H.A.; Baarends, W.M.; Brinkmann, A.O.; Grootegoed, J.A.; Trapman, J. Characterization of the Human Androgen Receptor Transcription Unit. J. Biol. Chem. 1991, 266, 10743–10749. [Google Scholar] [CrossRef]
- Tilley, W.D.; Wilson, C.M.; Marcelli, M.; McPhaul, M.J. Androgen receptor gene expression in human prostate carcinoma cell lines. Cancer Res. 1990, 50, 5382–5386. [Google Scholar]
- McEwan, I.J.; Brinkmann, A.O. Androgen Physiology: Receptor and Metabolic Disorders. In Endotext; Feingold, K.R., Ana-Walt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Simental, J.A.; Sar, M.; Lane, M.V.; French, F.S.; Wilson, E.M. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J. Biol. Chem. 1991, 266, 510–518. [Google Scholar] [CrossRef]
- Jenster, G.; van der Korput, H.; Trapman, J.; Brinkmann, A.O. Identification of Two Transcription Activation Units in the N-terminal Domain of the Human Androgen Receptor. J. Biol. Chem. 1995, 270, 7341–7346. [Google Scholar] [CrossRef] [Green Version]
- Callewaert, L.; van Tilborgh, N.; Claessens, F. Interplay between Two Hormone-Independent Activation Domains in the Androgen Receptor. Cancer Res. 2006, 66, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Dehm, S.M.; Regan, K.M.; Schmidt, L.J.; Tindall, D.J. Selective Role of an NH2-Terminal WxxLF Motif for Aberrant Androgen Receptor Activation in Androgen Depletion–Independent Prostate Cancer Cells. Cancer Res. 2007, 67, 10067–10077. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; McEwan, I.J. Allosteric Modulators of Steroid Hormone Receptors: Structural Dynamics and Gene Regulation. Endocr. Rev. 2012, 33, 271–299. [Google Scholar] [CrossRef] [Green Version]
- McEwan, I.J. Intrinsic disorder in the androgen receptor: Identification, characterisation and drugability. Mol. BioSyst. 2011, 8, 82–90. [Google Scholar] [CrossRef]
- Lavery, D.N.; McEwan, I.J. Structural Characterization of the Native NH2-Terminal Transactivation Domain of the Human Androgen Receptor: A Collapsed Disordered Conformation Underlies Structural Plasticity and Protein-Induced Folding. Biochemistry 2008, 47, 3360–3369. [Google Scholar] [CrossRef]
- Reid, J.; Kelly, S.M.; Watt, K.; Price, N.C.; McEwan, I.J. Conformational Analysis of the Androgen Receptor Amino-terminal Domain Involved in Transactivation. J. Biol. Chem. 2002, 277, 20079–20086. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.; Watt, K.; Kelly, S.; Clark, C.; Price, N.C.; McEwan, I.J. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J. Mol. Endocrinol. 2008, 41, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Wang, Y.-H.; Pérez-Escrivà, P.; Kieffer, B. Backbone 1H, 15N, 13C NMR assignment of the 518–627 fragment of the androgen receptor encompassing N-terminal and DNA binding domains. Biomol. NMR Assign. 2016, 10, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Piai, A.; Chiesa, G.; Mungianu, D.; Garcia, J.; Pierattelli, R.; Felli, I.C.; Salvatella, X. Sequence Context Influences the Structure and Aggregation Behavior of a PolyQ Tract. Biophys. J. 2016, 110, 2361–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; White, J.T.; Saavedra, H.; Wrabl, J.O.; Motlagh, H.N.; Liu, K.; Sowers, J.L.; Schroer, T.A.; Thompson, E.; Hilser, V.J. Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. eLife 2017, 6, e30688. [Google Scholar] [CrossRef] [PubMed]
- Brodie, J.; McEwan, I.J. Intra-domain communication between the N-terminal and DNA-binding domains of the androgen receptor: Modulation of androgen response element DNA binding. J. Mol. Endocrinol. 2005, 34, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Dubbink, H.; Erik, J.; Hersmus, R.; Verma, C.; van der Korput, H.; Berrevoets, C.; van Tol, J.; der Made, A.Z.-V.; Brinkmann, A.; Pike, A.; et al. Distinct Recognition Modes of FXXLF and LXXLL Motifs by the Androgen Receptor. Mol. Endocrinol. 2004, 18, 2132–2150. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar] [CrossRef] [Green Version]
