
Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations

 , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Cisplatin
1.2. Nephrotoxicity
1.3. Pathophysiology of Cisplatin-Induced AKI
2. Pharmacological Approaches Targeting Cisplatin Cellular Uptake
2.1. Cellular Uptake Transporters of Cisplatin
2.2. Organic Cation Transporter 2 (OCT2)
2.3. Copper Transporter 1
2.4. OAT1/OAT3
3. Pharmacological Approaches Targeting Cisplatin Cellular Efflux
3.1. