
Cerebral Cavernous Malformation 1 Determines YAP/TAZ Signaling-Dependent Metastatic Hallmarks of Prostate Cancer Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
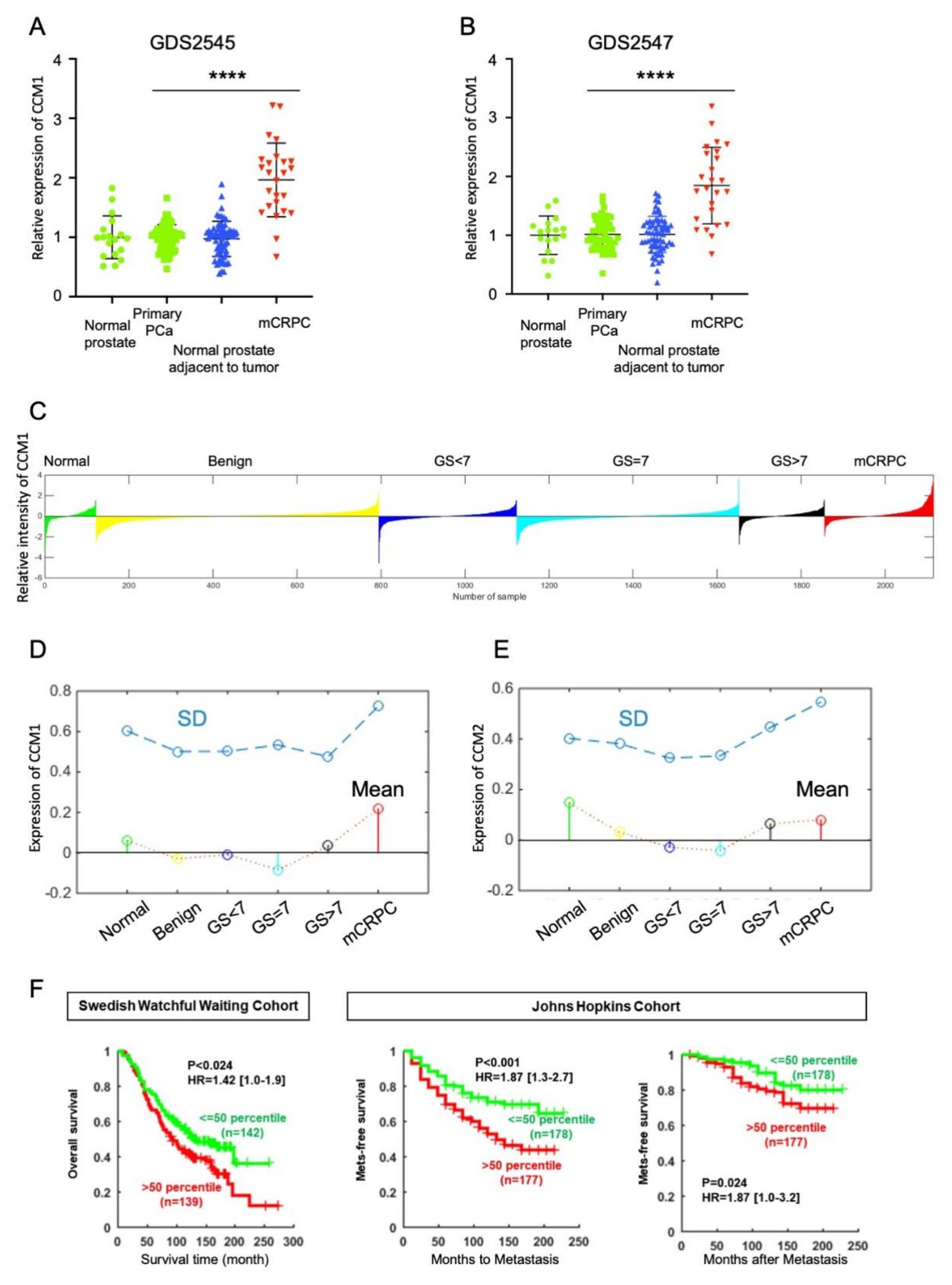
2.1. Higher Expression of Ccm1 at the Mcrpc Stage and Its Association with Poor Prognosis of Patients with PCa
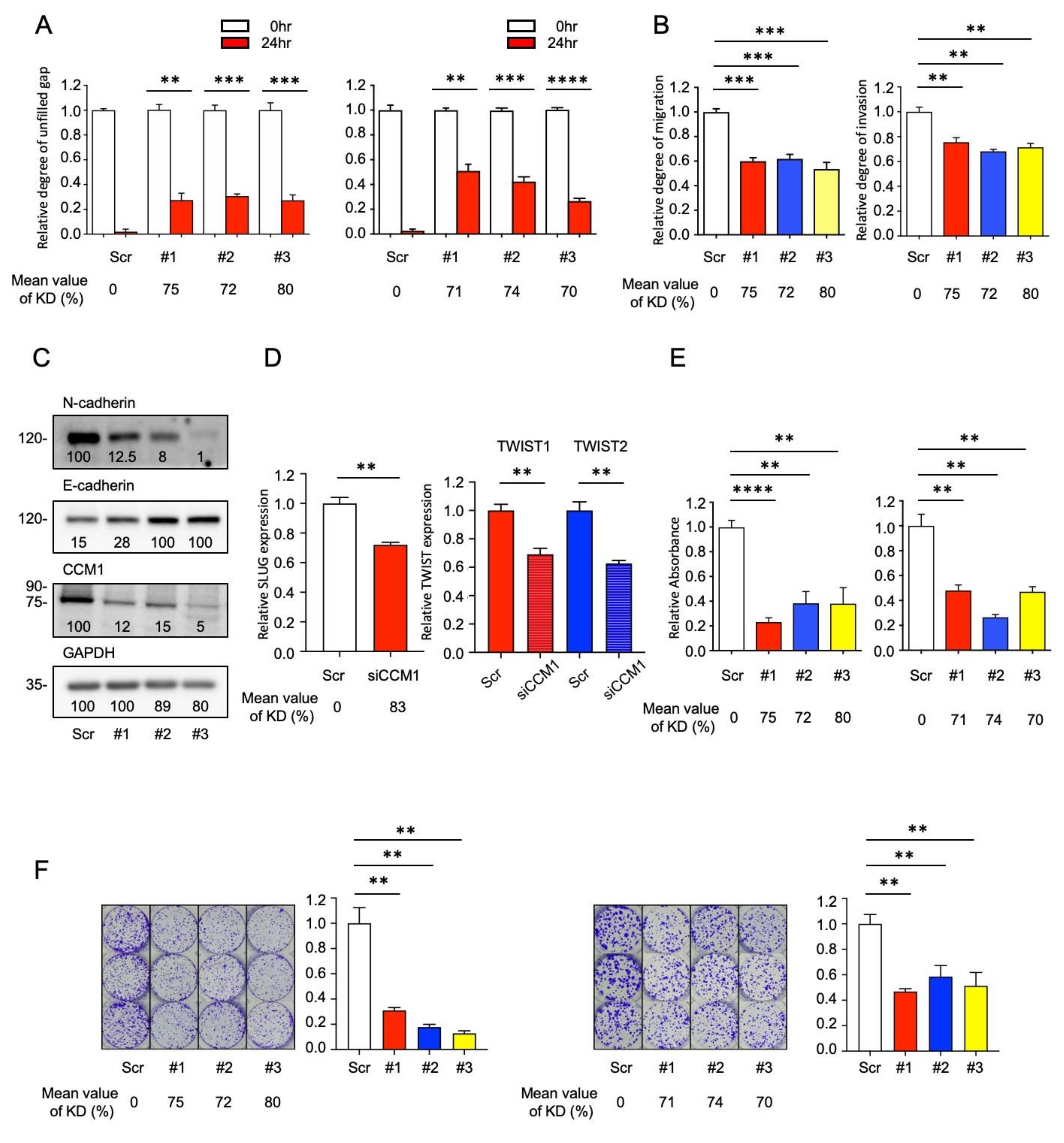
2.2. CCM1 Induces Metastatic Hallmarks of PCa Cells
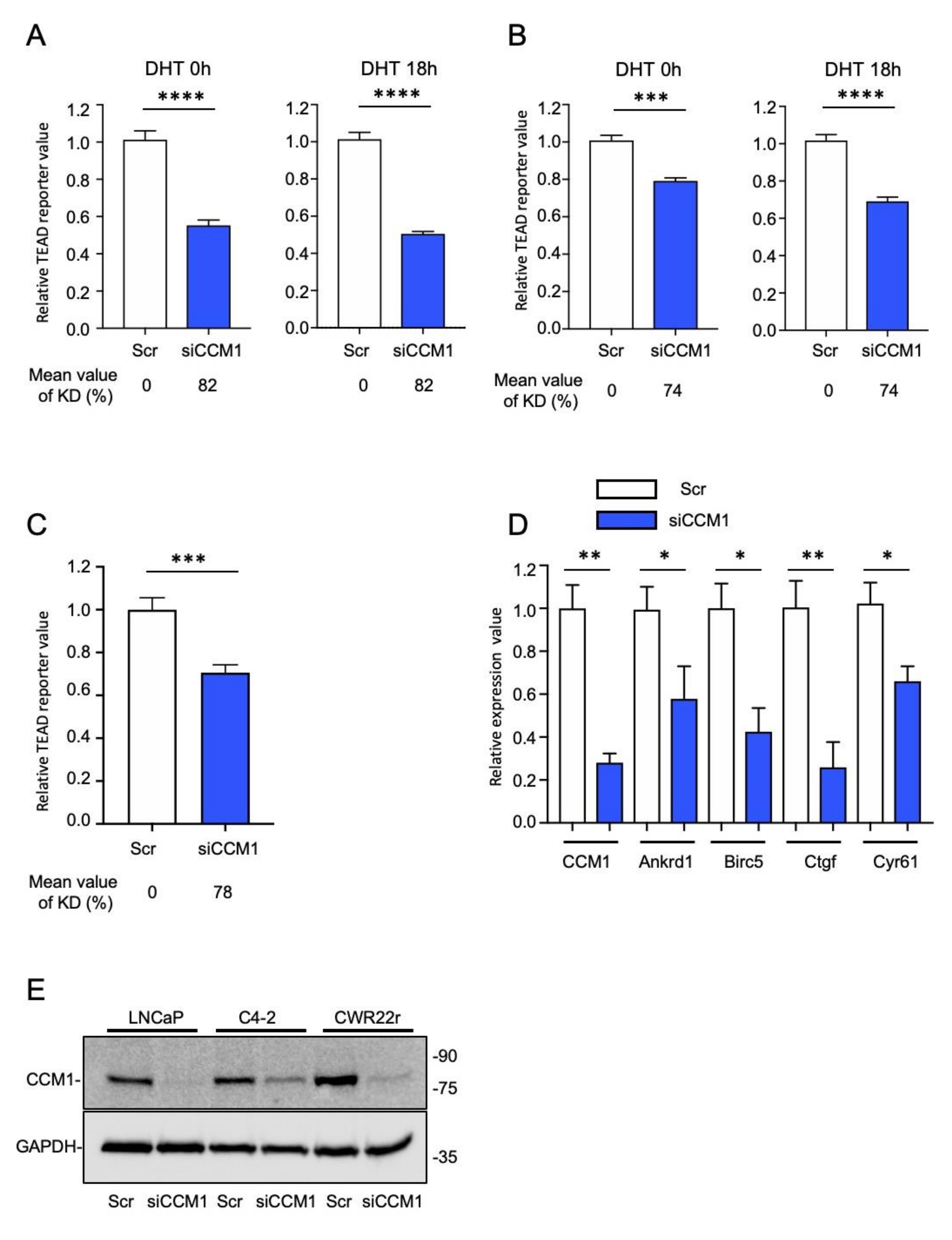
2.3. CCM1 Regulates YAP/TAZ Signaling
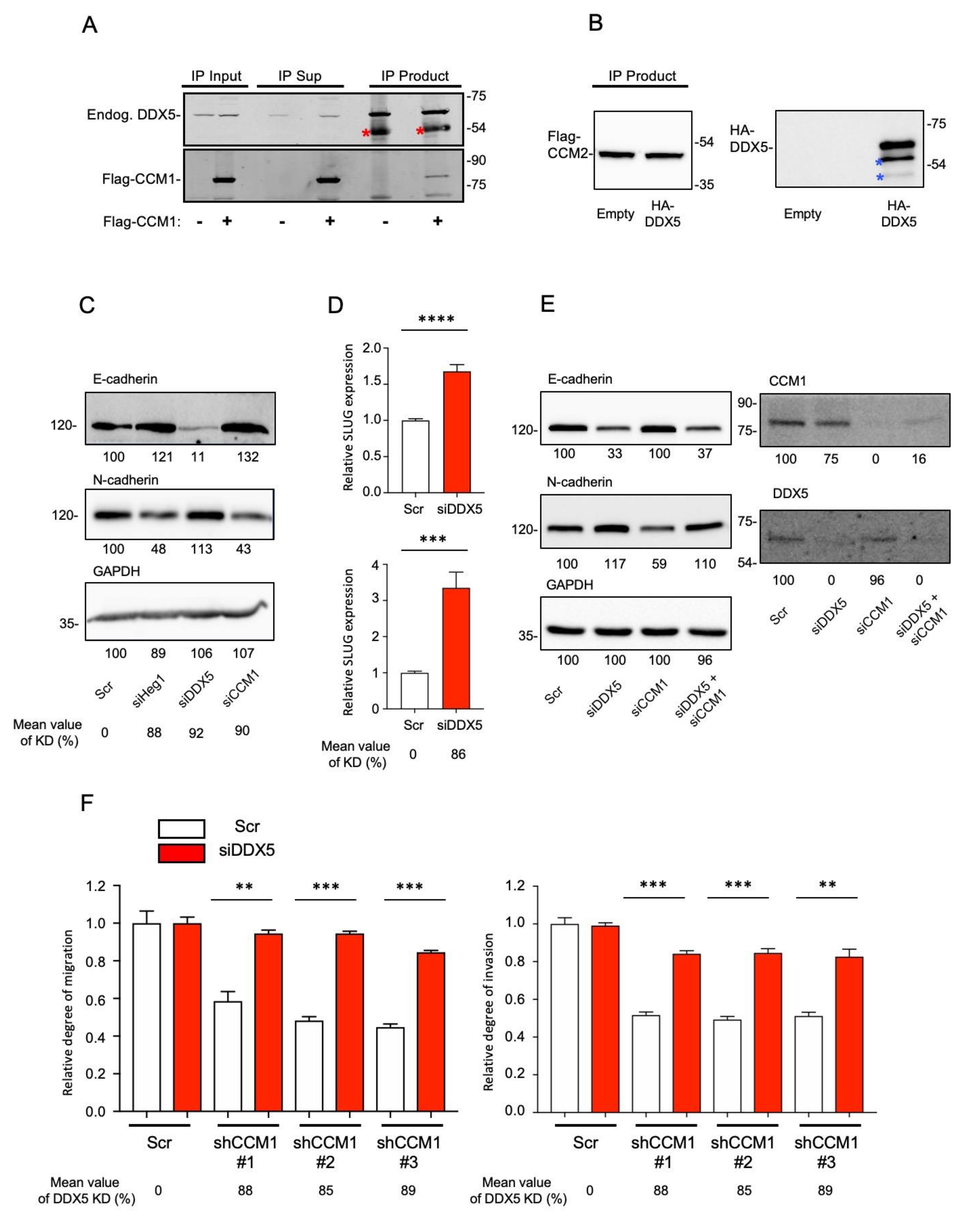
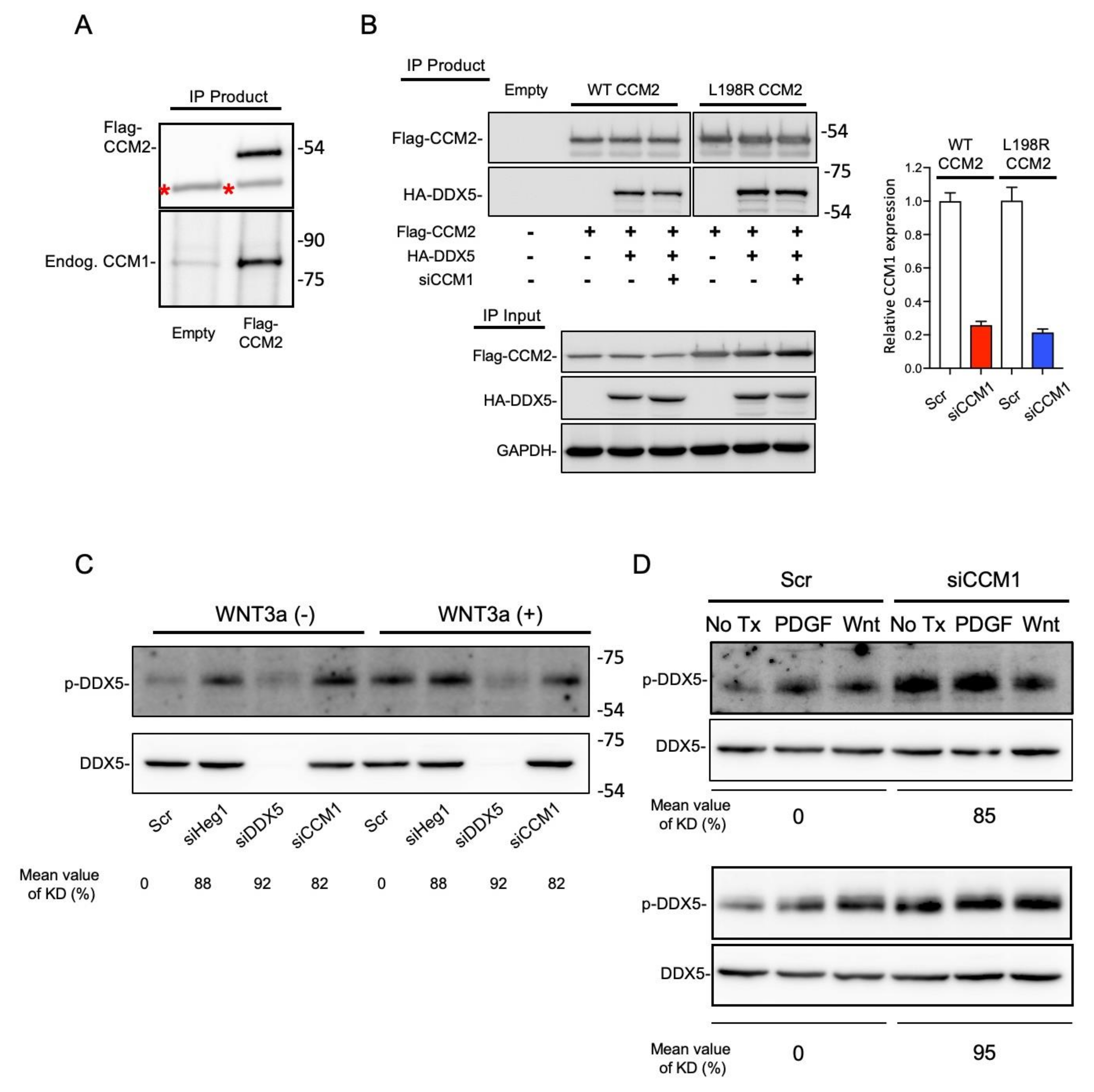
2.4. DDX5 Is a Functional Downstream Mediator of CCM1 in the Regulation of Metastatic Hallmarks
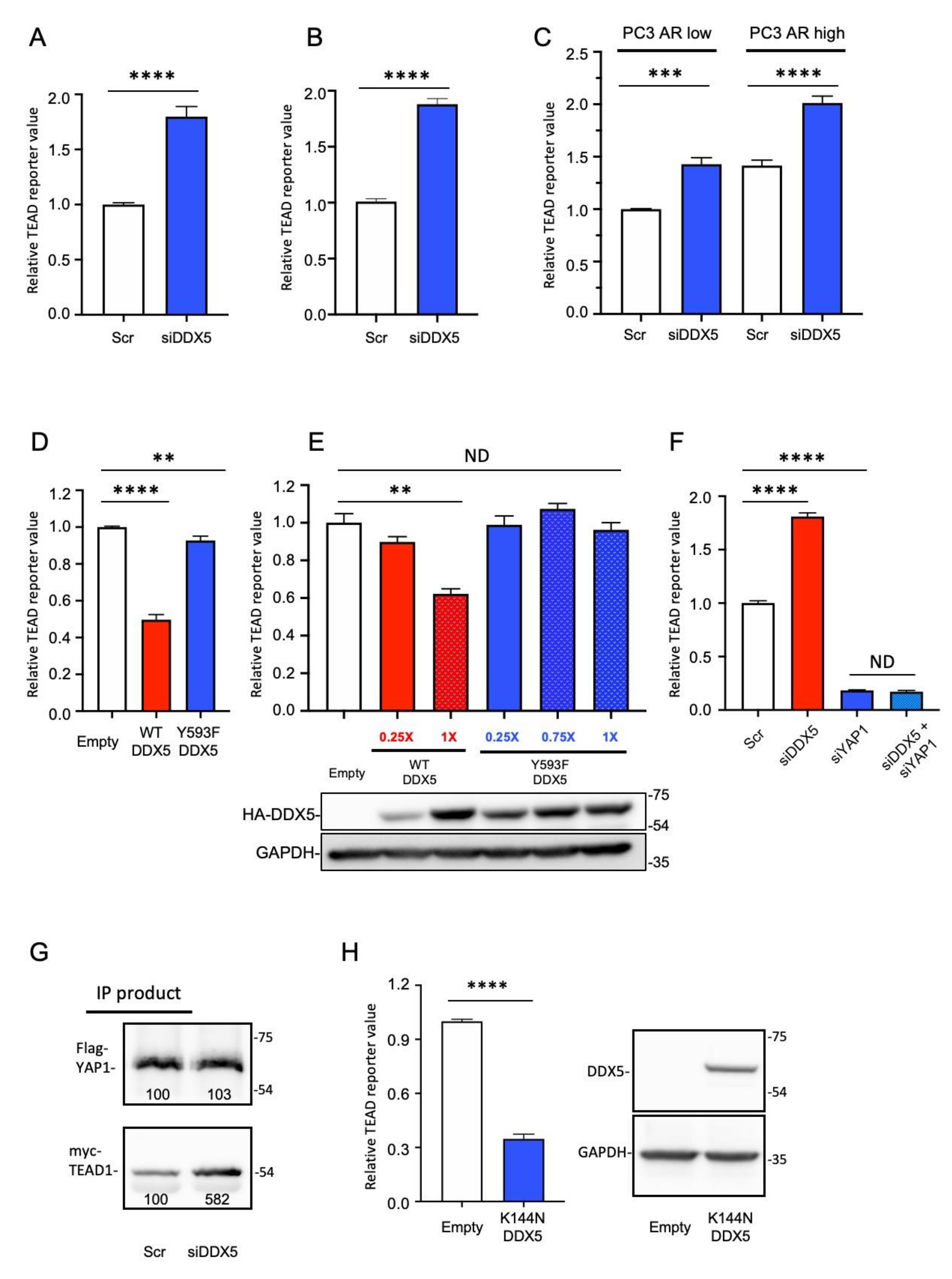
2.5. DDX5 Suppresses YAP/TAZ Signaling
2.6. CCM1 Regulates the Phosphorylation of DDX5 at Y593
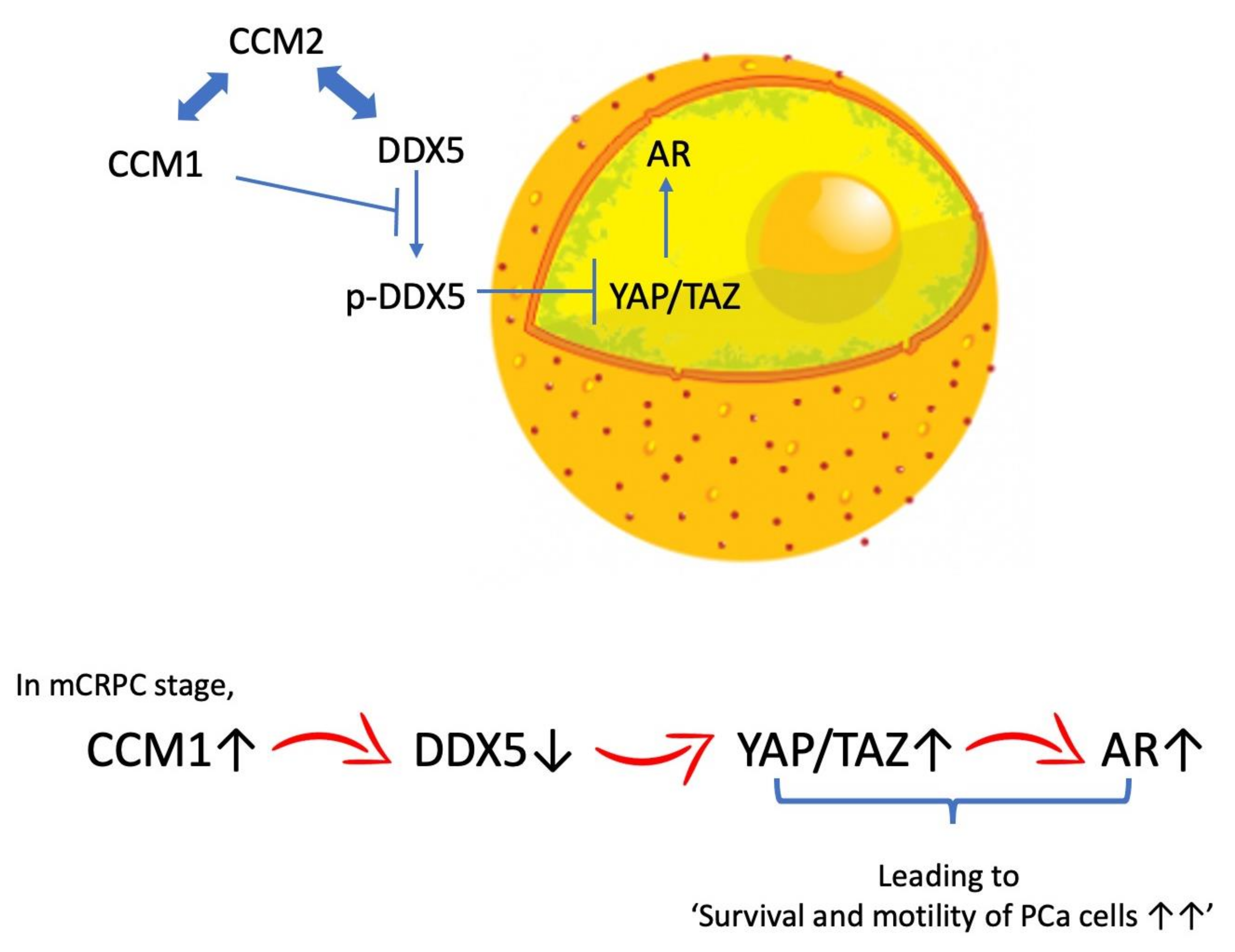
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Generation of Stable Cell Lines
4.3. Immunoblotting Assay
4.4. Real-Time PCR (qPCR) Analysis
4.5. Wound-Healing Assay
4.6. Transwell Invasion and Migration Assay
4.7. Soft Agar Assay
4.8. Hanging Drop Culture
4.9. Immunoprecipitation
4.10. Reporter Assay
4.11. Clonogenic Cell Survival Assay
4.12. Proliferation Assay and Cell Cycle Analysis
