
Detection of Tumor-Associated Membrane Receptors on Extracellular Vesicles from Non-Small Cell Lung Cancer Patients via Immuno-PCR

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Zx–DNA Conjugates Are Suitable Reagents for Immuno-PCR
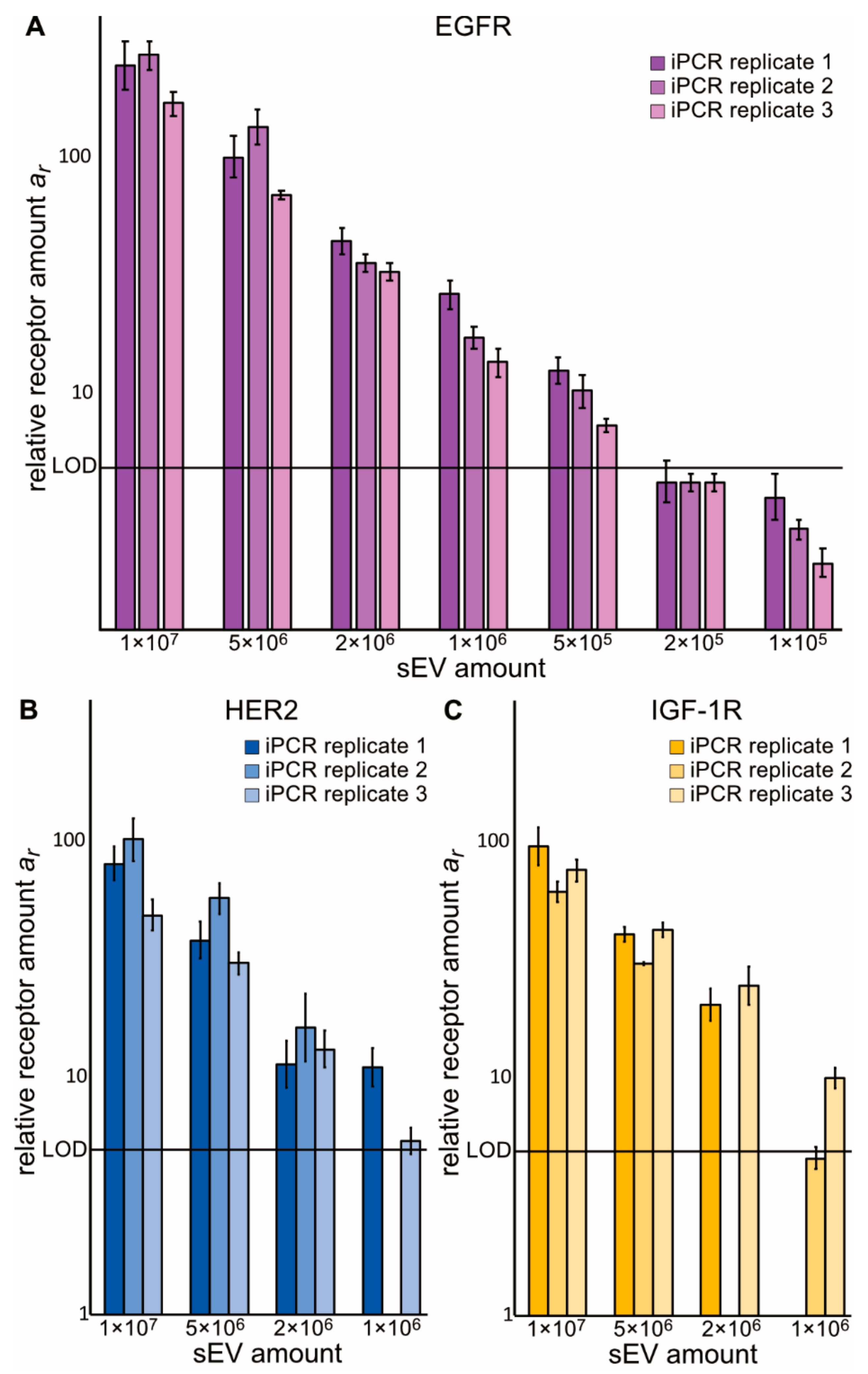
2.2. Zx–DNA Conjugates towards EGFR, IGF-1R, HER2, and HER3 Can Monitor Protein Expression on sEVs from NSCLC Cells
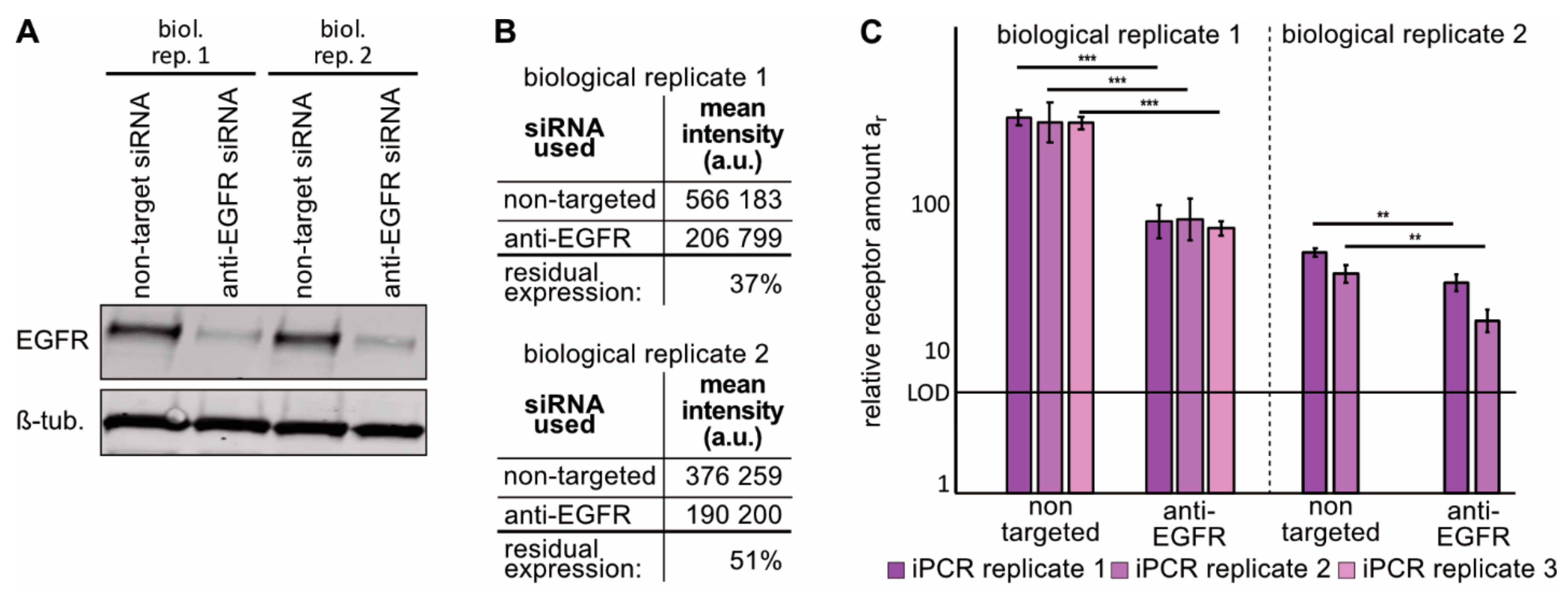
2.3. Immuno-PCR with the ZEGFR–DNA Conjugate towards EGFR Can Monitor Alterations in EGFR Expression in sEVs from NSCLC Cells
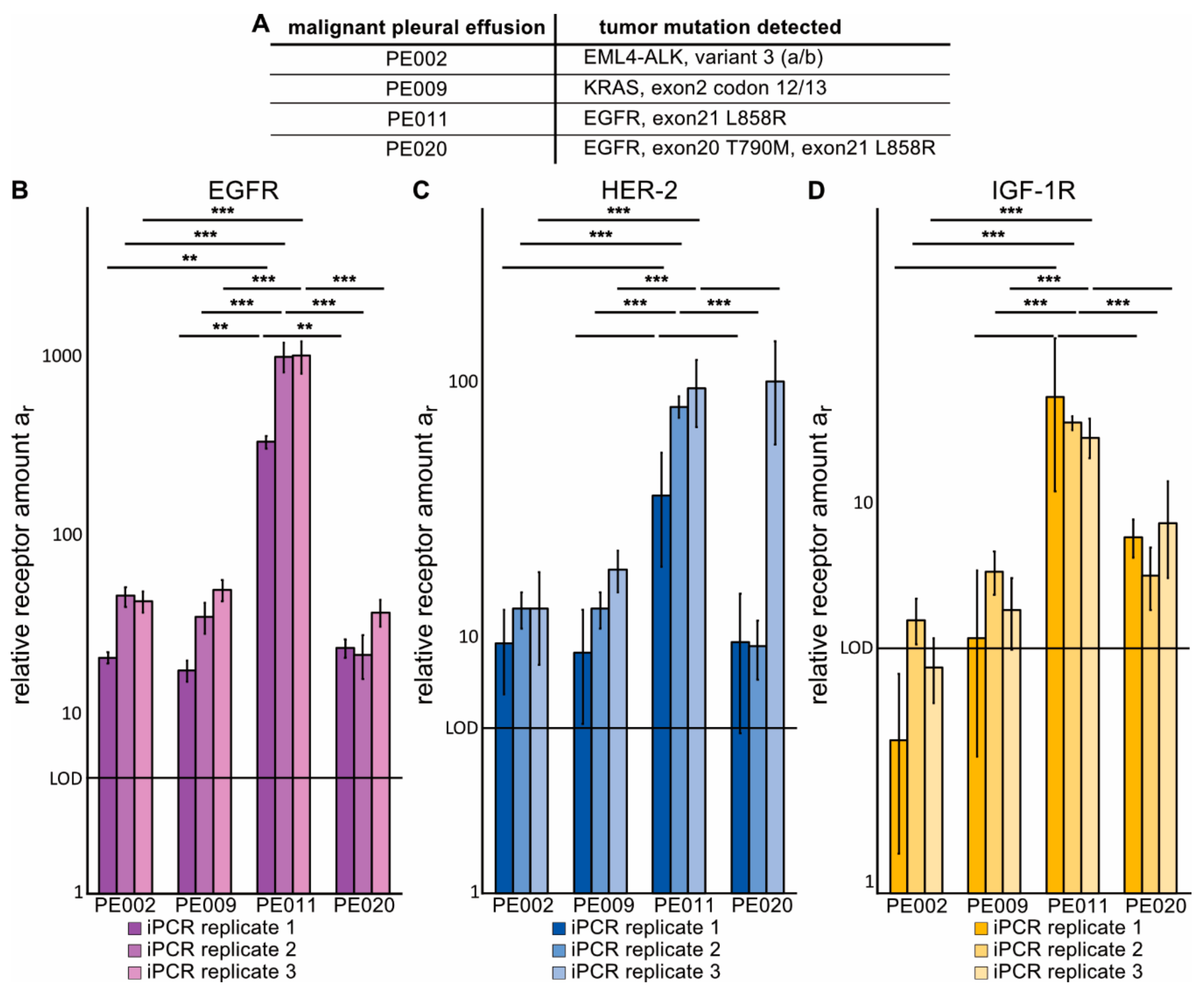
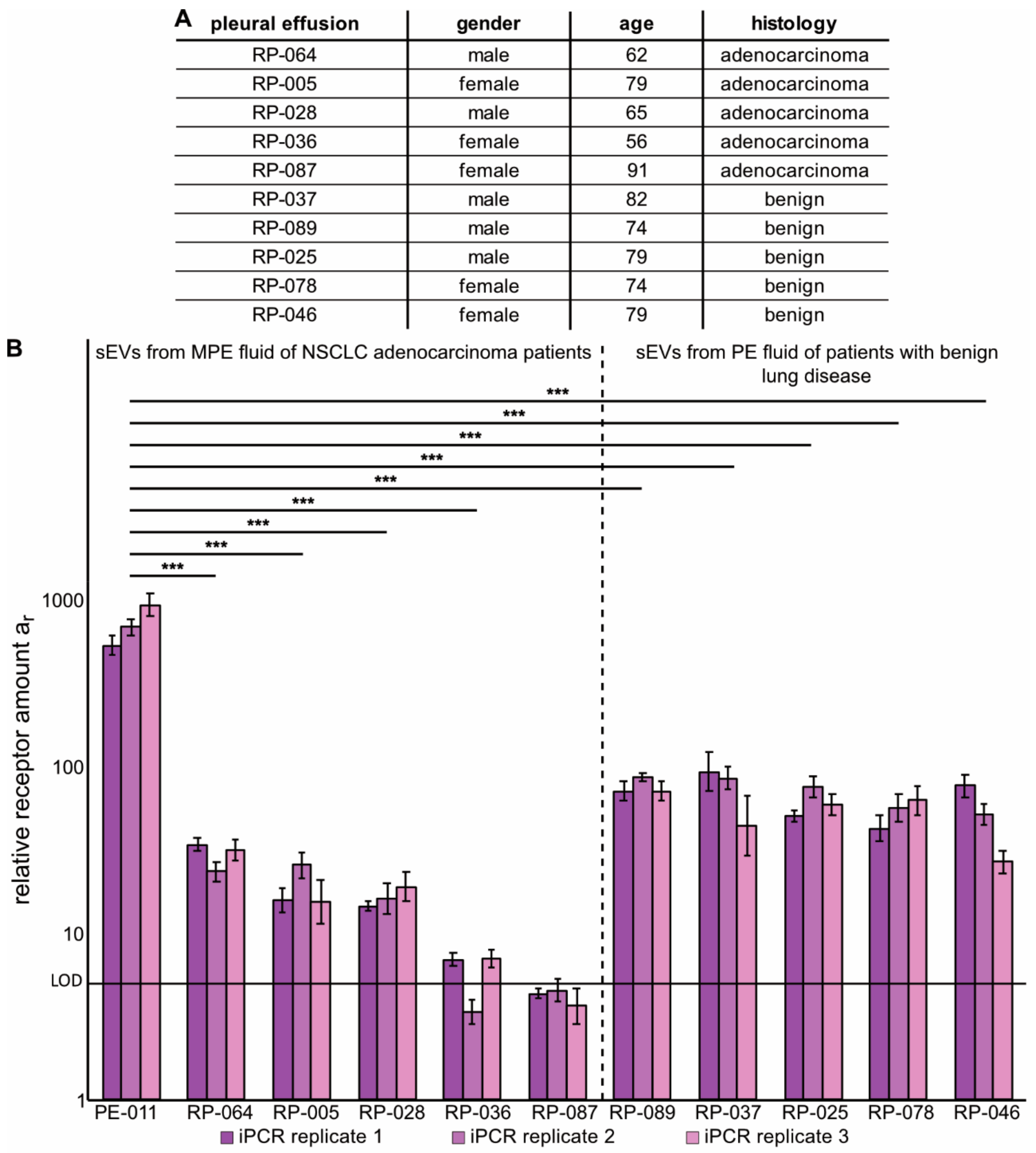
2.4. Immuno-PCR with Zx–DNA-Conjugates towards EGFR, HER2 and IGF-1R Can Monitor Protein Expression in sEVs from Liquid Biopsies of NSCLC Patients
3. Discussion
4. Material and Methods
4.1. Cell Culture Conditions
4.2. EGFR siRNA Transfection
4.3. Isolation of sEVs from H1975 Cells
4.4. Collection and Isolation of sEVs from Malignant Pleural Effusion (MPE) Fluid of Advanced NSCLC Patients
4.5. Isolation of sEVs from MPE Fluid Samples from NSCLC Adenocarcinoma Patients or from PE Fluid of Patients with Benign Lung Disease
4.6. Analyses of sEV Size and Amount by Nanoparticle Tracking Analysis (NTA)
4.7. Western Blot Analyses of sEVs
4.8. FACS Assessment of EGFR Expression After siRNA Treatment
4.9. Production of Zx–DNA-Conjugates
4.10. Measurement of Amplification Efficiencies for Zx–DNAs via RT-PCR
4.11. Detection of sEVs via Immuno-PCR
4.12. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santarpia, M.; Liguori, A.; D’Aveni, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffina, M.G.; Lazzari, C.; Altavilla, G.; Rosell, R. Liquid biopsy for lung cancer early detection. J. Thorac. Dis. 2018, 10, S882–S897. [Google Scholar] [CrossRef] [Green Version]
