
Tumor Immune Evasion Induced by Dysregulation of Erythroid Progenitor Cells Development



Abstract
:Simple Summary
Abstract

1. Introduction
2. Regulation of Erythropoiesis
3. Erythroid Progenitor Cells as Immune Regulators
4. The Role of Erythroid Progenitor Cells in Cancer
Tumor-Promoting Role of CD45− EPCs
5. Expansion of Erythroid Progenitor Cells
6. Cancer-Induced Dysregulation of Erythropoiesis
6.1. Dysregulation of Hematopoietic Stem and Progenitor Cells Differentiation
6.2. Disruption of Hematopoietic Stem and Progenitor Cells Niche
6.3. Suppression of Erythroid Differentiation of Hematopoiesis Stem Cells
6.4. Chronic Erythropoietin Production
6.5. Induction of Erythroid Cell Apoptosis
6.6. Transforming Growth Factor β
6.7. Iron Restriction
6.8. Pro-Inflammatory Cytokine-Driven Erythropoiesis Impairment
6.9. Cancer-Secreted Chemokines
6.10. Induction of Extramedullary Stress Erythropoiesis
6.11. Chemotherapy-Induced Impairment of Erythropoiesis
7. Modulation of EPCs to Inhibit Their Tumor-Promoting Effects
7.1. Modulation of EPCs Immunosuppressive Mechanisms
7.1.1. Reactive Oxygen Species
7.1.2. IL-10
7.1.3. PD-L1/PD-1 Axis
7.1.4. TGF-β
7.2. Anti-Artemin Therapy
7.3. Treating Anemia to Prevent EPC Expansion
7.4. Targeting Ineffective Erythropoiesis to Decrease EPC Expansion
7.5. Splenectomy
8. Clinical Consequences of Tumor-Induced Anemia and EPC Expansion
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Vervoort, S.J.; Hogg, S.J.; Ramsbottom, K.M.; Freeman, A.J.; Lalaoui, N.; Pijpers, L.; Michie, J.; Brown, K.K.; Knight, D.A.; et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Van Elsas, M.J.; van Hall, T.; van der Burg, S.H. Future Challenges in Cancer Resistance to Immunotherapy. Cancers 2020, 12, 935. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.A.; Metheny, H.; Iida, N.; Smith, L.; Hanson, M.; Steinhagen, F.; Leighty, R.M.; Roers, A.; Karp, C.L.; Müller, W.; et al. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J. Clin. Invest. 2013, 123, 4859–4874. [Google Scholar] [CrossRef]
- Busse, D.; de la Rosa, M.; Hobiger, K.; Thurley, K.; Flossdorf, M.; Scheffold, A.; Höfer, T. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc. Natl. Acad. Sci. USA 2010, 107, 3058–3063. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.L.; Chen, L.; Li, M.X.; Dong, P.; Xue, J.; Wang, J.; Zhang, T.T.; Wang, X.A.; Zhang, F.M.; Ge, H.L.; et al. Elevated expression of Foxp3 in tumor-infiltrating Treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-dependent manner. Clin. Immunol. 2010, 134, 277–288. [Google Scholar] [CrossRef]
- Sundström, P.; Stenstad, H.; Langenes, V.; Ahlmanner, F.; Theander, L.; Ndah, T.G.; Fredin, K.; Börjesson, L.; Gustavsson, B.; Bastid, J.; et al. Regulatory T Cells from Colon Cancer Patients Inhibit Effector T-cell Migration through an Adenosine-Dependent Mechanism. Cancer Immunol. Res. 2016, 4, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Ramakrishnan, R.; Altiok, S.; Youn, J.-I.; Cheng, P.; Celis, E.; Pisarev, V.; Sherman, S.; Sporn, M.B.; Gabrilovich, D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J. Clin. Invest. 2011, 121, 4015–4029. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Chittezhath, M.; Dhillon, M.K.; Lim, J.Y.; Laoui, D.; Shalova, I.N.; Teo, Y.L.; Chen, J.; Kamaraj, R.; Raman, L.; Lum, J.; et al. Molecular Profiling Reveals a Tumor-Promoting Phenotype of Monocytes and Macrophages in Human Cancer Progression. Immunity 2014, 41, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Vom Berg, J.; Vrohlings, M.; Haller, S.; Haimovici, A.; Kulig, P.; Sledzinska, A.; Weller, M.; Becher, B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 2013, 210, 2803–2811. [Google Scholar] [CrossRef] [Green Version]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; Van de Velde, L.-A.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Michaeli, J.; Shaul, M.E.; Mishalian, I.; Hovav, A.H.; Levy, L.; Zolotriov, L.; Granot, Z.; Fridlender, Z.G. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology 2017, 6, e1356965. [Google Scholar] [CrossRef]
- Wang, T.T.; Zhao, Y.L.; Peng, L.S.; Chen, N.; Chen, W.; Lv, Y.P.; Mao, F.Y.; Zhang, J.Y.; Cheng, P.; Teng, Y.S.; et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut 2017, 66, 1900–1911. [Google Scholar] [CrossRef] [Green Version]
- Nazareth, M.R.; Broderick, L.; Simpson-Abelson, M.R.; Kelleher, R.J., Jr.; Yokota, S.J.; Bankert, R.B. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J. Immunol. 2007, 178, 5552–5562. [Google Scholar] [CrossRef] [Green Version]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8+ T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; He, R.; Long, H.; Guo, B.; Jia, Q.; Qin, D.; Liu, S.-Q.; Wang, Z.; Xiang, T.; Zhang, J.; et al. Late-stage tumors induce anemia and immunosuppressive extramedullary erythroid progenitor cells. Nat. Med. 2018, 24, 1536–1544. [Google Scholar] [CrossRef]
- Chen, J.; Qiao, Y.-D.; Li, X.; Xu, J.-L.; Ye, Q.-J.; Jiang, N.; Zhang, H.; Wu, X.-Y. Intratumoral CD45+ CD71+ erythroid cells induce immune tolerance and predict tumor recurrence in hepatocellular carcinoma. Cancer Lett. 2020. [Google Scholar] [CrossRef]
- Sano, Y.; Yoshida, T.; Choo, M.-K.; Jiménez-Andrade, Y.; Hill, K.R.; Georgopoulos, K.; Park, J.M. Multiorgan Signaling Mobilizes Tumor-Associated Erythroid Cells Expressing Immune Checkpoint Molecules. Mol. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Jung, S.K.; Kurre, P. Hematopoietic stem and progenitor cell signaling in the niche. Leukemia 2020, 34, 3136–3148. [Google Scholar] [CrossRef]
- Peter, V.; Guntram, B.; Igor, T.; Iris, Z.U.; Ulrich, G.; Reinhard, S.; Karl, S.; Wolfgang, F.; Peter, B.; Michael, P.; et al. Normal and pathological erythropoiesis in adults: From gene regulation to targeted treatment concepts. Haematologica 2018, 103, 1593–1603. [Google Scholar] [CrossRef]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [Green Version]
- Oburoglu, L.; Romano, M.; Taylor, N.; Kinet, S. Metabolic regulation of hematopoietic stem cell commitment and erythroid differentiation. Curr. Opin. Hematol. 2016, 23, 198–205. [Google Scholar] [CrossRef]
- Koury, M.J. Tracking erythroid progenitor cells in times of need and times of plenty. Exp. Hematol. 2016, 44, 653–663. [Google Scholar] [CrossRef]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-C.; Hsu, P.-Y.; Hsu, H.-Y. Stem cell factor produced by tumor cells expands myeloid-derived suppressor cells in mice. Sci. Rep. 2020, 10, 11257. [Google Scholar] [CrossRef]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchemore, K.A.; Anderson, R.L.; Hamilton, J.A. Neutrophils, G-CSF and their contribution to breast cancer metastasis. FEBS J. 2018, 285, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Dentelli, P.; Rosso, A.; Olgasi, C.; Camussi, G.; Brizzi, M.F. IL-3 is a novel target to interfere with tumor vasculature. Oncogene 2011, 30, 4930–4940. [Google Scholar] [CrossRef] [Green Version]
- Kasper, C.; Terhaar, A.; Fosså, A.; Welt, A.; Seeber, S.; Nowrousian, M.R. Recombinant human erythropoietin in the treatment of cancer-related anaemia. Eur. J. Haematol. 1997, 58, 251–256. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, Y.; Liu, D.; Liu, F.; Li, X.; Pan, L.; Pang, Y.; Chen, D. Role of growth differentiation factor 11 in development, physiology and disease. Oncotarget 2017, 8, 81604–81616. [Google Scholar] [CrossRef] [Green Version]
- Bashir, M.; Damineni, S.; Mukherjee, G.; Kondaiah, P. Activin-A signaling promotes epithelial–mesenchymal transition, invasion, and metastatic growth of breast cancer. NPJ Breast Cancer 2015, 1, 15007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gao, A.; Zhao, H.; Lu, P.; Cheng, H.; Dong, F.; Gong, Y.; Ma, S.; Zheng, Y.; Zhang, H.; et al. Leukemia cell infiltration causes defective erythropoiesis partially through MIP-1α/CCL3. Leukemia 2016, 30, 1897–1908. [Google Scholar] [CrossRef]
- Liu, L.; Yu, Z.; Cheng, H.; Mao, X.; Sui, W.; Deng, S.; Wei, X.; Lv, J.; Du, C.; Xu, J.; et al. Multiple myeloma hinders erythropoiesis and causes anaemia owing to high levels of CCL3 in the bone marrow microenvironment. Sci. Rep. 2020, 10, 20508. [Google Scholar] [CrossRef]