4.13. Molecular Cohort Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spratt, D.E.; Zumsteg, Z.S.; Feng, F.Y.; Tomlins, S.A. Translational and clinical implications of the genetic landscape of prostate cancer. Nat. Rev. Clin. Oncol. 2016, 13, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Gleave, M.E. Targeting the adaptive molecular landscape of castration-resistant prostate cancer. EMBO Mol. Med. 2015, 7, 878–894. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Bangma, C.H.; Bjartell, A.; Catto, J.W.; Culig, Z.; Gronberg, H.; Luo, J.; Visakorpi, T.; Rubin, M.A. The mutational landscape of prostate cancer. Eur. Urol. 2013, 64, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Kelly, S.P.; Anderson, W.F.; Rosenberg, P.S.; Cook, M.B. Past, Current, and Future Incidence Rates and Burden of Metastatic Prostate Cancer in the United States. Eur. Urol. Focus 2018, 4, 121–127. [Google Scholar] [CrossRef]
- Li, J.; Siegel, D.A.; King, J.B. Stage-specific incidence rates and trends of prostate cancer by age, race, and ethnicity, United States, 2004–2014. Ann. Epidemiol. 2018, 28, 328–330. [Google Scholar] [CrossRef]
- Augello, M.A.; Den, R.B.; Knudsen, K.E. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev. 2014, 33, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.; Schweizer, M.T. Targeting persistent androgen receptor signaling in castration-resistant prostate cancer. Med. Oncol. 2016, 33, 44. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Sarkar, F.H. Overview on the complexity of androgen receptor-targeted therapy for prostate cancer. Cancer Cell Int. 2015, 15, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The Hippo Pathway Effector YAP Regulates Motility, Invasion, and Castration-Resistant Growth of Prostate Cancer Cells. Mol. Cell Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinar, B.; Collak, F.K.; Lopez, D.; Akgul, S.; Mukhopadhyay, N.K.; Kilicarslan, M.; Gioeli, D.G.; Freeman, M.R. MST1 is a multifunctional caspase-independent inhibitor of androgenic signaling. Cancer Res. 2011, 71, 4303–4313. [Google Scholar] [CrossRef] [Green Version]
- Kim, J. Introduction to cerebral cavernous malformation: A brief review. BMB Rep. 2016, 49, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Yoruk, B.; Gillers, B.S.; Chi, N.C.; Scott, I.C. Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Dev. Biol. 2012, 362, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawistowski, J.S.; Stalheim, L.; Uhlik, M.T.; Abell, A.N.; Ancrile, B.B.; Johnson, G.L.; Marchuk, D.A. CCM1 and CCM2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 2005, 14, 2521–2531. [Google Scholar] [CrossRef] [Green Version]
- Fisher, O.S.; Liu, W.; Zhang, R.; Stiegler, A.L.; Ghedia, S.; Weber, J.L.; Boggon, T.J. Structural basis for the disruption of the cerebral cavernous malformations 2 (CCM2) interaction with Krev interaction trapped 1 (KRIT1) by disease-associated mutations. J. Biol. Chem. 2015, 290, 2842–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.C.; Drakos, S.G.; Ruiz, O.E.; Smith, A.C.; Gibson, C.C.; Ling, J.; Passi, S.F.; Stratman, A.N.; Sacharidou, A.; Revelo, M.P.; et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J. Clin. Invest. 