- Macias, M.; Alegre, E.; Diaz-Lagares, A.; Patino, A.; Perez-Gracia, J.L.; Sanmamed, M.; Lopez-Lopez, R.; Varo, N.; Gonzalez, A. Liquid Biopsy: From Basic Research to Clinical Practice. Adv. Clin. Chem. 2018, 83, 73–119. [Google Scholar] [CrossRef]
- Whiteside, T.L. The potential of tumor-derived exosomes for noninvasive cancer monitoring: An update. Expert Rev. Mol. Diagn. 2018, 18, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Riethdorf, S.; O’Flaherty, L.; Hille, C.; Pantel, K. Clinical applications of the CellSearch platform in cancer patients. Adv. Drug Deliv. Rev. 2018, 125, 102–121. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [Green Version]
- Fahrmann, J.F.; Mao, X.Y.; Irajizad, E.; Katayama, H.; Capello, M.; Tanaka, I.; Kato, T.; Wistuba, I.I.; Maitra, A.; Ostrin, E.J.; et al. Plasma-Derived Extracellular Vesicles Convey Protein Signatures That Reflect Pathophysiology in Lung and Pancreatic Adenocarcinomas. Cancers 2020, 12, 1147. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melosky, B. Rapidly changing treatment algorithms for metastatic nonsquamous non-small-cell lung cancer. Curr. Oncol. 2018, 25, S68–S76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, K.; Rivard, C.J.; Mitsudomi, T.; Hirsch, F.R. Overcoming resistance to EGFR tyrosine kinase inhibitors in lung cancer, focusing on non-T790M mechanisms. Expert Rev. Anticancer Ther. 2017, 17, 779–786. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Tsiara, A.; Tsironis, G.; Lykka, M.; Liontos, M.; Bamias, A.; Dimopoulos, M.A. Treating ALK-positive non-small cell lung cancer. Ann. Transl. Med. 2018, 6, 141. [Google Scholar] [CrossRef]
- Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers 2018, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Santarpia, M.; Liguori, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffina, M.G.; D’Aveni, A.; Marabello, G.; Altavilla, G.; Rosell, R. Osimertinib in the treatment of non-small-cell lung cancer: Design, development and place in therapy. Lung Cancer 2017, 8, 109–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Schmid, S.; Li, J.J.N.; Leighl, N.B. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer 2020, 147, 123–129. [Google Scholar] [CrossRef]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Krug, A.K.; Enderle, D.; Karlovich, C.; Priewasser, T.; Bentink, S.; Spiel, A.; Brinkmann, K.; Emenegger, J.; Grimm, D.G.; Castellanos-Rizaldos, E.; et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann. Oncol. 2018, 29, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Castellanos-Rizaldos, E.; Grimm, D.G.; Tadigotla, V.; Hurley, J.; Healy, J.; Neal, P.L.; Sher, M.; Venkatesan, R.; Karlovich, C.; Raponi, M.; et al. Exosome-Based Detection of EGFR T790M in Plasma from Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 2944–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandfeld-Paulsen, B.; Aggerholm-Pedersen, N.; Baek, R.; Jakobsen, K.R.; Meldgaard, P.; Folkersen, B.H.; Rasmussen, T.R.; Varming, K.; Jorgensen, M.M.; Sorensen, B.S. Exosomal proteins as prognostic biomarkers in non-small cell lung cancer. Mol. Oncol. 2016, 10, 1595–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, S.K.; Sanger, H.; Krawczyk, M.; Julich-Haertel, H.; Willms, A.; Ligocka, J.; Azkargorta, M.; Mocan, T.; Kahlert, C.; Kruk, B.; et al. Synergistic effects of extracellular vesicle phenotyping and AFP in hepatobiliary cancer differentiation. Liver Int. 