
Transformed Canine and Murine Mesenchymal Stem Cells as a Model for Sarcoma with Complex Genomics

 , , , ,
, , , ,  ,
, 
Abstract
:Simple Summary
Abstract
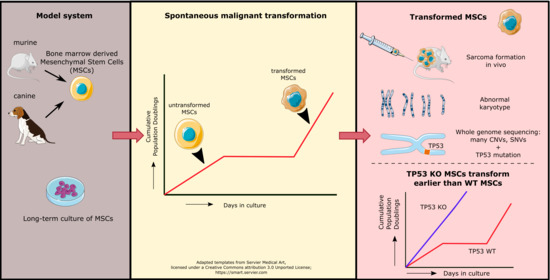
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mesenchymal Stem Cell Isolation and Cell Culture
2.2. Transformation Analysis of Mesenchymal Stem Cells
2.3. Whole Genome Sequencing and Data Analysis of Early Passage and Transformed Late Passage MSCs
2.4. Trilineage Differentiation
2.5. Reverse Transcriptase Quantitative PCR (RT-qPCR)
2.6. Cre-Mediated KO of Trp53 Exon 2–10 in Murine MSCs
2.7. Western Blotting
2.8. In Vivo Tumour Formation
2.9. Statistical Analysis
3. Results
3.1. All Murine MSCs Transform Spontaneously after Long-Term In Vitro Culture
3.2. Infrequent Spontaneous Transformation of Canine MSCs after Long-Term In Vitro Culture
3.3. Clonal Expansion of Murine MSCs Prior to Transformation Event
3.4. Transformed Murine and Canine MSCs Display Variable Mesenchymal Differentiation Capacity
3.5. Transformed Murine MSCs Form Tumours with Variable Growth Rate and Histology In Vivo
3.6. Transformed Murine and Canine MSCs Have Numerous Structural Variants and Copy Number Alterations
3.7. Cross-Species Analysis Reveal TP53/Trp53 Mutation as a Common Single-Nucleotide Variant in Transformed Murine and Canine MSCs
3.8. Murine MSCs with a KO of P53 Transform Earlier Compared to WT Murine MSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Classification of Tumours of Soft Tissue and Bone, 5th ed.; WHO Classification of Tumours Editorial Board: Lyon, France, 2020; Volume 3. [Google Scholar]
- Lam, S.W.; van Ijzendoorn, D.G.P.; Cleton-Jansen, A.M.; Szuhai, K.; Bovée, J.V.M.G. Molecular Pathology of Bone Tumors. J. Mol. Diagn. 2019, 21, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschini, N.; Lam, S.W.; Cleton-Jansen, A.M.; Bovée, J.V.M.G. What’s new in bone forming tumours of the skeleton? Virchows Arch. 2020, 476, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Xu, R.; Prieto, V.G.; Lee, P. Molecular classification of soft tissue sarcomas and its clinical applications. Int. J. Clin. Exp. Pathol. 2010, 3, 416–429. [Google Scholar] [PubMed]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, J.J.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.W.; Wood, G.A.; Lu, J.; Tang, Q.L.; Liu, J.; Molyneux, S.; Chen, Y.; Fang, H.; Adissu, H.; McKee, T.; et al. Cross-species genomics identifies DLG2 as a tumor suppressor in osteosarcoma. Oncogene 2019, 38, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Quindipan, C.; Parham, D.; Shen, L.; Ruble, D.; Bootwalla, M.; Maglinte, D.T.; Gai, X.; Saitta, S.C.; Biegel, J.A.; et al. Inherited germline ATRX mutation in two brothers with ATR-X syndrome and osteosarcoma. Am. J. Med. Genet. Part A 2017, 173, 1390–1395. [Google Scholar] [CrossRef]
- Smolle, M.A.; Heitzer, E.; Geigl, J.B.; Al Kaissi, A.; Liegl-Atzwanger, B.; Seidel, M.G.; Holzer, L.A.; Leithner, A. A novel mutation in ATRX associated with intellectual disability, syndromic features, and osteosarcoma. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Mejia-Guerrero, S.; Quejada, M.; Gokgoz, N.; Gill, M.; Parkes, R.K.; Wunder, J.S.; Andrulis, I.L. Characterization of the 12q15MDM2and 12q13-14CDK4amplicons and clinical correlations in osteosarcoma. Genes Chromosomes Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Movva, S.; Wen, W.; Chen, W.; Millis, S. Multi-platform profiling of over 2000 sarcomas: Identifcation of biomarkers and novel therapeutic targets. Oncotarget 2015, 6, 12234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, J.; Garg, S.; Lau, B.Y.; Dickson, B.C.; Traub, F.; Gokgoz, N.; Griffin, A.M.; Ferguson, P.C.; Andrulis, I.L.; Sim, H.W.; et al. Identifying actionable variants using next generation sequencing in patients with a historical diagnosis of undifferentiated pleomorphic sarcoma. Int. J. Cancer 2018, 142, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Qu, Y.; Wang, J.; Shi, Y.; Yan, W. Pathogenic and Targetable Genetic Alterations in Resected Recurrent Undifferentiated Pleomorphic Sarcomas Identified by Targeted Next-generation Sequencing. Cancer Genom. Proteom. 2019, 16, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, C.D.; Tarabichi, M.; Oukrif, D.; Webster, A.P.; Ye, H.; Fittall, M.; Lombard, P.; Martincorena, I.; Tarpey, P.S.; Collord, G.; et al. Undifferentiated Sarcomas Develop through Distinct Evolutionary Pathways. Cancer Cell 2019, 35, 441–456. [Google Scholar] [CrossRef] [Green Version]
- Mohseny, A.B.; Szuhai, K.; Romeo, S.; Buddingh, E.P.; Briaire-de Bruijn, I.; de Jong, D.; van Pel, M.; Cleton-Jansen, A.M.; Hogendoorn, P.C.W. Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. J. Pathol. 2009, 219, 294–305. [Google Scholar] [CrossRef]
- Xu, S.; De Becker, A.; De Raeve, H.; Van Camp, B.; Vanderkerken, K.; Van Riet, I. In vitro expanded bone marrow-derived murine (C57Bl/KaLwRij) mesenchymal stem cells can acquire CD34 expression and induce sarcoma formation in vivo. Biochem. Biophys. Res. Commun. 2012, 424, 391–397. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Bosch-Marce, M.; Okuyama, H.; Krishnamachary, B.; Kimura, H.; Zhang, L.; Huso, D.L.; Semenza, G.L. Spontaneous transformation of cultured mouse bone marrow-derived stromal cells. Cancer Res. 2006, 66, 10849–10854. [Google Scholar] [CrossRef] [Green Version]
- Tolar, J.; Nauta, A.J.; Osborn, M.J.; Panoskaltsis Mortari, A.; McElmurry, R.T.; Bell, S.; Xia, L.; Zhou, N.; Riddle, M.; Schroeder, T.M.; et al. Sarcoma Derived from Cultured Mesenchymal Stem Cells. Stem Cells 2007, 25, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Buddingh, E.P.; Ruslan, S.E.N.; Reijnders, C.M.A.; Szuhai, K.; Kuijjer, M.L.; Roelofs, H.; Hogendoorn, P.C.W.; Maarten Egeler, R.; Cleton-Jansen, A.M.; Lankester, A.C. Mesenchymal stromal cells of osteosarcoma patients do not show evidence of neoplastic changes during long-term culture. Clin. Sarcoma Res. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Withrow, S.J.; Wilkins, R.M. Cross talk from pets to people: Translational osteosarcoma treatments. ILAR J. 2010, 51, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Kirpensteijn, J.; Kik, M.; Teske, E.; Rutteman, G.R. TP53 gene mutations in canine osteosarcoma. Vet. Surg. 2008, 37, 454–460. [Google Scholar] [CrossRef]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef]