2011, 121, 1871–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Eng, M.; Ghabrial, A.S. Focal defects in single-celled tubes mutant for Cerebral cavernous malformation 3, GCKIII, or NSF2. Dev. Cell 2013, 25, 507–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wu, Q.; Xu, J.F.; Miller, D.; Sandalcioglu, I.E.; Zhang, J.M.; Sure, U. Differential angiogenesis function of CCM2 and CCM3 in cerebral cavernous malformations. Neurosurg. Focus 2010, 29, E1. [Google Scholar] [CrossRef]
- Glading, A.J.; Ginsberg, M.H. Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signaling. Dis. Models Mech. 2010, 3, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhao, H.; Shan, J.; Long, F.; Chen, Y.; Chen, Y.; Zhang, Y.; Han, X.; Ma, D. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol. Biol. Cell 2007, 18, 1965–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, U.R.; Ma, C.; Dhir, R.; Bisceglia, M.; Lyons-Weiler, M.; Liang, W.; Michalopoulos, G.; Becich, M.; Monzon, F.A. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 2007, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Al-Deen Ashab, H.; Davicioni, E.; Karnes, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sboner, A.; Demichelis, F.; Calza, S.; Pawitan, Y.; Setlur, S.R.; Hoshida, Y.; Perner, S.; Adami, H.O.; Fall, K.; Mucci, L.A.; et al. Molecular sampling of prostate cancer: A dilemma for predicting disease progression. BMC Med. Genom. 