- Haelens, A.; Tanner, T.; Denayer, S.; Callewaert, L.; Claessens, F. The Hinge Region Regulates DNA Binding, Nuclear Trans-location, and Transactivation of the Androgen Receptor. Cancer Res. 2007, 67, 4514–4523. [Google Scholar] [CrossRef] [Green Version]
- Sahu, B.; Pihlajamaa, P.; Dubois, V.; Kerkhofs, S.; Claessens, F.; Jänne, O.A. Androgen receptor uses relaxed response element stringency for selective chromatin binding and transcriptional regulation in vivo. Nucleic Acids Res. 2014, 42, 4230–4240. [Google Scholar] [CrossRef]
- Kerkhofs, S.; Dubois, V.; de Gendt, K.; Helsen, C.; Clinckemalie, L.; Spans, L.; Schuit, F.; Boonen, S.; Vanderschueren, D.; Saun ders, P.T.; et al. A role for selective androgen response elements in the development of the epididymis and the androgen control of the 5α reductase II gene. FASEB J. 2012, 26, 4360–4372. [Google Scholar]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kregel, S.; Bagamasbad, P.D.; He, S.; la Pensee, E.; Raji, Y.; Brogley, M.; Chinnaiyan, A.; Cieslik, M.; Robins, D.M. Differential modulation of the androgen receptor for prostate cancer therapy depends on the DNA response element. Nucleic Acids Res. 2020, 48, 4741–4755. [Google Scholar] [CrossRef] [PubMed]
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural Evidence for Ligand Specificity in the Binding Domain of the Human Androgen Receptor. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jésus-Tran, K.P.; Côté, P.-L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Rojas, A.; Hagler, A.T.; Estebanez-Perpina, E. Diversity of Quaternary Structures Regulates Nuclear Receptor Activities. Trends Biochem. Sci. 2019, 44, 2–6. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. A history of prostate cancer treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Smith, M.R.; Ryan, C.J.; Berry, W.R.; Shore, N.D.; Liu, G.; Higano, C.S.; Alumkal, J.J.; Hauke, R.; Tutrone, R.F.; et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann. Oncol. 2017, 28, 2264–2271. [Google Scholar] [CrossRef]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A Novel Antiandrogen for Prostate Cancer Treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Habenicht, U.F.; Schröder, H.; el Etreby, M.F.; Neumann, F. Advantages and disadvantages of pure antiandrogens and of antiandrogens of the cyproterone acetate-type in the treatment of prostatic cancer. Prog. Clin. Biol. Res. 1988, 260, 63–75. [Google Scholar]
- Torri, V.; Floriani, I. Cyproterone acetate in the therapy of prostate carcinoma. Arch. Ital. Urol. Androl. 2005, 77, 157–163. [Google Scholar] [PubMed]
- Raynaud, J.P.; Bonne, C.; Moguilewsky, M.; Lefebvre, F.A.; Bélanger, A.; Labrie, F. The pure antiandrogen ru 23908 (anandron®), a candidate of choice for the combined antihormonal treatment of prostatic cancer: A review. Prostate 1984, 5, 299–311. [Google Scholar] [CrossRef]
- Irwin, R.J.; Prout, G.R. A new antiprostatic agent for treatment of prostatic carcinoma. Surg. Forum 1973, 24, 536–537. [Google Scholar]
- Peets, E.A.; Henson, M.F.; Neri, R. On the Mechanism of the Anti-Androgenic Action of Flutamide (Alpha-Alpha-Alpha-Trifluoro-2-Methyl-4′-Nitro-M-Propionotoluidide) in the Rat. Endocrinology 1974, 94, 532–540. [Google Scholar] [CrossRef]
- Liao, S.; Howell, D.K.; Chang, T.M. Action of a Nonsteroidal Antiandrogen, Flutamide, on the Receptor Binding and Nuclear Retention of 5 Alpha-Dihydrotestosterone in Rat Ventral Prostate. Endocrinology 1974, 94, 1205–1209. [Google Scholar] [CrossRef]
- Sogani, P.; Ray, B.; Whitmore, W. Advanced prostatic carcinoma Flutamide therapy after conventional endocrine treatment. Urology 1975, 6, 164–166. [Google Scholar] [CrossRef]