- Silvestris, F.; Cafforio, P.; Tucci, M.; Dammacco, F. Negative regulation of erythroblast maturation by Fas-L+/TRAIL+ highly malignant plasma cells: A major pathogenetic mechanism of anemia in multiple myeloma. Blood 2002, 99, 1305–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hexner, E.O.; Serdikoff, C.; Jan, M.; Swider, C.R.; Robinson, C.; Yang, S.; Angeles, T.; Emerson, S.G.; Carroll, M.; Ruggeri, B.; et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood 2008, 111, 5663–5671. [Google Scholar] [CrossRef]
- Khalil, S.; Delehanty, L.; Grado, S.; Holy, M.; White, Z., III; Freeman, K.; Kurita, R.; Nakamura, Y.; Bullock, G.; Goldfarb, A. Iron modulation of erythropoiesis is associated with Scribble-mediated control of the erythropoietin receptor. J. Exp. Med. 2017, 215, 661–679. [Google Scholar] [CrossRef]
- Han, Y.; Liu, Q.; Hou, J.; Gu, Y.; Zhang, Y.; Chen, Z.; Fan, J.; Zhou, W.; Qiu, S.; Zhang, Y.; et al. Tumor-Induced Generation of Splenic Erythroblast-like Ter-Cells Promotes Tumor Progression. Cell 2018, 173, 634–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestris, F.; Tucci, M.; Cafforio, P.; Dammacco, F. Fas-L up-regulation by highly malignant myeloma plasma cells: Role in the pathogenesis of anemia and disease progression. Blood 2001, 97, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Gilreath, J.A.; Stenehjem, D.D.; Rodgers, G.M. Diagnosis and treatment of cancer-related anemia. Am. J. Hematol. 2014, 89, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Zowczak, M.; Iskra, M.; Torliński, L.; Cofta, S. Analysis of serum copper and zinc concentrations in cancer patients. Biol. Trace Elem. Res. 2001, 82, 1–8. [Google Scholar] [CrossRef]
- Gilreath, J.A.; Rodgers, G.M. How I treat cancer-associated anemia. Blood 2020, 136, 801–813. [Google Scholar] [CrossRef]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef]
- Vela, D.; Vela-Gaxha, Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp. Mol. Med. 2018, 50, e436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Mei, Y.; Liu, Y.; Ji, P. Understanding terminal erythropoiesis: An update on chromatin condensation, enucleation, and reticulocyte maturation. Blood Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bain, B.J. The bone marrow aspirate of healthy subjects. Br. J. Haematol. 1996, 94, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, S.; Kramer, M.; Weller, S.; Schuler, U.; Ordemann, R.; Rall, G.; Schaich, M.; Bornhäuser, M.; Ehninger, G.; Kroschinsky, F. Reevaluation of reference values for bone marrow differential counts in 236 healthy bone marrow donors. Ann. Hematol. 2020, 99, 2723–2729. [Google Scholar] [CrossRef]
- Goodman, J.W.; Hall, E.A.; Miller, K.L.; Shinpock, S.G. Interleukin 3 promotes erythroid burst formation in “serum-free” cultures without detectable erythropoietin. Proc. Natl. Acad. Sci. USA 1985, 82, 3291–3295. [Google Scholar] [CrossRef] [Green Version]
- Emerson, S.G.; Thomas, S.; Ferrara, J.L.; Greenstein, J.L. Developmental regulation of erythropoiesis by hematopoietic growth factors: Analysis on populations of BFU-E from bone marrow, peripheral blood, and fetal liver. Blood 1989, 74, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Muta, K.; Krantz, S.B.; Bondurant, M.C.; Wickrema, A. Distinct roles of erythropoietin, insulin-like growth factor I, and stem cell factor in the development of erythroid progenitor cells. J. Clin. Invest. 1994, 94, 34–43. [Google Scholar] [CrossRef] [Green Version]
- De Maria, R.; Testa, U.; Luchetti, L.; Zeuner, A.; Stassi, G.; Pelosi, E.; Riccioni, R.; Felli, N.; Samoggia, P.; Peschle, C. Apoptotic role of Fas/Fas ligand system in the regulation of erythropoiesis. Blood 1999, 93, 796–803. [Google Scholar] [CrossRef]
- Bhoopalan, S.V.; Huang, L.J.; Weiss, M.J. Erythropoietin regulation of red blood cell production: From bench to bedside and back. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-Y.; Gao, X.; Barrasa, M.I.; Li, H.; Elmes, R.R.; Peters, L.L.; Lodish, H.F. PPAR-α and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature 2015, 522, 474–477. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, L.; Caballero, N.; Fernández-Calleja, L.; Karkoulia, E.; Strouboulis, J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life 2020, 72, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Pevny, L.; Simon, M.C.; Robertson, E.; Klein, W.H.; Tsai, S.F.; D’Agati, V.; Orkin, S.H.; Costantini, F. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 1991, 349, 257–260. [Google Scholar] [CrossRef]
- De Maria, R.; Zeuner, A.; Eramo, A.; Domenichelli, C.; Bonci, D.; Grignani, F.; Srinivasula, S.M.; Alnemri, E.S.; Testa, U.; Peschle, C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 1999, 401, 489–493. [Google Scholar] [CrossRef]
- Han, X.; Zhang, J.; Peng, Y.; Peng, M.; Chen, X.; Chen, H.; Song, J.; Hu, X.; Ye, M.; Li, J.; et al. Unexpected role for p19INK4d in posttranscriptional regulation of GATA1 and modulation of human terminal erythropoiesis. Blood 2017, 129, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Ribeil, J.-A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Cruz Moura, I.; Zeuner, A.; Kirkegaard-Sørensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef]
- Elahi, S. Neglected Cells: Immunomodulatory Roles of CD71+ Erythroid Cells. Trends Immunol. 2019, 40, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S.; Ertelt, J.M.; Kinder, J.M.; Jiang, T.T.; Zhang, X.; Xin, L.; Chaturvedi, V.; Strong, B.S.; Qualls, J.E.; Steinbrecher, K.A.; et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature 2013, 504, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Shahbaz, S.; Bozorgmehr, N.; Koleva, P.; Namdar, A.; Jovel, J.; Fava, R.A.; Elahi, S. CD71+VISTA+ erythroid cells promote the development and function of regulatory T cells through TGF-β. PLoS Biol. 2018, 16, e2006649. [Google Scholar] [CrossRef] [Green Version]
- Delyea, C.; Bozorgmehr, N.; Koleva, P.; Dunsmore, G.; Shahbaz, S.; Huang, V.; Elahi, S. CD71+ Erythroid Suppressor Cells Promote Fetomaternal Tolerance through Arginase-2 and PDL-1. J. Immunol. 2018, 200, 4044–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, Y.A.; Weliwitigoda, A.; Campbell, T.; Dosanjh, M.; Johnson, P. Splenic erythroid progenitors decrease TNF-α production by macrophages and reduce systemic inflammation in a mouse model of T cell-induced colitis. Eur. J. Immunol. 2020. [Google Scholar] [CrossRef]
- Namdar, A.; Dunsmore, G.; Shahbaz, S.; Koleva, P.; Xu, L.; Jovel, J.; Houston, S.; Elahi, S. CD71+ Erythroid Cells Exacerbate HIV-1 Susceptibility, Mediate trans-Infection, and Harbor Infective Viral Particles. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, J.P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J.I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, 1296–1314. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Sosnowska, A.; Rydzynska, Z.; Lazniewski, M.; Plewczynski, D.; Klicka, K.; Malecka, M.; Rodziewicz-Lurzynska, A.; Ciepiela, O.; Justyniarska, M.; et al. Potent but transient immunosuppression of T-cells is a general feature of erythroid progenitor cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dunsmore, G.; Koleva, P.; Ghobakhloo, N.; Sutton, R.; Ambrosio, L.; Meng, X.; Hotte, N.; Nguyen, V.; Madsen, K.L.; Dieleman, L.A.; et al. Lower Abundance and Impaired Function of CD71+ Erythroid Cells in Inflammatory Bowel Disease Patients during Pregnancy. J. Crohns Colitis 2019, 13, 230–244. [Google Scholar] [CrossRef]
- Elahi, S.; Vega-López, M.A.; Herman-Miguel, V.; Ramírez-Estudillo, C.; Mancilla-Ramírez, J.; Motyka, B.; West, L.; Oyegbami, O. CD71+ Erythroid Cells in Human Neonates Exhibit Immunosuppressive Properties and Compromise Immune Response Against Systemic Infection in Neonatal Mice. Front. Immunol. 2020, 11, 597433. [Google Scholar] [CrossRef]
- Shahbaz, S.; Xu, L.; Osman, M.; Sligl, W.; Shields, J.; Joyce, M.; Tyrrell, L.; Oyegbami, O.; Elahi, S. Erythroid precursors and progenitors suppress adaptive immunity and get invaded by SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lindsey Robert, B.; Sankar, S.; Michael, A.; Gayle, B.; Bernard, C.C.; Corey, C.; Brenda, C.; Erik, R.D.; Ashley Morris, E.; Alison, G.F.; et al. Prevention and Treatment of Cancer-Related Infections, Version 2.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 882–913. [Google Scholar] [CrossRef]
- Allen, B.M.; Hiam, K.J.; Burnett, C.E.; Venida, A.; DeBarge, R.; Tenvooren, I.; Marquez, D.M.; Cho, N.W.; Carmi, Y.; Spitzer, M.H. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 2020, 26, 1125–1134. [Google Scholar] [CrossRef]
- Wu, C.; Hua, Q.; Zheng, L. Generation of Myeloid Cells in Cancer: The Spleen Matters. Front. Immunol. 2020, 11, 1126. [Google Scholar] [CrossRef]
- Strauss, L.; Guarneri, V.; Gennari, A.; Sica, A. Implications of metabolism-driven myeloid dysfunctions in cancer therapy. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Gillespie, M.A.; Palii, C.G.; Sanchez-Taltavull, D.; Shannon, P.; Longabaugh, W.J.R.; Downes, D.J.; Sivaraman, K.; Espinoza, H.M.; Hughes, J.R.; Price, N.D.; et al. Absolute Quantification of Transcription Factors Reveals Principles of Gene Regulation in Erythropoiesis. Mol. Cell 2020. [Google Scholar] [CrossRef]
- An, X.; Schulz, V.P.; Li, J.; Wu, K.; Liu, J.; Xue, F.; Hu, J.; Mohandas, N.; Gallagher, P.G. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014, 123, 3466–3477. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hale, J.; Bhagia, P.; Xue, F.; Chen, L.; Jaffray, J.; Yan, H.; Lane, J.; Gallagher, P.G.; Mohandas, N.; et al. Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E. Blood 2014, 124, 3636–3645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Hale, J.; Jaffray, J.; Li, J.; Wang, Y.; Huang, Y.; An, X.; Hillyer, C.; Wang, N.; Kinet, S.; et al. Developmental differences between neonatal and adult human erythropoiesis. Am. J. Hematol. 2018, 93, 494–503. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Chang, K.-H.; Qu, H.; Zhang, Z.; Xiong, Q.; Qi, H.; Cui, P.; Lin, Q.; Ruan, X.; et al. Transcriptome dynamics during human erythroid differentiation and development. Genomics 2013, 102, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Lin, Y.-H.; Sierant, M.C.; Zhu, F.; Cui, S.; Guan, Y.; Sartor, M.A.; Tanabe, O.; Lim, K.-C.; Engel, J.D. Developmental transcriptome analysis of human erythropoiesis. Hum. Mol. Genet. 2014, 23, 4528–4542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Y.; Ni, M.; Cao, H.; Signer, R.A.J.; Li, D.; Li, M.; Gu, Z.; Hu, Z.; Dickerson, K.E.; et al. Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat. Cell Biol. 2017, 19, 626–638. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Zhao, Y.; Zhong, J.; Zhang, X.; Liu, Q.; Qiu, X.; Chen, S.; Yan, H.; Hillyer, C.; Mohandas, N.; et al. Putative regulators for the continuum of erythroid differentiation revealed by single-cell transcriptome of human BM and UCB cells. Proc. Natl. Acad. Sci. USA 2020, 201915085. [Google Scholar] [CrossRef]
- Gautier, E.-F.; Ducamp, S.; Leduc, M.; Salnot, V.; Guillonneau, F.; Dussiot, M.; Hale, J.; Giarratana, M.-C.; Raimbault, A.; Douay, L.; et al. Comprehensive Proteomic Analysis of Human Erythropoiesis. Cell Rep. 2016, 16, 1470–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amon, S.; Meier-Abt, F.; Gillet, L.C.; Dimitrieva, S.; Theocharides, A.P.A.; Manz, M.G.; Aebersold, R. Sensitive Quantitative Proteomics of Human Hematopoietic Stem and Progenitor Cells by Data-independent Acquisition Mass Spectrometry. Mol. Cell Proteom. 2019, 18, 1454–1467. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.; Ranish, J.A.; Kummer, N.T.; Hamilton, J.; Igarashi, K.; Francastel, C.; Chi, T.H.; Crabtree, G.R.; Aebersold, R.; Groudine, M. Dynamic changes in transcription factor complexes during erythroid differentiation revealed by quantitative proteomics. Nat. Struct. Mol. Biol. 2004, 11, 73–80. [Google Scholar] [CrossRef]
- Jassinskaja, M.; Johansson, E.; Kristiansen, T.A.; Åkerstrand, H.; Sjöholm, K.; Hauri, S.; Malmström, J.; Yuan, J.; Hansson, J. Comprehensive Proteomic Characterization of Ontogenic Changes in Hematopoietic Stem and Progenitor Cells. Cell Rep. 2017, 21, 3285–3297. [Google Scholar] [CrossRef] [Green Version]
- Mello, F.V.; Land, M.G.P.; Costa, E.S.; Teodósio, C.; Sanchez, M.-L.; Bárcena, P.; Peres, R.T.; Pedreira, C.E.; Alves, L.R.; Orfao, A. Maturation-associated gene expression profiles during normal human bone marrow erythropoiesis. Cell Death Discov. 2019, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Sainan, Y.; Xin, P.; Silian, H.; Zailin, Y.; Xiaomin, S.; Wen, C.; Yong, Z. CD45+ Erythroid Progenitor Cell Contribute to Antiangiogenic Drug Resistance Through Reactive Oxygen Species in Lymphoma. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity-From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 4, eaau6085. [Google Scholar] [CrossRef]
- Imai, T.; Ishida, H.; Suzue, K.; Taniguchi, T.; Okada, H.; Shimokawa, C.; Hisaeda, H. Cytotoxic activities of CD8+ T cells collaborate with macrophages to protect against blood-stage murine malaria. Elife 2015, 4. [Google Scholar] [CrossRef]
- Ilieva, M.; Nielsen, J.; Korshunova, I.; Gotfryd, K.; Bock, E.; Pankratova, S.; Michel, T.M. Artemin and an Artemin-Derived Peptide, Artefin, Induce Neuronal Survival, and Differentiation Through Ret and NCAM. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, H.; Cai, J.; Du, S.; Xin, B.; Wei, W.; Zhang, T.; Shen, X. Artemin regulates CXCR4 expression to induce migration and invasion in pancreatic cancer cells through activation of NF-κB signaling. Exp. Cell Res. 2018, 365, 12–23. [Google Scholar] [CrossRef]
- Song, Z.; Yang, F.; Du, H.; Li, X.; Liu, J.; Dong, M.; Xu, X. Role of artemin in non-small cell lung cancer. Thorac. Cancer 2018, 9, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.-J.; Li, H.; Zhang, W.-H.; Xu, S.-S.; Jiang, W.; Li, S.; Gao, H.-L.; Han, X.; Xu, H.-X.; Wu, C.-T.; et al. Human splenic TER cells: A relevant prognostic factor acting via the artemin-GFRα3-ERK pathway in pancreatic ductal adenocarcinoma. Int. J. Cancer 2020. [Google Scholar] [CrossRef]
- Dzierzak, E.; Philipsen, S. Erythropoiesis: Development and differentiation. Cold Spring Harb. Perspect. Med. 2013, 3, a011601. [Google Scholar] [CrossRef]
- Ileana, C.; Sjaak, P. Flicking the switch: Adult hemoglobin expression in erythroid cells derived from cord blood and human induced pluripotent stem cells. Haematologica 2014, 99, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Baron, M.H.; Isern, J.; Fraser, S.T. The embryonic origins of erythropoiesis in mammals. Blood 2012, 119, 4828–4837. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.S.; Ben-Ezra, J.M.; Goel, R.; Tidwell, A. Reticulocytes and reticulocyte enumeration. J. Clin. Lab. Anal. 2001, 15, 267–294. [Google Scholar] [CrossRef]
- Paulson, R.F.; Hariharan, S.; Little, J.A. Stress erythropoiesis: Definitions and models for its study. Exp. Hematol. 2020, 89, 43–54. [Google Scholar] [CrossRef]
- Paulson, R.F.; Ruan, B.; Hao, S.; Chen, Y. Stress Erythropoiesis is a Key Inflammatory Response. Cells 2020, 9, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.; Nanton, M.R.; O’Donnell, H.; Akue, A.D.; McSorley, S.J. Innate immune activation during Salmonella infection initiates extramedullary erythropoiesis and splenomegaly. J. Immunol. 2010, 185, 6198–6204. [Google Scholar] [CrossRef] [Green Version]
- Rinchai, D.; Altman, M.C.; Konza, O.; Hässler, S.; Martina, F.; Toufiq, M.; Garand, M.; Kabeer, B.S.A.; Palucka, K.; Mejias, A.; et al. Definition of erythroid cell-positive blood transcriptome phenotypes associated with severe respiratory syncytial virus infection. Clin. Transl. Med. 2020, 10, e244. [Google Scholar] [CrossRef]
- Tabares Calvache, E.; Tabares Calvache, A.D.; Faulhaber, G.A.M. Systematic review about etiologic association to the leukoerythroblastic reaction. Int. J. Lab. Hematol. 2020, 42, 495–500. [Google Scholar] [CrossRef]
- Delsol, G.; Guiu-Godfrin, B.; Guiu, M.; Pris, J.; Corberand, J.; Fabre, J. Leukoerythroblastosis and cancer frequency, prognosis, and physiopathologic significance. Cancer 1979, 44, 1009–1013. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Cohen, A.C.; Soper, D.; Huang, Y.-W.; Cesano, A. Age-related changes of healthy bone marrow cell signaling in response to growth factors provide insight into low risk MDS. Cytom. Part B Clin. Cytom. 2014, 86, 383–396. [Google Scholar] [CrossRef]