2010, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Tosoian, J.J.; Trock, B.J.; Landis, P.; Feng, Z.; Epstein, J.I.; Partin, A.W.; Walsh, P.C.; Carter, H.B. Active surveillance program for prostate cancer: An update of the Johns Hopkins experience. J. Clin. Oncol. 2011, 29, 2185–2190. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, X.; Rycaj, K.; Chao, H.P.; Deng, Q.; Jeter, C.; Liu, C.; Honorio, S.; Li, H.; Davis, T.; et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget 2015, 6, 23959–23986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussemakers, M.J.; Van Bokhoven, A.; Tomita, K.; Jansen, C.F.; Schalken, J.A. Complex cadherin expression in human prostate cancer cells. Int. J. Cancer 2000, 85, 446–450. [Google Scholar] [CrossRef]
- Ganesan, R.; Mallets, E.; Gomez-Cambronero, J. The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol. Oncol. 2016, 10, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Zhang, S.; Dong, X.; Tian, D.; Cui, Z.; Qiu, X. Prognostic significance of twist and N-cadherin expression in NSCLC. PLoS ONE 2013, 8, e62171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jividen, K.; Kedzierska, K.Z.; Yang, C.S.; Szlachta, K.; Ratan, A.; Paschal, B.M. Genomic analysis of DNA repair genes and androgen signaling in prostate cancer. BMC Cancer 2018, 18, 960. [Google Scholar] [CrossRef] [Green Version]