- Katchen, B.; Buxbaum, S. Disposition of a New, Nonsteroid, Antiandrogen, Alpha,Alpha,Alpha-Trifluoro-2-Methyl-4′-Nitro-M-Propionotoluidide (Flutamide), in Men Following a Single Oral 200 mg Dose. J. Clin. Endocrinol. Metab. 1975, 41, 373–379. [Google Scholar] [CrossRef]
- Caine, M.; Perlberg, S.; Gordon, R. The Treatment of Benign Prostatic Hypertrophy with Flutamide (SCH 13521): A Placebo-Controlled Study. J. Urol. 1975, 114, 564–568. [Google Scholar] [CrossRef]
- Boris, A.; Scott, J.W.; de Martino, L.; Cox, D.C. Endocrine Profile of a Nonsteroidal Antiandrogen N-(3,5-Dimethyl-4-Isoxazolylmethyl)Phthalimide (DIMP). Acta Endocrinol. Eur. J. Endocrinol. 1973, 72, 604–614. [Google Scholar] [CrossRef]
- Furr, B.J.; Valcaccia, B.; Curry, B.; Woodburn, J.R.; Chesterson, G.; Tucker, H. ICI 176,334: A Novel Non-Steroidal, Peripheral-ly Selective Antiandrogen. J. Endocrinol. 1987, 113, R7–R9. [Google Scholar] [CrossRef]
- Freeman, S.N.; Mainwaring, W.I.; Furr, B.J. A possible explanation for the peripheral selectivity of a novel non-steroidal pure antiandrogen, Casodex (ICI 176,334). Br. J. Cancer 1989, 60, 664–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunglmayr, G. Casodex (ICI 176,334), a New, Non-Steroidal Anti-Androgen. Early Clinical Results. Horm. Res. 1989, 32 (Suppl. 1), 77–81. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Miyazaki, J.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel Mutations of Androgen Receptor: A Possible Mechanism of Bicalutamide Withdrawal Syndrome. Cancer Res. 2003, 63, 149–153. [Google Scholar]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A Clinically Relevant Androgen Receptor Mutation Confers Resistance to Second-Generation Antiandrogens Enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; A Watson, P.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013, 2, e00499. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef]
- Wang, R.; Sun, Y.; Li, L.; Niu, Y.; Lin, W.; Lin, C.; Antonarakis, E.S.; Luo, J.; Yeh, S.; Chang, C. Preclinical Study using Malat1 Small Interfering RNA Or Androgen Receptor Splicing Variant 7 Degradation Enhancer ASC-J9((R)) to Suppress Enzalutamide-Resistant Prostate Cancer Progression. Eur. Urol. 2017, 72, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, T.; Baumgart, S.J.; Nevedomskaya, E.; Reichert, K.; Steuber, H.; Lejeune, P.; Mumberg, D.; Haendler, B. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int. J. Cancer 2019, 145, 1382–1394. [Google Scholar] [CrossRef]
- Montgomery, B.; Tretiakova, M.S.; Joshua, A.M.; Gleave, M.E.; Fleshner, N.; Bubley, G.J.; Mostaghel, E.A.; Chi, K.N.; Lin, D.W.; Sanda, M.; et al. Neoadjuvant Enzalutamide Prior to Prostatectomy. Clin. Cancer Res. 2017, 23, 2169–2176. [Google Scholar] [CrossRef] [Green Version]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy with Enzalutamide or Placebo in Men with Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Nonmetastatic, Castration-Resistant Prostate Cancer and Survival with Darolutamide. N. Engl. J. Med. 2020, 383, 1040–1049. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, A.; Cyrus, K.; Salcius, M.; Kim, K.; Crews, C.M.; Deshaies, R.J.; Sakamoto, K.M. Targeting Steroid Hormone Receptors for Ubiquitination and Degradation in Breast and Prostate Cancer. Oncogene 2008, 27, 7201–7211. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.B.; Hines, J.; McDonnell, D.P.; et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef]