- Oikonomidou, P.R.; Rivella, S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018, 32, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Manso, B.A.; Zhang, H.; Mikkelson, M.G.; Gwin, K.A.; Secreto, C.R.; Ding, W.; Parikh, S.A.; Kay, N.E.; Medina, K.L. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia 2019, 33, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Stefanie, G.; Manuel, R.-P.; Paul, J.; Annemarie, K.; Felix, B.; Julian, G.; Christoph, Z.; Ulrich, G.; Guido, K.; Frank, L.; et al. Transforming growth factor β1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Zhao, M.; Yang, W.; Gao, A.; Yin, X.; Hu, L.; Wang, X.; Xu, J.; Hao, S.; Cheng, T.; et al. Megakaryocyte-derived excessive transforming growth factor β1 inhibits proliferation of normal hematopoietic stem cells in acute myeloid leukemia. Exp. Hematol. 2018, 60, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Bruns, I.; Cadeddu, R.-P.; Brueckmann, I.; Fröbel, J.; Geyh, S.; Büst, S.; Fischer, J.C.; Roels, F.; Wilk, C.M.; Schildberg, F.A.; et al. Multiple myeloma–related deregulation of bone marrow–derived CD34+ hematopoietic stem and progenitor cells. Blood 2012, 120, 2620–2630. [Google Scholar] [CrossRef] [Green Version]
- Waclawiczek, A.; Hamilton, A.; Rouault-Pierre, K.; Abarrategi, A.; Albornoz, M.G.; Miraki-Moud, F.; Bah, N.; Gribben, J.; Fitzgibbon, J.; Taussig, D.; et al. Mesenchymal niche remodeling impairs hematopoiesis via stanniocalcin 1 in acute myeloid leukemia. J. Clin. Invest. 2020, 130, 3038–3050. [Google Scholar] [CrossRef]
- Wu, W.-C.; Sun, H.-W.; Chen, H.-T.; Liang, J.; Yu, X.-J.; Wu, C.; Wang, Z.; Zheng, L. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl. Acad. Sci. USA 2014, 111, 4221–4226. [Google Scholar] [CrossRef] [Green Version]
- Al Sayed, M.F.; Amrein, M.A.; Bührer, E.D.; Huguenin, A.-L.; Radpour, R.; Riether, C.; Ochsenbein, A.F. T-cell–Secreted TNFα Induces Emergency Myelopoiesis and Myeloid-Derived Suppressor Cell Differentiation in Cancer. Cancer Res. 2019, 79, 346–359. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Ning, H.; Liu, M.; Lin, J.; Luo, S.; Zhu, W.; Xu, J.; Wu, W.-C.; Liang, J.; Shao, C.-K.; et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Invest. 2018, 128, 3425–3438. [Google Scholar] [CrossRef] [Green Version]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre–metastatic Niche. Cancer Res. 2016, 76, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Glait-Santar, C.; Desmond, R.; Feng, X.; Bat, T.; Chen, J.; Heuston, E.; Mizukawa, B.; Mulloy, J.C.; Bodine, D.M.; Larochelle, A.; et al. Functional Niche Competition Between Normal Hematopoietic Stem and Progenitor Cells and Myeloid Leukemia Cells. Stem Cells 2015, 33, 3635–3642. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, R.K.; Falcon, B.; Hanson, J.; Goldstein, W.E.; Perruzzi, C.; Rafii, S.; Aird, W.C.; Benjamin, L.E. VEGF-A/VEGFR Inhibition Restores Hematopoietic Homeostasis in the Bone Marrow and Attenuates Tumor Growth. Cancer Res. 2016, 76, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, S.; Zhu, R.; Li, H.; Han, Q.; Zhao, R.C. Lung tumor exosomes induce a pro-inflammatory phenotype in mesenchymal stem cells via NFκB-TLR signaling pathway. J. Hematol. Oncol. 2016, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.; Duarte, T.L.; Teles, M.J.; Mosteo, L.; Chacim, S.; Aguiar, E.; Pereira-Reis, J.; Oliveira, M.; Silva, A.M.N.; Gonçalves, N.; et al. Loss of erythroblasts in acute myeloid leukemia causes iron redistribution with clinical implications. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cheng, H.; Hao, S.; Liu, Y.; Pang, Y.; Ma, S.; Dong, F.; Xu, J.; Zheng, G.; Li, S.; Yuan, W.; et al. Leukemic marrow infiltration reveals a novel role for Egr3 as a potent inhibitor of normal hematopoietic stem cell proliferation. Blood 2015, 126, 1302–1313. [Google Scholar] [CrossRef]
- Bouchnita, A.; Eymard, N.; Moyo, T.K.; Koury, M.J.; Volpert, V. Bone marrow infiltration by multiple myeloma causes anemia by reversible disruption of erythropoiesis. Am. J. Hematol. 2016, 91, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Haase, V.H. Hypoxic regulation of erythropoiesis and iron metabolism. Am. J. Physiol. Ren. Physiol. 2010, 299, F1–F13. [Google Scholar] [CrossRef] [Green Version]
- Dev, A.; Fang, J.; Sathyanarayana, P.; Pradeep, A.; Emerson, C.; Wojchowski, D.M. During EPO or anemia challenge, erythroid progenitor cells transit through a selectively expandable proerythroblast pool. Blood 2010, 116, 5334–5346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Kut, C.; Mac Gabhann, F.; Popel, A.S. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br. J. Cancer 2007, 97, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Lim, S.; Yang, Y.; Wang, Z.; Jensen, L.D.E.; Hedlund, E.-M.; Andersson, P.; Sasahara, M.; Larsson, O.; Galter, D.; et al. PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat. Med. 2012, 18, 100–110. [Google Scholar] [CrossRef]
- Greenwald, A.C.; Licht, T.; Kumar, S.; Oladipupo, S.S.; Iyer, S.; Grunewald, M.; Keshet, E. VEGF expands erythropoiesis via hypoxia-independent induction of erythropoietin in noncanonical perivascular stromal cells. J. Exp. Med. 2019, 216, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Pop, R.; Sadegh, C.; Brugnara, C.; Haase, V.H.; Socolovsky, M. Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood 2006, 108, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Bordini, J.; Bertilaccio, M.T.; Ponzoni, M.; Fermo, I.; Chesi, M.; Bergsagel, P.L.; Camaschella, C.; Campanella, A. Erythroblast apoptosis and microenvironmental iron restriction trigger anemia in the VK*MYC model of multiple myeloma. Haematologica 2015, 100, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Testa, U. Apoptotic mechanisms in the control of erythropoiesis. Leukemia 2004, 18, 1176–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, T.; Yu, C.; Ma, A.; Orkin, S.H.; Blobel, G.A.; Weiss, M.J. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 1999, 94, 87–96. [Google Scholar] [CrossRef]
- Tanaka, H.; Matsumura, I.; Nakajima, K.; Daino, H.; Sonoyama, J.; Yoshida, H.; Oritani, K.; Machii, T.; Yamamoto, M.; Hirano, T.; et al. GATA-1 blocks IL-6-induced macrophage differentiation and apoptosis through the sustained expression of cyclin D1 and bcl-2 in a murine myeloid cell line M1. Blood 2000, 95, 1264–1273. [Google Scholar] [CrossRef]
- Spierings, D.C.J.; de Vries, E.G.E.; Timens, W.; Groen, H.J.M.; Boezen, H.M.; de Jong, S. Expression of TRAIL and TRAIL Death Receptors in Stage III Non-Small Cell Lung Cancer Tumors. Clin. Cancer Res. 2003, 9, 3397–3405. [Google Scholar]
- Secchiero, P.; Melloni, E.; Heikinheimo, M.; Mannisto, S.; Di Pietro, R.; Iacone, A.; Zauli, G. TRAIL regulates normal erythroid maturation through an ERK-dependent pathway. Blood 2004, 103, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Zamai, L.; Secchiero, P.; Pierpaoli, S.; Bassini, A.; Papa, S.; Alnemri, E.S.; Guidotti, L.; Vitale, M.; Zauli, G. TNF-related apoptosis-inducing ligand (TRAIL) as a negative regulator of normal human erythropoiesis. Blood 2000, 95, 3716–3724. [Google Scholar]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- González-Santiago, A.E.; Mendoza-Topete, L.A.; Sánchez-Llamas, F.; Troyo-Sanromán, R.; Gurrola-Díaz, C.M. TGF-β1 serum concentration as a complementary diagnostic biomarker of lung cancer: Establishment of a cut-point value. J. Clin. Lab. Anal. 2011, 25, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Kawata, S.; Tamura, S.; Ito, N.; Tsushima, H.; Takaishi, K.; Kiso, S.; Matsuzawa, Y. Plasma transforming growth factor-beta 1 in patients with hepatocellular carcinoma. Comparison with chronic liver diseases. Cancer 1994, 73, 2275–2279. [Google Scholar] [CrossRef]
- Zermati, Y.; Fichelson, S.; Valensi, F.; Freyssinier, J.M.; Rouyer-Fessard, P.; Cramer, E.; Guichard, J.; Varet, B.; Hermine, O. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp. Hematol. 2000, 28, 885–894. [Google Scholar] [CrossRef]
- Kuhikar, R.; Khan, N.; Philip, J.; Melinkeri, S.; Kale, V.; Limaye, L. Transforming growth factor β1 accelerates and enhances in vitro red blood cell formation from hematopoietic stem cells by stimulating mitophagy. Stem Cell Res. Ther. 2020, 11, 71. [Google Scholar] [CrossRef]