- Dai, T.-Y.; Cao, L.; Yang, Z.-C.; Li, Y.-S.; Tan, L.; Ran, X.-Z.; Shi, C.-M. P68 RNA helicase as a molecular target for cancer therapy. J. Exp. Clin. Cancer Res. 2014, 33, 64. [Google Scholar] [CrossRef]
- Fuller-Pace, F.V. DEAD box RNA helicase functions in cancer. RNA Biol. 2013, 10, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, C.; Liu, Z.R. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell 2006, 127, 139–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingras, A.R.; Puzon-McLaughlin, W.; Ginsberg, M.H. The structure of the ternary complex of Krev interaction trapped 1 (KRIT1) bound to both the Rap1 GTPase and the heart of glass (HEG1) cytoplasmic tail. J. Biol. Chem. 2013, 288, 23639–23649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleaveland, B.; Zheng, X.; Liu, J.J.; Blum, Y.; Tung, J.J.; Zou, Z.; Sweeney, S.M.; Chen, M.; Guo, L.; Lu, M.M.; et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 2009, 15, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyamao, R.M.; Wu, J.; Yu, L.; Xiao, X.; Zhang, F.M. Roles of DDX5 in the tumorigenesis, proliferation, differentiation, metastasis and pathway regulation of human malignancies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espiritu, S.M.G.; Liu, L.Y.; Rubanova, Y.; Bhandari, V.; Holgersen, E.M.; Szyca, L.M.; Fox, N.S.; Chua, M.L.K.; Yamaguchi, T.N.; Heisler, L.E.; et