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, J.; Xu, T.; Ahmadinejad, N.; Hess, K.; Lin, S.H.; Zhang, J.; Liu, X.; Liu, L.; Ning, B.; et al. Extracellular vesicle tetraspanin-8 level predicts distant metastasis in non-small cell lung cancer after concurrent chemoradiation. Sci. Adv. 2020, 6, eaaz6162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, H.; Im, H.; Castro, C.M.; Breakefield, X.; Weissleder, R.; Lee, H. New Technologies for Analysis of Extracellular Vesicles. Chem. Rev. 2018, 118, 1917–1950. [Google Scholar] [CrossRef]
- Wang, S.; Khan, A.; Huang, R.; Ye, S.; Di, K.; Xiong, T.; Li, Z. Recent advances in single extracellular vesicle detection methods. Biosens. Bioelectron. 2020, 154, 112056. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiller, C.; Aghelpasand, H.; Frick, T.; Westerlund, K.; Ahmadian, A.; Karlstrom, A.E. Fast and Efficient Fc-Specific Photoaffinity Labeling To Produce Antibody-DNA Conjugates. Bioconjug. Chem. 2019, 30, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Wikman, M.; Steffen, A.C.; Gunneriusson, E.; Tolmachev, V.; Adams, G.P.; Carlsson, J.; Stahl, S. Selection and characterization of HER2/neu-binding affibody ligands. Protein Eng. Des. Sel. 2004, 17, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Friedman, M.; Orlova, A.; Johansson, E.; Eriksson, T.L.; Hoiden-Guthenberg, I.; Tolmachev, V.; Nilsson, F.Y.; Stahl, S. Directed evolution to low nanomolar affinity of a tumor-targeting epidermal growth factor receptor-binding affibody molecule. J. Mol. Biol. 2008, 376, 1388–1402. [Google Scholar] [CrossRef]
- Li, J.; Lundberg, E.; Vernet, E.; Larsson, B.; Hoiden-Guthenberg, I.; Graslund, T. Selection of affibody molecules to the ligand-binding site of the insulin-like growth factor-1 receptor. Biotechnol. Appl. Biochem. 2010, 55, 99–109. [Google Scholar] [CrossRef]