- Malagola, E.; Teunissen, M.; van der Laan, L.J.; Verstegen, M.M.; Schotanus, B.A.; van Steenbeek, F.G.; Penning, L.C.; van Wolferen, M.E.; Tryfonidou, M.A.; Spee, B. Characterization and Comparison of Canine Multipotent Stromal Cells Derived from Liver and Bone Marrow. Stem Cells Dev. 2016, 25, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.F.; Hazewinkel, H.A.; Grinwis, G.C.; Wolschrijn, C.F.; Siebelt, M.; Vernooij, J.C.; Voorhout, G.; Tryfonidou, M.A. Delayed endochondral ossification in early medial coronoid disease (MCD): A morphological and immunohistochemical evaluation in growing Labrador retrievers. Vet. J. 2013, 197, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, M.; Verseijden, F.; Riemers, F.M.; van Osch, G.J.V.M.; Tryfonidou, M.A. The lower in vitro chondrogenic potential of canine adipose tissue-derived mesenchymal stromal cells (MSC) compared to bone marrow-derived MSC is not improved by BMP-2 or BMP-6. Vet. J. 2021, 269. [Google Scholar] [CrossRef] [PubMed]
- Szuhai, K.; Tanke, H.J. COBRA: Combined binary ratio labeling of nucleic-acid probes for multi-color fluorescence in situ hybridization karyotyping. Nat. Protoc. 2006, 1, 264–275. [Google Scholar] [CrossRef]
- Vindelov, L.L.; Christensen, I.J.; Nissen, N.I. A Detergent-Trypsin Method for the Preparation of Nuclei for Flow Cytometric DNA analysis. Cytometry 1983, 3, 323–327. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17. [Google Scholar] [CrossRef]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files, Version 1.33. [Software]; 2011. Available online: https://github.com/najoshi/sickle (accessed on 12 November 2020).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- COBALT. Available online: https://www.ncbi.nlm.nih.gov/tools/cobalt/re_cobalt.cgi (accessed on 12 November 2020).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 12 November 2020).
- Seshan, V.E.; Olshen, A. DNAcopy: DNA Copy Number Data Analysis. R Package Version 1.56.0. 2019. Available online: https://software.pureos.net/package/bin/amber/r-bioc-dnacopy (accessed on 12 November 2020).
- R Core Team. R: A Language and Environment for Statistical Computing. R Found. Stat. Comput. 2014. Available online: https://www.R-project.org/ (accessed on 12 November 2020).
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2015, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bähler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061. Available online: https://www.nature.com/articles/ncomms14061#supplementary-information (accessed on 12 November 2020). [CrossRef] [PubMed] [Green Version]
- Tomayko, M.M.; Reynold, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- IARC TP53 Database. Available online: https://p53.iarc.fr/ (accessed on 7 January 2021).
- Gardner, H.L.; Sivaprakasam, K.; Briones, N.; Zismann, V.; Perdigones, N.; Drenner, K.; Facista, S.; Richholt, R.; Liang, W.; Aldrich, J.; et al. Canine osteosarcoma genome sequencing identifies recurrent mutations in DMD and the histone methyltransferase gene SETD2. Commun. Biol. 2019, 2, 266. [Google Scholar] [CrossRef] [Green Version]
- Sakthikumar, S.; Elvers, I.; Kim, J.; Arendt, M.L.; Thomas, R.; Turner-Maier, J.; Swofford, R.; Johnson, J.; Schumacher, S.E.; Alfoldi, J.; et al. SETD2 Is Recurrently Mutated in Whole-Exome Sequenced Canine Osteosarcoma. Cancer Res. 2018, 78, 3421–3431. [Google Scholar] [CrossRef] [Green Version]
- Cochet-Bissuel, M.; Lory, P.; Monteil, A. The sodium leak channel, NALCN, in health and disease. Front. Cell Neurosci. 2014, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Shi, C.; Jiang, H.; Qin, S. Identification of novel therapeutic target genes and pathway in pancreatic cancer by integrative analysis. Medicine 2017, 96, e8261. [Google Scholar] [CrossRef]