al. The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression. Cell 2018, 173, 1003–1013. [Google Scholar] [CrossRef]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Gao, X.; Yang, J.J.; Liu, Z.R. Interaction between p68 RNA helicase and Ca2+-calmodulin promotes cell migration and metastasis. Nat. Commun. 2013, 4, 1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collak, F.K.; Demir, U.; Ozkanli, S.; Kurum, E.; Zerk, P.E. Increased expression of YAP1 in prostate cancer correlates with extraprostatic extension. Cancer Biol. Med. 2017, 14, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakula, H.; Xiang, D.; Li, Z. A Tale of Two Signals: AR and WNT in Development and Tumorigenesis of Prostate and Mammary Gland. Cancers 2017, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Linja, M.J.; Savinainen, K.J.; Saramaki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001, 61, 3550–3555. [Google Scholar] [PubMed]
- Antonarakis, E.S. Current understanding of resistance to abiraterone and enzalutamide in advanced prostate cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 316–319. [Google Scholar]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, W.B.; Xu, J. Current progress and questions in germline genetics of prostate cancer. Asian J. Urol. 2019, 6, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Nelson, P.; Vasioukhin, V. Recent advances in prostate cancer research: Large-scale genomic analyses reveal novel driver mutations and DNA repair defects. F1000Res. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kokolo, M.; Bach-Elias, M. Downregulation of p68 RNA Helicase (DDX5) Activates a Survival Pathway Involving mTOR and MDM2 Signals. Folia Biol. 2017, 63, 52–59. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Lee, H.-Y.; Kim, J.; Park, H.; Ju, Y.S.; Kim, E.-G.; Kim, J. Cerebral Cavernous Malformation 1 Determines YAP/TAZ Signaling-Dependent Metastatic Hallmarks of Prostate Cancer Cells. Cancers 2021, 13, 1125. https://doi.org/10.3390/cancers13051125
Park S, Lee H-Y, Kim J, Park H, Ju YS, Kim E-G, Kim J. Cerebral Cavernous Malformation 1 Determines YAP/TAZ Signaling-Dependent Metastatic Hallmarks of Prostate Cancer Cells. Cancers. 2021; 13(5):1125. https://doi.org/10.3390/cancers13051125
Chicago/Turabian StylePark, Sangryong, Ho-Young Lee, Jayoung Kim, Hansol Park, Young Seok Ju, Eung-Gook Kim, and Jaehong Kim. 2021. "Cerebral Cavernous Malformation 1 Determines YAP/TAZ Signaling-Dependent Metastatic Hallmarks of Prostate Cancer Cells" Cancers 13, no. 5: 1125. https://doi.org/10.3390/cancers13051125
APA StylePark, S., Lee, H. -Y., Kim, J., Park, H., Ju, Y. S., Kim, E. -G., & Kim, J. (2021). Cerebral Cavernous Malformation 1 Determines YAP/TAZ Signaling-Dependent Metastatic Hallmarks of Prostate Cancer Cells. Cancers, 13(5), 1125. https://doi.org/10.3390/cancers13051125