- Tolmachev, V.; Malmberg, J.; Hofstrom, C.; Abrahmsen, L.; Bergman, T.; Sjoberg, A.; Sandstrom, M.; Graslund, T.; Orlova, A. Imaging of insulinlike growth factor type 1 receptor in prostate cancer xenografts using the affibody molecule 111In-DOTA-ZIGF1R:4551. J. Nucl. Med. 2012, 53, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Orlova, A.; Magnusson, M.; Eriksson, T.L.; Nilsson, M.; Larsson, B.; Hoiden-Guthenberg, I.; Widstrom, C.; Carlsson, J.; Tolmachev, V.; Stahl, S.; et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006, 66, 4339–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malm, M.; Kronqvist, N.; Lindberg, H.; Gudmundsdotter, L.; Bass, T.; Frejd, F.Y.; Hoiden-Guthenberg, I.; Varasteh, Z.; Orlova, A.; Tolmachev, V.; et al. Inhibiting HER3-mediated tumor cell growth with affibody molecules engineered to low picomolar affinity by position-directed error-prone PCR-like diversification. PLoS ONE 2013, 8, e62791. [Google Scholar] [CrossRef]
- Tolmachev, V.; Rosik, D.; Wallberg, H.; Sjoberg, A.; Sandstrom, M.; Hansson, M.; Wennborg, A.; Orlova, A. Imaging of EGFR expression in murine xenografts using site-specifically labelled anti-EGFR 111In-DOTA-Z EGFR:2377 Affibody molecule: Aspect of the injected tracer amount. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 613–622. [Google Scholar] [CrossRef]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Baburaj, G.; Damerla, R.R.; Udupa, K.S.; Parida, P.; Munisamy, M.; Kolesar, J.; Rao, M. Liquid biopsy approaches for pleural effusion in lung cancer patients. Mol. Biol. Rep. 2020, 47, 8179–8187. [Google Scholar] [CrossRef]
- Tong, L.; Ding, N.; Tong, X.; Li, J.; Zhang, Y.; Wang, X.; Xu, X.; Ye, M.; Li, C.; Wu, X.; et al. Tumor-derived DNA from pleural effusion supernatant as a promising alternative to tumor tissue in genomic profiling of advanced lung cancer. Theranostics 2019, 9, 5532–5541. [Google Scholar] [CrossRef]