- Huxley, M.; Sanchez-Cano, C.; Browning, M.J.; Navarro-Ranninger, C.; Quiroga, A.G.; Rodger, A.; Hannon, M.J. An Androgenic Steroid Delivery Vector that Imparts Activity to a Non-Conventional Platinum(II) Metallo-Drug. Dalton Trans. 2010, 39, 11353–11364. [Google Scholar]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Gandhi, J.G.; Clark, W.R.; Heath, E.; Lin, J.; Oh, W.K.; Agus, D.B.; Carthon, B.; Moran, S.; Kong, N.; et al. Phase 1/2 Study of Orteronel (TAK-700), an Investigational 17,20-Lyase Inhibitor, with Docetaxel-Prednisone in Metastatic Castration-Resistant Prostate Cancer. Invest. New Drugs 2015, 33, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; A Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef] [Green Version]
- Petrylak, D.P.; Gao, X.; Vogelzang, N.J.; Garfield, M.H.; Taylor, I.; Moore, M.D.; Peck, R.A.; Burris, H.A. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). J. Clin. Oncol. 2020, 38, 3500. [Google Scholar] [CrossRef]
- Yu, Z.; Cai, C.; Gao, S.; Simon, N.I.; Shen, H.C.; Balk, S.P. Galeterone Prevents Androgen Receptor Binding to Chromatin and Enhances Degradation of Mutant Androgen Receptor. Clin. Cancer Res. 2014, 20, 4075–4085. [Google Scholar] [CrossRef] [Green Version]
- Kwegyir-Afful, A.K.; Ramalingam, S.; Purushottamachar, P.; Ramamurthy, V.P.; Njar, V.C.O. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget 2015, 6, 27440–27460. [Google Scholar] [CrossRef] [Green Version]
- Alyamani, M.; Li, Z.; Berk, M.; Li, J.; Tang, J.; Upadhyay, S.; Auchus, R.J.; Sharifi, N. Steroidogenic Metabolism of Galeterone Reveals a Diversity of Biochemical Activities. Cell Chem. Biol. 2017, 24, 825–832.e6. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; A Bastos, D. Galeterone for the treatment of advanced prostate cancer: The evidence to date. Drug Des. Dev. Ther. 2016, 10, 2289–2297. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.; Lim, A.C.; Hay, C.W.; Taylor, A.E.; Wingate, A.; Nowakowska, K.; Pezaro, C.; Carreira, S.; Goodall, J.; Arlt, W.; et al. Interactions of Abiraterone, Eplerenone, and Prednisolone with Wild-type and Mutant Androgen Receptor: A Rationale for Increasing Abiraterone Exposure or Combining with MDV3100. Cancer Res. 2012, 72, 2176–2182. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bishop, A.C.; Alyamani, M.; Garcia, J.A.; Dreicer, R.; Bunch, D.; Liu, J.; Upadhyay, S.K.; Auchus, R.J.; Sharifi, N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nat. Cell Biol. 2015, 523, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Coss, C.C.; Thiyagarajan, T.; Watts, K.; Hwang, D.-J.; Christopher, L.; Selth, L.A.; McEwan, I.J.; Duke, C.B.; Pagadala, J.; et al. Novel Selective Agents for the Degradation of Androgen Receptor Variants to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 6282–6298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnusamy, S.; He, Y.; Hwang, D.-J.; Thiyagarajan, T.; Houtman, R.; Bocharova, V.; Sumpter, B.G.; Fernandez, E.; Johnson, D.L.; Du, Z.; et al. Orally Bioavailable Androgen Receptor Degrader, Potential Next-Generation Therapeutic for Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 6764–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loddick, S.A.; Ross, S.J.; Thomason, A.G.; Robinson, D.M.; Walker, G.E.; Dunkley, T.P.; Brave, S.R.; Broadbent, N.; Stratton, N.C.; Trueman, D.; et al. AZD3514: A Small Molecule That Modulates Androgen Receptor Signaling and Function In Vitro and In Vivo. Mol. Cancer Ther. 2013, 12, 1715–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omlin, A.; Jones, R.J.; van der Noll, R.; Satoh, T.; Niwakawa, M.; Smith, S.A.; Graham, J.; Ong, M.; Finkelman, R.D.; Schellens, J.H.; et al. AZD3514, an Oral Selective Androgen Receptor Down-Regulator in Patients with Castration-Resistant Prostate Cancer—Results of Two Parallel First-in-Human Phase I Studies. Invest. New Drugs 2015, 33, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases is Associated with Castration-Resistance and Short Survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, J.-K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef] [Green Version]