- Akel, S.; Petrow-Sadowski, C.; Laughlin, M.J.; Ruscetti, F.W. Neutralization of autocrine transforming growth factor-beta in human cord blood CD34+CD38−Lin− cells promotes stem-cell-factor-mediated erythropoietin-independent early erythroid progenitor development and reduces terminal differentiation. Stem Cells 2003, 21, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lee, H.Y.; da Rocha, E.L.; Zhang, C.; Lu, Y.F.; Li, D.; Feng, Y.; Ezike, J.; Elmes, R.R.; Barrasa, M.I.; et al. TGF-β inhibitors stimulate red blood cell production by enhancing self-renewal of BFU-E erythroid progenitors. Blood 2016, 128, 2637–2641. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Elhance, A.; Van Duzer, A.; Kumar, S.; Leitenberger, J.J.; Oshimori, N. Tumor-initiating cells establish an IL-33–TGF-β niche signaling loop to promote cancer progression. Science 2020, 369, eaay1813. [Google Scholar] [CrossRef]
- Fournié, J.-J.; Poupot, M. The Pro-tumorigenic IL-33 Involved in Antitumor Immunity: A Yin and Yang Cytokine. Front. Immunol. 2018, 9, 2506. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.W.; Koneva, L.A.; Regan-Komito, D.; Sansom, S.N.; Powrie, F.; Griseri, T. IL-33 promotes anemia during chronic inflammation by inhibiting differentiation of erythroid progenitors. J. Exp. Med. 2020, 217, e20200164. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.V.S.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.A.; Li, R.; Ramanathan, H.N.; Bhasin, M.; Pearsall, R.S.; Kumar, R.; Suragani, R.N.V.S. Smad2/3-pathway ligand trap luspatercept enhances erythroid differentiation in murine β-thalassaemia by increasing GATA-1 availability. J. Cell. Mol. Med. 2020, 24, 6162–6177. [Google Scholar] [CrossRef] [PubMed]
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat. Med. 2014, 20, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, H.; Shao, Z. GDF11 Increased in Patients with Myelodysplastic Syndrome. Blood 2015, 126, 5224. [Google Scholar] [CrossRef]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef]
- Lenox, L.E.; Perry, J.M.; Paulson, R.F. BMP4 and Madh5 regulate the erythroid response to acute anemia. Blood 2005, 105, 2741–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.M.; Harandi, O.F.; Paulson, R.F. BMP4, SCF, and hypoxia cooperatively regulate the expansion of murine stress erythroid progenitors. Blood 2007, 109, 4494–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.; Nemeth, E. New insights into iron regulation and erythropoiesis. Curr. Opin. Hematol. 2015, 22, 199–205. [Google Scholar] [CrossRef]
- Clara, C.; Antonella, N.; Laura, S. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, H.; Müldür, E.; Endler, G.; Hübl, W. Prevalence of iron deficiency across different tumors and its association with poor performance status, disease status and anemia. Ann. Oncol. 2013, 24, 1886–1892. [Google Scholar] [CrossRef]
- Liu, S.; Bhattacharya, S.; Han, A.; Suragani, R.N.; Zhao, W.; Fry, R.C.; Chen, J.J. Haem-regulated eIF2alpha kinase is necessary for adaptive gene expression in erythroid precursors under the stress of iron deficiency. Br. J. Haematol. 2008, 143, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Masahiro, K.; Hiroki, K.; Hiroshi, H.; Ari, I.-N.; Tohru, F.; Akihiko, M.; Yukihiro, I.; Kenji, I.; Wataru, H.; Naohisa, T.; et al. Iron-heme-Bach1 axis is involved in erythroblast adaptation to iron deficiency. Haematologica 2017, 102, 454–465. [Google Scholar] [CrossRef] [Green Version]
- Bullock, G.C.; Delehanty, L.L.; Talbot, A.-L.; Gonias, S.L.; Tong, W.-H.; Rouault, T.A.; Dewar, B.; Macdonald, J.M.; Chruma, J.J.; Goldfarb, A.N. Iron control of erythroid development by a novel aconitase-associated regulatory pathway. Blood 2010, 116, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Schranzhofer, M.; Schifrer, M.; Cabrera, J.A.; Kopp, S.; Chiba, P.; Beug, H.; Müllner, E.W. Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood 2006, 107, 4159–4167. [Google Scholar] [CrossRef]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef]
- Pfeifhofer-Obermair, C.; Tymoszuk, P.; Petzer, V.; Weiss, G.; Nairz, M. Iron in the Tumor Microenvironment—Connecting the Dots. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Anemia of Inflammation. N. Engl. J. Med. 2019, 381, 1148–1157. [Google Scholar] [CrossRef]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Tyrkalska, S.D.; Pérez-Oliva, A.B.; Rodríguez-Ruiz, L.; Martínez-Morcillo, F.J.; Alcaraz-Pérez, F.; Martínez-Navarro, F.J.; Lachaud, C.; Ahmed, N.; Schroeder, T.; Pardo-Sánchez, I.; et al. Inflammasome Regulates Hematopoiesis through Cleavage of the Master Erythroid Transcription Factor GATA1. Immunity 2019, 51, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Osswald, L.; Saller, B.S.; Unger, S.; De Feo, D.; Vinnakota, J.M.; Konantz, M.; Uhl, F.M.; Becker, H.; Lübbert, M.; et al. Oncogenic KrasG12D causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 2020, 11, 1659. [Google Scholar] [CrossRef] [Green Version]
- Prince, O.D.; Langdon, J.M.; Layman, A.J.; Prince, I.C.; Sabogal, M.; Mak, H.H.; Berger, A.E.; Cheadle, C.; Chrest, F.J.; Yu, Q.; et al. Late stage erythroid precursor production is impaired in mice with chronic inflammation. Haematologica 2012, 97, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Libregts, S.F.; Gutiérrez, L.; de Bruin, A.M.; Wensveen, F.M.; Papadopoulos, P.; van Ijcken, W.; Ozgür, Z.; Philipsen, S.; Nolte, M.A. Chronic IFN-γ production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood 2011, 118, 2578–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Krantz, S.B. Interferon γ Induces Upregulation and Activation of Caspases 1, 3, and 8 to Produce Apoptosis in Human Erythroid Progenitor Cells. Blood 1999, 93, 3309–3316. [Google Scholar] [CrossRef]
- Wang, C.Q.; Udupa, K.B.; Lipschitz, D.A. Interferon-γ exerts its negative regulatory effect primarily on the earliest stages of murine erythroid progenitor cell development. J. Cell. Physiol. 1995, 162, 134–138. [Google Scholar] [CrossRef]
- Dai, C.-H.; Price, J.O.; Brunner, T.; Krantz, S.B. Fas Ligand Is Present in Human Erythroid Colony-Forming Cells and Interacts with Fas Induced by Interferon γ to Produce Erythroid Cell Apoptosis. Blood 1998, 91, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Zhang, X.; Iwama, A.; Yu, C.; Smith, K.A.; Mueller, B.U.; Narravula, S.; Torbett, B.E.; Orkin, S.H.; Tenen, D.G. PU. 1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 2000, 96, 2641–2648. [Google Scholar] [CrossRef]
- Yamada, T.; Kondoh, N.; Matsumoto, M.; Yoshida, M.; Maekawa, A.; Oikawa, T. Overexpression of PU.1 induces growth and differentiation inhibition and apoptotic cell death in murine erythroleukemia cells. Blood 1997, 89, 1383–1393. [Google Scholar] [CrossRef]
- Burda, P.; Laslo, P.; Stopka, T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia 2010, 24, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Rusten, L.S.; Jacobsen, S.E. Tumor necrosis factor (TNF)-alpha directly inhibits human erythropoiesis in vitro: Role of p55 and p75 TNF receptors. Blood 1995, 85, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsopra, O.A.; Ziros, P.G.; Lagadinou, E.D.; Symeonidis, A.; Kouraklis-Symeonidis, A.; Thanopoulou, E.; Angelopoulou, M.K.; Vassilakopoulos, T.P.; Pangalis, G.A.; Zoumbos, N.C. Disease-related anemia in chronic lymphocytic leukemia is not due to intrinsic defects of erythroid precursors: A possible pathogenetic role for tumor necrosis factor-alpha. Acta Haematol. 2009, 121, 187–195. [Google Scholar] [CrossRef]
- Papadaki, H.A.; Kritikos, H.D.; Valatas, V.; Boumpas, D.T.; Eliopoulos, G.D. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: Improvement following anti–tumor necrosis factor-α antibody therapy. Blood 2002, 100, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Jacobs-Helber, S.M.; Roh, K.-H.; Bailey, D.; Dessypris, E.N.; Ryan, J.J.; Chen, J.; Wickrema, A.; Barber, D.L.; Dent, P.; Sawyer, S.T. Tumor necrosis factor-alpha expressed constitutively in erythroid cells or induced by erythropoietin has negative and stimulatory roles in normal erythropoiesis and erythroleukemia. Blood 2003, 101, 524–531. [Google Scholar] [CrossRef]