- Hydbring, P.; De Petris, L.; Zhang, Y.; Branden, E.; Koyi, H.; Novak, M.; Kanter, L.; Haag, P.; Hurley, J.; Tadigotla, V.; et al. Exosomal RNA-profiling of pleural effusions identifies adenocarcinoma patients through elevated miR-200 and LCN2 expression. Lung Cancer 2018, 124, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.; Weissleder, R. Modeling EV Kinetics for Use in Early Cancer Detection. Adv. Biosyst. 2020, e1900305. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, U.; Perret-Liaudet, A.; van Waalwijk van Doorn, L.J.; Blennow, K.; Chiasserini, D.; Engelborghs, S.; Fladby, T.; Genc, S.; Kruse, N.; Kuiperij, H.B.; et al. A Practical Guide to Immunoassay Method Validation. Front. Neurol. 2015, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.Y.; Lee, J.S.; Kim, I.A.; Kim, H.J.; Kim, W.S.; Lee, K.Y. Extracellular vesicle-based EGFR genotyping in bronchoalveolar lavage fluid from treatment-naive non-small cell lung cancer patients. Transl. Lung Cancer Res. 2019, 8, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.Y.; Kim, H.J.; Lee, J.S.; Choi, C.M.; Lee, J.C.; Jung, M.K.; Pack, C.G.; Lee, K.Y. Extracellular vesicle-derived DNA for performing EGFR genotyping of NSCLC patients. Mol. Cancer 2018, 17, 15. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Du, R.; Jiang, S.; Wu, C.; Barkauskas, D.S.; Richey, J.; Molter, J.; Lam, M.; Flask, C.; Gerson, S.; et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br. J. Cancer 2008, 99, 911–922. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, S.; Horak, J.; Haag, P.; Gupta, D.; Stiller, C.; Sahu, S.S.; Gorgens, A.; Gatty, H.K.; Viktorsson, K.; El Andaloussi, S.; et al. Label-Free Surface Protein Profiling of Extracellular Vesicles by an Electrokinetic Sensor. ACS Sens. 2019, 4, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Haag, P.; Viktorsson, K.; Karolinska Institute, Stockholm, Sweden. Personal communication, 2021.





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiller, C.; Viktorsson, K.; Paz Gomero, E.; Hååg, P.; Arapi, V.; Kaminskyy, V.O.; Kamali, C.; De Petris, L.; Ekman, S.; Lewensohn, R.; et al. Detection of Tumor-Associated Membrane Receptors on Extracellular Vesicles from Non-Small Cell Lung Cancer Patients via Immuno-PCR. Cancers 2021, 13, 922. https://doi.org/10.3390/cancers13040922
Stiller C, Viktorsson K, Paz Gomero E, Hååg P, Arapi V, Kaminskyy VO, Kamali C, De Petris L, Ekman S, Lewensohn R, et al. Detection of Tumor-Associated Membrane Receptors on Extracellular Vesicles from Non-Small Cell Lung Cancer Patients via Immuno-PCR. Cancers. 2021; 13(4):922. https://doi.org/10.3390/cancers13040922
Chicago/Turabian StyleStiller, Christiane, Kristina Viktorsson, Elizabeth Paz Gomero, Petra Hååg, Vasiliki Arapi, Vitaliy O. Kaminskyy, Caroline Kamali, Luigi De Petris, Simon Ekman, Rolf Lewensohn, and et al. 2021. "Detection of Tumor-Associated Membrane Receptors on Extracellular Vesicles from Non-Small Cell Lung Cancer Patients via Immuno-PCR" Cancers 13, no. 4: 922. https://doi.org/10.3390/cancers13040922
APA StyleStiller, C., Viktorsson, K., Paz Gomero, E., Hååg, P., Arapi, V., Kaminskyy, V. O., Kamali, C., De Petris, L., Ekman, S., Lewensohn, R., & Karlström, A. E. (2021). Detection of Tumor-Associated Membrane Receptors on Extracellular Vesicles from Non-Small Cell Lung Cancer Patients via Immuno-PCR. Cancers, 13(4), 922. https://doi.org/10.3390/cancers13040922