- Brand, L.J.; Olson, M.E.; Ravindranathan, P.; Guo, H.; Kempema, A.M.; Andrews, T.E.; Chen, X.; Raj, G.V.; Harki, D.A.; Dehm, S.M. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget 2015, 6, 3811–3824. [Google Scholar] [CrossRef] [Green Version]
- Sadar, M.D. Discovery of drugs that directly target the intrinsically disordered region of the androgen receptor. Expert Opin. Drug Discov. 2020, 15, 551–560. [Google Scholar] [CrossRef]
- Banuelos, C.A.; Ito, Y.; Obst, J.K.; Mawji, N.R.; Wang, J.; Hirayama, Y.; Leung, J.K.; Tam, T.; Tien, A.H.; Andersen, R.J.; et al. Ralaniten Sensitizes Enzalutamide-Resistant Prostate Cancer to Ionizing Radiation in Prostate Cancer Cells that Express Androgen Receptor Splice Variants. Cancers 2020, 12, 1991. [Google Scholar] [CrossRef]
- Banuelos, C.A.; Tavakoli, I.; Tien, A.H.; Caley, D.P.; Mawji, N.R.; Li, Z.; Wang, J.; Yang, Y.C.; Imamura, Y.; Yan, L.; et al. Sintokamide A Is a Novel Antagonist of Androgen Receptor That Uniquely Binds Activation Function-1 in Its Amino-terminal Domain. J. Biol. Chem. 2016, 291, 22231–22243. [Google Scholar] [CrossRef] [Green Version]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Van Soest, R.; Andersen, R.J. ChemInform Abstract: Sintokamide A to E, Chlorinated Peptides from the Sponge Dysidea sp. that Inhibit Transactivation of the N-Terminus of the Androgen Receptor in Prostate Cancer Cells. Chemin 2009, 40, 4947–4950. [Google Scholar] [CrossRef]
- Banuelos, C.A.; Lal, A.; Tien, A.H.; Shah, N.; Yang, Y.C.; Mawji, N.R.; Meimetis, L.G.; Park, J.; Kunzhong, J.; Andersen, R.J.; et al. Characterization of Niphatenones that Inhibit Androgen Receptor N-Terminal Domain. PLoS ONE 2014, 9, e107991. [Google Scholar] [CrossRef] [Green Version]
- Meimetis, L.G.; Williams, D.E.; Mawji, N.R.; Banuelos, C.A.; Lal, A.A.; Park, J.J.; Tien, A.H.; Fernandez, J.G.; de Voogd, N.J.; Sadar, M.D.; et al. Niphatenones, Glycerol Ethers from the Sponge Niphates Digitalis Block Androgen Receptor Transcriptional Activity in Prostate Cancer Cells: Structure Elucidation, Synthesis, and Biological Activity. J. Med. Chem. 2012, 55, 503–514. [Google Scholar] [CrossRef]
- Quayle, S.N.; Mawji, N.R.; Wang, J.; Sadar, M.D. Androgen receptor decoy molecules block the growth of prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 1331–1336. [Google Scholar] [CrossRef] [Green Version]
- Brooke, G.N.; Powell, S.M.; Lavery, D.N.; Waxman, J.; Buluwela, L.; Ali, S.; Bevan, C.L. Engineered repressors are potent inhibitors of androgen receptor activity. Oncotarget 2014, 5, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Buzon, V.; Carbo, L.R.; Estruch, S.B.; Fletterick, R.J.; Estebanez-Perpina, E. A Conserved Surface on the Ligand Binding Domain of Nuclear Receptors for Allosteric Control. Mol. Cell. Endocrinol. 2012, 348, 394–402. [Google Scholar] [CrossRef]
- Estébanez-Perpiñá, E.; Arnold, A.A.; Nguyen, P.; Rodrigues, E.D.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.K.; et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. USA 2007, 104, 16074–16079. [Google Scholar] [CrossRef] [Green Version]
- Lallous, N.; Leblanc, E.; Munuganti, R.S.; Hassona, M.D.; Al Nakouzi, N.; Awrey, S.; Morin, H.; Roshan-Moniri, M.; Singh, K.; Lawn, S.; et al. Targeting Binding Function-3 of the Androgen Receptor Blocks Its Co-Chaperone Interactions, Nuclear Translocation, and Activation. Mol. Cancer Ther. 2016, 15, 2936–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lack, N.A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F.Q.; Chan, K.H.; Feau, C.; Leblanc, E.; Guns, E.T.; Guy, R.K.; Rennie, P.S.; et al. Targeting the Binding Function 3 (BF3) Site of the Human Androgen Receptor through Virtual Screening. J. Med. Chem. 2011, 54, 8563–8573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munuganti, R.S.; Hassona, M.D.; Leblanc, E.; Frewin, K.; Singh, K.; Ma, D.; Ban, F.; Hsing, M.; Adomat, H.; Lallous, N.; et al. Identification of a Potent Antiandrogen that Targets the BF3 Site of the Androgen Receptor and Inhibits Enzalutamide-Resistant Prostate Cancer. Chem. Biol. 2014, 21, 1476–1485. [Google Scholar] [CrossRef] [Green Version]