- Bibikova, E.; Youn, M.-Y.; Danilova, N.; Ono-Uruga, Y.; Konto-Ghiorghi, Y.; Ochoa, R.; Narla, A.; Glader, B.; Lin, S.; Sakamoto, K.M. TNF-mediated inflammation represses GATA1 and activates p38 MAP kinase in RPS19-deficient hematopoietic progenitors. Blood 2014, 124, 3791–3798. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hernandez, A.; Ray, P.; Litos, G.; Ciro, M.; Ottolenghi, S.; Beug, H.; Boyes, J. Acetylation and MAPK phosphorylation cooperate to regulate the degradation of active GATA-1. EMBO J. 2006, 25, 3264–3274. [Google Scholar] [CrossRef] [Green Version]
- Manso, B.A.; Krull, J.E.; Gwin, K.A.; Lothert, P.K.; Welch, B.M.; Novak, A.J.; Parikh, S.A.; Kay, N.E.; Medina, K.L. Chronic lymphocytic leukemia B-cell-derived TNFα impairs bone marrow myelopoiesis. iScience 2021, 24, 101994. [Google Scholar] [CrossRef] [PubMed]
- Schooley, J.C.; Kullgren, B.; Allison, A.C. Inhibition by interleukin-1 of the action of erythropoietin on erythroid precursors and its possible role in the pathogenesis of hypoplastic anaemias. Br. J. Haematol. 1987, 67, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.B.; Sworder, B.; Bouladoux, N.; Roy, C.N.; Uchida, A.M.; Grigg, M.; Robey, P.G.; Belkaid, Y. Stromal-derived IL-6 alters the balance of myeloerythroid progenitors during Toxoplasma gondii infection. J. Leukoc. Biol. 2012, 92, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Gołab, J.; Zagozdzon, R.; Stokłosa, T.; Jakóbisiak, M.; Pojda, Z.; Machaj, E. Erythropoietin prevents the development of interleukin-12-induced anemia and thrombocytopenia but does not decrease its antitumor activity in mice. Blood 1998, 91, 4387–4388. [Google Scholar] [CrossRef]
- De la Fuente López, M.; Landskron, G.; Parada, D.; Dubois-Camacho, K.; Simian, D.; Martinez, M.; Romero, D.; Roa, J.C.; Chahuán, I.; Gutiérrez, R.; et al. The relationship between chemokines CCL2, CCL3, and CCL4 with the tumor microenvironment and tumor-associated macrophage markers in colorectal cancer. Tumor Biol. 2018, 40, 1010428318810059. [Google Scholar] [CrossRef] [Green Version]
- Buck, I.; Morceau, F.; Cristofanon, S.; Heintz, C.; Chateauvieux, S.; Reuter, S.; Dicato, M.; Diederich, M. Tumor necrosis factor α inhibits erythroid differentiation in human erythropoietin-dependent cells involving p38 MAPK pathway, GATA-1 and FOG-1 downregulation and GATA-2 upregulation. Biochem. Pharmacol. 2008, 76, 1229–1239. [Google Scholar] [CrossRef]
- Liao, C.; Prabhu, K.S.; Paulson, R.F. Monocyte-derived macrophages expand the murine stress erythropoietic niche during the recovery from anemia. Blood 2018, 132, 2580–2593. [Google Scholar] [CrossRef] [Green Version]
- Millot, S.; Andrieu, V.; Letteron, P.; Lyoumi, S.; Hurtado-Nedelec, M.; Karim, Z.; Thibaudeau, O.; Bennada, S.; Charrier, J.L.; Lasocki, S.; et al. Erythropoietin stimulates spleen BMP4-dependent stress erythropoiesis and partially corrects anemia in a mouse model of generalized inflammation. Blood 2010, 116, 6072–6081. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.F.; Liao, C.; Quickel, M.D.; Yeoh, B.S.; Vijay-Kumar, M.; Hankey-Giblin, P.; Prabhu, K.S.; Paulson, R.F. Inflammation induces stress erythropoiesis through heme-dependent activation of SPI-C. Sci. Signal. 2019, 12, eaap7336. [Google Scholar] [CrossRef]
- Corazza, F.; Beguin, Y.; Bergmann, P.; André, M.; Ferster, A.; Devalck, C.; Fondu, P.; Buyse, M.; Sariban, E. Anemia in children with cancer is associated with decreased erythropoietic activity and not with inadequate erythropoietin production. Blood 1998, 92, 1793–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Xiang, J.; Qian, F.; Diwakar, B.T.; Ruan, B.; Hao, S.; Prabhu, K.S.; Paulson, R.F. Epo receptor signaling in macrophages alters the splenic niche to promote erythroid differentiation. Blood 2020, 136, 235–246. [Google Scholar] [CrossRef]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ Immature Myeloid Cells Mediate Suppression of T Cells in Mice Bearing Tumors of IL-1β-Secreting Cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, W.; Guo, X.; Qin, F.; Li, Y.; Wang, G.; Bi, Y.; Jin, X.; Han, L.; Dong, X.; Zhao, Y. G-CSF shifts erythropoiesis from bone marrow into spleen in the setting of systemic inflammation. Life Sci. Alliance 2021, 4, e202000737. [Google Scholar] [CrossRef] [PubMed]
- McKim, D.B.; Yin, W.; Wang, Y.; Cole, S.W.; Godbout, J.P.; Sheridan, J.F. Social Stress Mobilizes Hematopoietic Stem Cells to Establish Persistent Splenic Myelopoiesis. Cell Rep. 2018, 25, 2552–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamo, I.G.; Kannan, K.B.; Loftus, T.J.; Ramos, H.; Efron, P.A.; Mohr, A.M. Severe trauma and chronic stress activates extramedullary erythropoiesis. J. Trauma Acute Care Surg. 2017, 83, 144–150. [Google Scholar] [CrossRef]
- Zeuner, A.; Pedini, F.; Signore, M.; Testa, U.; Pelosi, E.; Peschle, C.; De Maria, R. Stem cell factor protects erythroid precursor cells from chemotherapeutic agents via up-regulation of BCL-2 family proteins. Blood 2003, 102, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Bartucci, M.; Dattilo, R.; Martinetti, D.; Todaro, M.; Zapparelli, G.; Di Virgilio, A.; Biffoni, M.; De Maria, R.; Zeuner, A. Prevention of Chemotherapy-Induced Anemia and Thrombocytopenia by Constant Administration of Stem Cell Factor. Clin. Cancer Res. 2011, 17, 6185–6191. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re308. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Infante, J.R.; Papadopoulos, K.P.; Chan, I.H.; Shen, C.; Ratti, N.P.; Rojo, B.; Autio, K.A.; Wong, D.J.; Patel, M.R.; et al. PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8+ T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell 2018, 34, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Vahl, J.M.; Friedrich, J.; Mittler, S.; Trump, S.; Heim, L.; Kachler, K.; Balabko, L.; Fuhrich, N.; Geppert, C.-I.; Trufa, D.I.; et al. Interleukin-10-regulated tumour tolerance in non-small cell lung cancer. Br. J. Cancer 2017, 117, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Liu, Z.; Dong, C.; Luan, Y.; Zhang, A.; Moore, C.; Fu, K.; Peng, J.; Wang, Y.; Ren, Z.; et al. Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8+ T Cell Apoptosis. Cancer Cell 2019, 35, 901–915. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumm, J.B.; Emmerich, J.; Zhang, X.; Chan, I.; Wu, L.; Mauze, S.; Blaisdell, S.; Basham, B.; Dai, J.; Grein, J.; et al. IL-10 elicits IFNγ-dependent tumor immune surveillance. Cancer Cell 2011, 20, 781–796. [Google Scholar] [CrossRef] [Green Version]
- Oft, M. Immune regulation and cytotoxic T cell activation of IL-10 agonists—Preclinical and clinical experience. Semin. Immunol. 2019, 44, 101325. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Chamoto, K.; Hatae, R.; Honjo, T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int. J. Clin. Oncol. 2020, 25, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Li, S.; Liu, M.; Do, M.H.; Chou, C.; Stamatiades, E.G.; Nixon, B.G.; Shi, W.; Zhang, X.; Li, P.; Gao, S.; et al. Cancer immunotherapy via targeted TGF-β signalling blockade in TH cells. Nature 2020, 587, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeneveldt, C.; van Hall, T.; van der Burg, S.H.; Ten Dijke, P.; van Montfoort, N. Immunotherapeutic Potential of TGF-β Inhibition and Oncolytic Viruses. Trends Immunol. 2020, 41, 406–420. [Google Scholar] [CrossRef]
- DeBerry, J.J.; Saloman, J.L.; Dragoo, B.K.; Albers, K.M.; Davis, B.M. Artemin Immunotherapy Is Effective in Preventing and Reversing Cystitis-Induced Bladder Hyperalgesia via TRPA1 Regulation. J. Pain 2015, 16, 628–636. [Google Scholar] [CrossRef] [Green Version]
- Bohlius, J.; Bohlke, K.; Lazo-Langner, A. Management of Cancer-Associated Anemia With Erythropoiesis-Stimulating Agents: ASCO/ASH Clinical Practice Guideline Update. J. Oncol. Pract. 2019, 15, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Aapro, M.; Beguin, Y.; Bokemeyer, C.; Dicato, M.; Gascón, P.; Glaspy, J.; Hofmann, A.; Link, H.; Littlewood, T.; Ludwig, H.; et al. Management of anaemia and iron deficiency in patients with cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. 2018, 29, iv96–iv110. [Google Scholar] [CrossRef]