- Requicha, J.F.; Carvalho, P.P.; Anjos Pires, M.; Isabel Dias, M. Evaluation of Canine Adipose-derived Stem Cells in a Healthy Mice Subcutaneous Model. J. Stem Cell Res. Ther. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.C.; Tomiyasu, H.; Garbe, J.R.; Cornax, I.; Amaya, C.; O’Sullivan, M.G.; Subramanian, S.; Bryan, B.A.; Modiano, J.F. Heterotypic mouse models of canine osteosarcoma recapitulate tumor heterogeneity and biological behavior. Dis. Model. Mech. 2016, 9, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Walerych, D.; Napoli, M.; Collavin, L.; Del Sal, G. The rebel angel: Mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 2012, 33, 2007–2017. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Ruijs, M.W.G.; Verhoef, S.; Rookus, M.A.; Pruntel, R.; van der Hout, A.H.; Hogervorst, F.B.L.; Kluijt, I.; Sijmons, R.H.; Aalfs, C.M.; Wagner, A.; et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: Mutation detection rate and relative frequency of cancers in different familial phenotypes. J. Med. Genet. 2010, 47, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermsen, R.; Toonen, P.; Kuijk, E.; Youssef, S.A.; Kuiper, R.; van Heesch, S.; de Bruin, A.; Cuppen, E.; Simonis, M. Lack of major genome instability in tumors of p53 null rats. PLoS ONE 2015, 10, e0122066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.; Sato, M.; Aldape, K.; Mason, C.C.; Alfaro-Munoz, K.; Heathcock, L.; South, S.T.; Abegglen, L.M.; Schiffman, J.D.; Colman, H. DNA copy number analysis of Grade II-III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol. Commun. 2015, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehine, M.; Kaasinen, E.; Makinen, N.; Katainen, R.; Kampjarvi, K.; Pitkanen, E.; Heinonen, H.R.; Butzow, R.; Kilpivaara, O.; Kuosmanen, A.; et al. Characterization of uterine leiomyomas by whole-genome sequencing. N. Engl. J. Med. 2013, 369, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Walkley, C.R.; Qudsi, R.; Sankaran, V.G.; Perry, J.A.; Gostissa, M.; Roth, S.I.; Rodda, S.J.; Snay, E.; Dunning, P.; Fahey, F.H.; et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008, 22, 1662–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franceschini, N.; Verbruggen, B.; Tryfonidou, M.A.; Kruisselbrink, A.B.; Baelde, H.; de Visser, K.E.; Szuhai, K.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. Transformed Canine and Murine Mesenchymal Stem Cells as a Model for Sarcoma with Complex Genomics. Cancers 2021, 13, 1126. https://doi.org/10.3390/cancers13051126
Franceschini N, Verbruggen B, Tryfonidou MA, Kruisselbrink AB, Baelde H, de Visser KE, Szuhai K, Cleton-Jansen A-M, Bovée JVMG. Transformed Canine and Murine Mesenchymal Stem Cells as a Model for Sarcoma with Complex Genomics. Cancers. 2021; 13(5):1126. https://doi.org/10.3390/cancers13051126
Chicago/Turabian StyleFranceschini, Natasja, Bas Verbruggen, Marianna A. Tryfonidou, Alwine B. Kruisselbrink, Hans Baelde, Karin E. de Visser, Karoly Szuhai, Anne-Marie Cleton-Jansen, and Judith V. M. G. Bovée. 2021. "Transformed Canine and Murine Mesenchymal Stem Cells as a Model for Sarcoma with Complex Genomics" Cancers 13, no. 5: 1126. https://doi.org/10.3390/cancers13051126
APA StyleFranceschini, N., Verbruggen, B., Tryfonidou, M. A., Kruisselbrink, A. B., Baelde, H., de Visser, K. E., Szuhai, K., Cleton-Jansen, A. -M., & Bovée, J. V. M. G. (2021). Transformed Canine and Murine Mesenchymal Stem Cells as a Model for Sarcoma with Complex Genomics. Cancers, 13(5), 1126. https://doi.org/10.3390/cancers13051126