- Badders, N.M.; Korff, A.; Miranda, H.C.; Vuppala, P.K.; Smith, R.B.; Winborn, B.J.; Quemin, E.R.; Sopher, B.L.; Dearman, J.; Messing, J.; et al. Selective Modulation of the Androgen Receptor AF2 Domain Rescues Degeneration in Spinal Bulbar Muscular Atrophy. Nat. Med. 2018, 24, 427–437. [Google Scholar] [CrossRef]
- Lee, G.T.; Nagaya, N.; DeSantis, J.; Madura, K.; E Sabaawy, H.; Kim, W.-J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC resistant to Second-Line Antiandrogen Therapy. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef]
- Lim, M.; Otto-Duessel, M.; He, M.; Su, L.; Nguyen, D.; Chin, E.; Alliston, T.; Jones, J.O. Ligand-Independent and Tissue-Selective Androgen Receptor Inhibition by Pyrvinium. ACS Chem. Biol. 2014, 9, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ban, F.; Dalal, K.; Leblanc, E.; Frewin, K.; Ma, D.; Adomat, H.; Rennie, P.S.; Cherkasov, A. Discovery of Small-Molecule Inhibitors Selectively Targeting the DNA-Binding Domain of the Human Androgen Receptor. J. Med. Chem. 2014, 57, 6458–6467. [Google Scholar] [CrossRef] [PubMed]
- Darshan, M.S.; Loftus, M.S.; Thadani-Mulero, M.; Levy, B.P.; Escuin, D.; Zhou, X.K.; Gjyrezi, A.; Chanel-Vos, C.; Shen, R.; Tagawa, S.T.; et al. Taxane-Induced Blockade to Nuclear Accumulation of the Androgen Receptor Predicts Clinical Responses in Metastatic Prostate Cancer. Cancer Res. 2011, 71, 6019–6029. [Google Scholar] [CrossRef] [Green Version]
- Thadani-Mulero, M.; Portella, L.; Sun, S.; Sung, M.; Matov, A.; Vessella, R.L.; Corey, E.; Nanus, D.M.; Plymate, S.R.; Giannakakou, P. Androgen Receptor Splice Variants Determine Taxane Sensitivity in Prostate Cancer. Cancer Res. 2014, 74, 2270–2282. [Google Scholar] [CrossRef] [Green Version]
- Krause, W.C.; Shafi, A.A.; Nakka, M.; Weigel, N.L. Androgen Receptor and its Splice Variant, AR-V7, Differentially Regulate FOXA1 Sensitive Genes in LNCaP Prostate Cancer Cells. Int. J. Biochem. Cell Biol. 2014, 54, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.K.; Banuelos, C.A.; Sadar, M.D.; Kyprianou, N. N-terminal targeting of androgen receptor variant enhances response of castration resistant prostate cancer to taxane chemotherapy. Mol. Oncol. 2014, 9, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Tamada, S.; Kato, M.; Hirayama, Y.; Takeyama, Y.; Iguchi, T.; Sadar, M.D.; Nakatani, T. Androgen Receptor Splice Variant 7 Drives the Growth of Castration Resistant Prostate Cancer without Being Involved in the Efficacy of Taxane Chemotherapy. J. Clin. Med. 2018, 7, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients with Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estébanez-Perpiñá, E.; Bevan, C.L.; McEwan, I.J. Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes on. Cancers 2021, 13, 509. https://doi.org/10.3390/cancers13030509
Estébanez-Perpiñá E, Bevan CL, McEwan IJ. Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes on. Cancers. 2021; 13(3):509. https://doi.org/10.3390/cancers13030509
Chicago/Turabian StyleEstébanez-Perpiñá, Eva, Charlotte L. Bevan, and Iain J. McEwan. 2021. "Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes on" Cancers 13, no. 3: 509. https://doi.org/10.3390/cancers13030509
APA StyleEstébanez-Perpiñá, E., Bevan, C. L., & McEwan, I. J. (2021). Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes on. Cancers, 13(3), 509. https://doi.org/10.3390/cancers13030509