- Katsarou, A.; Pantopoulos, K. Hepcidin Therapeutics. Pharmaceuticals 2018, 11, 127. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Nguyen, A.N.; Sohal, D.; Ying Ma, J.; Pahanish, P.; Gundabolu, K.; Hayman, J.; Chubak, A.; Mo, Y.; Bhagat, T.D.; et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood 2008, 112, 3434–3443. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.; Suragani, R.N.V.S.; Aluri, S.; Shah, N.; Bhagat, T.D.; Alexander, M.J.; Komrokji, R.; Kumar, R. Biological basis for efficacy of activin receptor ligand traps in myelodysplastic syndromes. J. Clin. Invest. 2020, 130, 582–589. [Google Scholar] [CrossRef]
- Teixeira, A.F.; ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Xue, F.; Halverson, G.; Reid, M.; Guo, A.; Chen, L.; Raza, A.; Galili, N.; Jaffray, J.; et al. Isolation and functional characterization of human erythroblasts at distinct stages: Implications for understanding of normal and disordered erythropoiesis in vivo. Blood 2013, 121, 3246–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvarsdóttir, E.M.; Mortera-Blanco, T.; Dimitriou, M.; Bouderlique, T.; Jansson, M.; Hofman, I.J.F.; Conte, S.; Karimi, M.; Sander, B.; Douagi, I.; et al. A three-dimensional in vitro model of erythropoiesis recapitulates erythroid failure in myelodysplastic syndromes. Leukemia 2020, 34, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.-K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Porter, J.; Origa, R.; Forni, G.L.; Voskaridou, E.; Galactéros, F.; Taher, A.T.; Arlet, J.-B.; Ribeil, J.-A.; Garbowski, M.; et al. Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: A phase II, open-label, dose-finding study. Haematologica 2019, 104, 477–484. [Google Scholar] [CrossRef]
- Raftopoulos, H.; Laadem, A.; Hesketh, P.J.; Goldschmidt, J.; Gabrail, N.; Osborne, C.; Ali, M.; Sherman, M.L.; Wang, D.; Glaspy, J.A.; et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: Results from two phase 2 studies. Support. Care Cancer 2016, 24, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Morrell, N.W.; Bloch, D.B.; ten Dijke, P.; Goumans, M.-J.T.H.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Steinbicker, A.U.; Sachidanandan, C.; Vonner, A.J.; Yusuf, R.Z.; Deng, D.Y.; Lai, C.S.; Rauwerdink, K.M.; Winn, J.C.; Saez, B.; Cook, C.M.; et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011, 117, 4915–4923. [Google Scholar] [CrossRef] [Green Version]
- Mayeur, C.; Kolodziej, S.A.; Wang, A.; Xu, X.; Lee, A.; Yu, P.B.; Shen, J.; Bloch, K.D.; Bloch, D.B. Oral administration of a bone morphogenetic protein type I receptor inhibitor prevents the development of anemia of inflammation. Haematologica 2015, 100, e68–e71. [Google Scholar] [CrossRef] [Green Version]
- Wannamaker, W.; Davies, R.; Namchuk, M.; Pollard, J.; Ford, P.; Ku, G.; Decker, C.; Charifson, P.; Weber, P.; Germann, U.A.; et al. (S)-1-((S)-2-{[1 -(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J. Pharm. Exp. Ther. 2007, 321, 509–516. [Google Scholar] [CrossRef]
- Hu, P.; Nebreda, A.R.; Hanenberg, H.; Kinnebrew, G.H.; Ivan, M.; Yoder, M.C.; Filippi, M.-D.; Broxmeyer, H.E.; Kapur, R. P38α/JNK signaling restrains erythropoiesis by suppressing Ezh2-mediated epigenetic silencing of Bim. Nat. Commun. 2018, 9, 3518. [Google Scholar] [CrossRef]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Sudo, T.; Senftleben, U.; Dadak, A.M.; Johnson, R.; Karin, M. Requirement for p38α in Erythropoietin Expression: A Role for Stress Kinases in Erythropoiesis. Cell 2000, 102, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Kuhrt, D.; Wojchowski, D.M. Emerging EPO and EPO receptor regulators and signal transducers. Blood 2015, 125, 3536–3541. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Libani, I.V.; Guy, E.C.; Melchiori, L.; Schiro, R.; Ramos, P.; Breda, L.; Scholzen, T.; Chadburn, A.; Liu, Y.; Kernbach, M.; et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood 2008, 112, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talpaz, M.; Kiladjian, J.-J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Javadian, M.; Babaloo, Z.; Baradaran, B. Janus kinase inhibitors: A therapeutic strategy for cancer and autoimmune diseases. J. Cell. Physiol. 2020, 235, 5903–5924. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Campreciós, G.; Rimmelé, P.; Liang, R.; Yalcin, S.; Mungamuri, S.K.; Barminko, J.; D’Escamard, V.; Baron, M.H.; Brugnara, C.; et al. FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am. J. Hematol. 2014, 89, 954–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, S.S.; De Falco, L.; Ghaffari, S.; Brugnara, C.; Sinclair, D.A.; Matte, A.; Iolascon, A.; Mohandas, N.; Bertoldi, M.; An, X.; et al. Resveratrol accelerates erythroid maturation by activation of FoxO3 and ameliorates anemia in beta-thalassemic mice. Haematologica 2014, 99, 267–275. [Google Scholar] [CrossRef]
- Sibon, D.; Coman, T.; Rossignol, J.; Lamarque, M.; Kosmider, O.; Bayard, E.; Fouquet, G.; Rignault, R.; Topçu, S.; Bonneau, P.; et al. Enhanced Renewal of Erythroid Progenitors in Myelodysplastic Anemia by Peripheral Serotonin. Cell Rep. 2019, 26, 3246–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amireault, P.; Hatia, S.; Bayard, E.; Bernex, F.; Collet, C.; Callebert, J.; Launay, J.-M.; Hermine, O.; Schneider, E.; Mallet, J.; et al. Ineffective erythropoiesis with reduced red blood cell survival in serotonin-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 13141–13146. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Dutta, R.; Zhang, T.Y.; Köhnke, T.; Thomas, D.; Linde, M.; Gars, E.; Stafford, M.; Kaur, S.; Nakauchi, Y.; Yin, R.; et al. Enasidenib drives human erythroid differentiation independently of isocitrate dehydrogenase 2. J. Clin. Invest. 2020, 130, 1843–1849. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhu, F.; Xu, C.; Xiao, P.; Wen, J.; Zhang, X.; Wu, B. Dangguibuxue decoction abolishes abnormal accumulation of erythroid progenitor cells induced by melanoma. J. Ethnopharmacol. 2019, 242, 112035. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Romano, M.; Yan, H.; Deshmukh, R.; Papoin, J.; Oburoglu, L.; Daumur, M.; Dumé, A.S.; Phadke, I.; Mongellaz, C.; et al. An IDH1-vitamin C crosstalk drives human erythroid development by inhibiting pro-oxidant mitochondrial metabolism. Cell Rep. 2021, 34, 108723. [Google Scholar] [CrossRef]
- Steenbrugge, J.; De Jaeghere, E.A.; Meyer, E.; Denys, H.; De Wever, O. Splenic Hematopoietic and Stromal Cells in Cancer Progression. Cancer Res. 2021, 81, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Mishalian, I.; Bayuch, R.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Splenectomy inhibits non-small cell lung cancer growth by modulating anti-tumor adaptive and innate immune response. OncoImmunology 2015, 4, e998469. [Google Scholar] [CrossRef] [Green Version]
- Sano, T.; Sasako, M.; Mizusawa, J.; Yamamoto, S.; Katai, H.; Yoshikawa, T.; Nashimoto, A.; Ito, S.; Kaji, M.; Imamura, H.; et al. Randomized Controlled Trial to Evaluate Splenectomy in Total Gastrectomy for Proximal Gastric Carcinoma. Ann. Surg. 2017, 265, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Olszewski, A.J. Diagnostic and therapeutic splenectomy for splenic lymphomas: Analysis of the National Cancer Data Base. Hematology 2019, 24, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Choi, G.S.; Chung, H.Y. Randomized clinical trial of splenectomy versus splenic preservation in patients with proximal gastric cancer. Br. J. Surg. 2006, 93, 559–563. [Google Scholar] [CrossRef]
- Crawford, J.; Cella, D.; Cleeland, C.S.; Cremieux, P.Y.; Demetri, G.D.; Sarokhan, B.J.; Slavin, M.B.; Glaspy, J.A. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer 2002, 95, 888–895. [Google Scholar] [CrossRef]
- Caro, J.J.; Salas, M.; Ward, A.; Goss, G. Anemia as an independent prognostic factor for survival in patients with cancer: A systemic, quantitative review. Cancer 2001, 91, 2214–2221. [Google Scholar] [CrossRef]
- Ludwig, H.; Van Belle, S.; Barrett-Lee, P.; Birgegård, G.; Bokemeyer, C.; Gascón, P.; Kosmidis, P.; Krzakowski, M.; Nortier, J.; Olmi, P.; et al. The European Cancer Anaemia Survey (ECAS): A large, multinational, prospective survey defining the prevalence, incidence, and treatment of anaemia in cancer patients. Eur. J. Cancer 2004, 40, 2293–2306. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef] [Green Version]
- Lucotti, S.; Muschel, R.J. Platelets and Metastasis: New Implications of an Old Interplay. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Slowikowski, K.; Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 2020, 52, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Wynn, J.L.; Scumpia, P.O.; Stocks, B.T.; Romano-Keeler, J.; Alrifai, M.W.; Liu, J.-H.; Kim, A.S.; Alford, C.E.; Matta, P.; Weitkamp, J.-H.; et al. Neonatal CD71+ Erythroid Cells Do Not Modify Murine Sepsis Mortality. J. Immunol. 2015, 195, 1064–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sennikov, S.V.; Injelevskaya, T.V.; Krysov, S.V.; Silkov, A.N.; Kovinev, I.B.; Dyachkova, N.J.; Zenkov, A.N.; Loseva, M.I.; Kozlov, V.A. Production of hemo- and immunoregulatory cytokines by erythroblast antigen+ and glycophorin A+ cells from human bone marrow. BMC Cell Biol. 2004, 5, 39. [Google Scholar] [CrossRef] [Green Version]





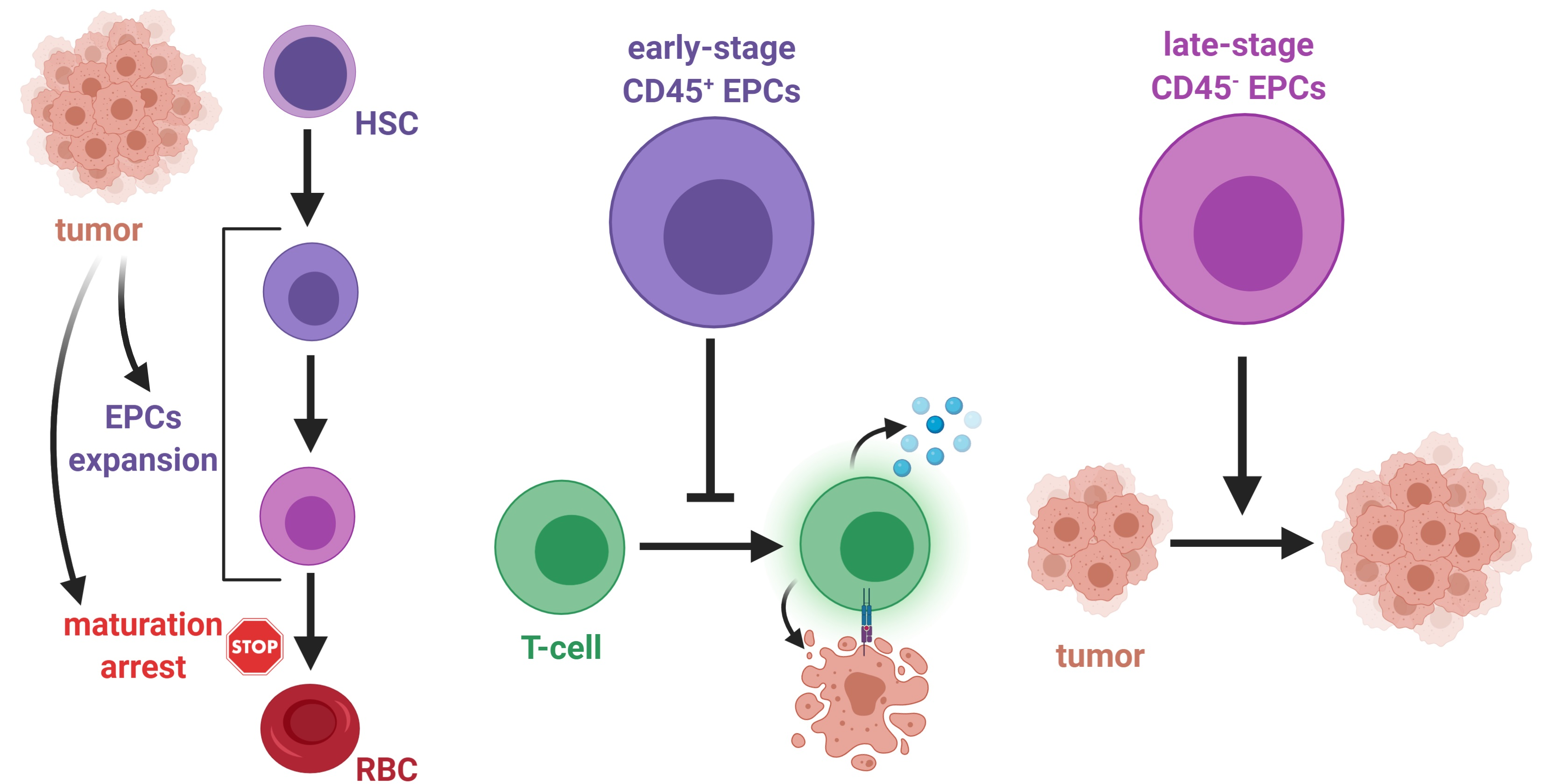{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Mechanisms | Effects | Ref |
|---|---|---|---|
| Regulatory T-cells (Tregs) | IL-10 | T-cell suppression | [14] |
| IL-2 consumption | T-cell suppression | [15] | |
| COX-2 and PGE2 | T-cell suppression | [16] | |
| Adenosine | T-cell suppression | [17] | |
| Myeloid-derived suppressor cells (MDSCs) | ARG1 | T-cell suppression | [18] |
| IDO | T-cell suppression Tregs induction NK cell suppression | [19,20] | |
| PD-L1/PD-1 | T-cell suppression | [21] | |
| IL-10 | Tregs induction | [22] | |
| TGF-β | Tregs induction | [22] | |
| CD40/CD40L | Tregs activation | [23] | |
| Depletion of cystine and cysteine | T-cell suppression | [24] | |
| ROS | T-cell suppression | [25] | |
| Free radical peroxynitrite | Resistance to cytotoxic T-cells | [26] | |
| Tumor associated macrophages (TAMs) | PD-L1/PD-1 | Decreased phagocytosis | [27] |
| ARG1 | T-cell suppression | [28] | |
| IL-10 | T-cell suppression | [29] | |
| IL-1β | MDSC infiltration Induction of the protumor phenotype | [30,31] | |
| IL-12 | Induction of T-cell response | [32] | |
| TNF-α | Induction of anti-tumor response | [33] | |
| Tumor associated neutrophils (TANs) | ARG1 | T-cell suppression | [18,28] |
| NOS | T-cell suppression T-cell apoptosis | [34,35] | |
| PD-L1/PD-1 | T-cell suppression | [36] | |
| Cancer associated fibroblasts (CAFs) | PD-L1/PD-1 | T-cell suppression | [37] |
| FasL, PD-L2 | T-cell suppression | [38] | |
| IL-6 | Induction of PD-L1+ TANs | [39] | |
| Chemokines | MDSC infiltration | [40] | |
| Erythroid progenitor cells (EPCs) | ROS | T-cell suppression | [41,42] |
| IL-10 | T-cell suppression | [42] | |
| PD-L1/PD-1 | T-cell suppression | [43] | |
| TGF-β | T-cell suppression | [42] |
| Factor | Role in Erythropoiesis | Dysregulation in Cancer | References |
|---|---|---|---|
| SCF | Growth factors regulating early stages of erythropoiesis | Production in TME Increased serum concentration | [50,51] |
| G-CSF | [52] | ||
| IL-3 | [53] | ||
| EPO | Growth factors regulating late stages of erythropoiesis | Increased serum concentration | [54] |
| GDF11 | Production in TME | [55] | |
| Activin A | Production in TME | [56] | |
| GATA1 | Crucial TFs regulating erythropoiesis | Decreased expression in EPCs in cancer | [57,58,59] |
| STAT5 | Increased in EPCs in MPNs Decreased in EPCs in iron deficiency | [60,61] | |
| MCL-1 | Survival factors for erythroid cells | ||
| BCL-xL | |||
| HSP70 | |||
| TGF-β | Negative regulators of erythropoiesis | Production in TME Increased concentration | [62] |
| SMAD signaling | Increased level in EPCs in cancer | [62] | |
| FasL | High expression on cancer cells | [59,63] | |
| Fas | Increased level in EPCs in cancer | [59,63] | |
| Vitamin B12 | Essential vitamins, trace elements, and iron-metabolism proteins | Decreased in a subset of patients | [64] |
| Folic Acid | Decreased in a subset of patients | [64] | |
| Copper | Increased concentration | [65] | |
| Iron | Decreased in a subset of patients | [66] | |
| Ferritin | Decreased or increased | [66] | |
| Transferrin | Decreased in a subset of patients | [66] | |
| Ferroportin | Decreased expression | [67] | |
| Hepcidin | Increased concentration | [68] |
| Source | Mechanism | Effect | Mouse | Humans | Ref. |
|---|---|---|---|---|---|
| Neonates | ARG2 | ↓cytokine production bymyeloid cells | + | + | [86,93] |
| TGF-β | ↑Tregs differentiation | + | + | [87] | |
| ROS | ↓cytokine production bymyeloid cells ↓cytokine production by T-cells | - | + | [94] | |
| PD-1/PD-L1 | ↓cytokine production by T-cells | + | + | [88] | |
| Pregnancy | ARG2 | ↓cytokine production bymyeloid cells | + | + | [24,93] |
| TGF-β | ↑Tregs differentiation | n.d. | + | [93] | |
| ROS | ↓cytokine production bymyeloid cells ↓cytokine production by T-cells | n.d. | + | [93] | |
| PD-1/PD-L1 | ↓cytokine production by T-cells | + | + | [88] | |
| Inflammatory diseases | EPCs phagocytosis | ↓cytokine production byred pulp macrophages | + | n.d. | [89] |
| HIV-infected patients | ROS | ↑HIV replication in T-cells ↑HIV trans-infection | n.d. | + | [90] |
| COVID-19 patients | ARG1 | ↓cytokine production by T-cells ↓T-cell proliferation | n.d. | + | [95] |
| ARG2 | ↓cytokine production by T-cells ↓T-cell proliferation | n.d. | + | [95] | |
| ROS | ↓cytokine production by T-cells ↓T-cell proliferation | n.d. | + | [95] | |
| Anemia | ARG1 | ↓cytokine production by T-cells ↓T-cell proliferation | - | + | [92] |
| ARG2 | ↓cytokine production by T-cells ↓T-cell proliferation | + | + | [92] | |
| ROS | ↓cytokine production by T-cells ↓T-cell proliferation | + | + | [92] | |
| Cancer | TGF-β | ↓T-cells proliferation↓cytokine production by T-cells | n.d. | + | [42] |
| ROS | ↓T-cell proliferation↓cytokine production by T-cells | + | + | [41,42] | |
| PD-L1/PD-1 | ↓cytokine production by T-cells | + | + | [43] | |
| IL-10 | ↓T-cell proliferation↓cytokine production by T-cells | n.d. | + | [42] |
| Feature | Early-Stage EPCs (CD45+) | Late-Stage EPCs (CD45-) |
|---|---|---|
| ROS level | ↑ | ↓ |
| IL-10 | ↑ | ↓ |
| TGF-β | ↑ | ↓ |
| ROS pathway | ↑ | ↓ |
| IL-10 pathway | ↑ | ↓ |
| TGF-β pathway | ↑ | ↓ |
| PD-1/PD-L1 | n.d. | n.d. |
| ARG2 | n.d. | n.d. |
| Process | Early-Stage EPCs (CD45+) | Late-Stage EPCs (CD45-) |
|---|---|---|
| T-cell proliferation | ↓ suppressed | ↔ no effect |
| Production of IFN-γ by T-cells | ↓ suppressed | ↔ no effect |
| Production of TNF-α by T-cells | ↓ suppressed | ↔ no effect |
| CD8+ T-cells cytotoxicity | ↓ suppressed | ↔ no effect |
| Dendritic cells activation | n.d. | ↔ no effect |
| Production of IL-6 and IL-12 by dendritic cells | n.d. | ↔ no effect |
| Tregs induction | n.d. | ↔ no effect |
| Anti-tumor immune response | ↓ suppressed | ↔ no effect |
| Activation of signaling pathways in tumor cells | n.d. | ↑ promoted |
| Regulation of cancer cell metabolism | ↑ promoted | n.d. |
| Tumor cells proliferation | n.d. | ↑ promoted |
| Tumor cells invasiveness | n.d. | ↑ promoted |
| Tumor growth | ↑ promoted | ↑ promoted |
| Organ | Mice | Humans | Ref. | ||
|---|---|---|---|---|---|
| Healthy | Tumor-Bearing | Healthy | Cancer Patient | ||
| Peripheral blood | 5% | 60% | 0.13% | 2–4.25% | [41,42] |
| Spleen | 5% | 20–50% | 0.02% | 0.15% | [41,43,62,123] |
| Bone marrow | 15–20% | 55% | 14% | n.d. | [41,43,134] |
| Liver | 10% | 2–30% | 2.5% | 10% | [41,42,62] |
| Lymph node | 1% | 1% | n.d | n.d | [41,62] |
| Tumor | - | 2–10% | - | 10% | [41,42,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzywa, T.M.; Justyniarska, M.; Nowis, D.; Golab, J. Tumor Immune Evasion Induced by Dysregulation of Erythroid Progenitor Cells Development. Cancers 2021, 13, 870. https://doi.org/10.3390/cancers13040870
Grzywa TM, Justyniarska M, Nowis D, Golab J. Tumor Immune Evasion Induced by Dysregulation of Erythroid Progenitor Cells Development. Cancers. 2021; 13(4):870. https://doi.org/10.3390/cancers13040870
Chicago/Turabian StyleGrzywa, Tomasz M., Magdalena Justyniarska, Dominika Nowis, and Jakub Golab. 2021. "Tumor Immune Evasion Induced by Dysregulation of Erythroid Progenitor Cells Development" Cancers 13, no. 4: 870. https://doi.org/10.3390/cancers13040870
APA StyleGrzywa, T. M., Justyniarska, M., Nowis, D., & Golab, J. (2021). Tumor Immune Evasion Induced by Dysregulation of Erythroid Progenitor Cells Development. Cancers, 13(4), 870. https://doi.org/10.3390/cancers13040870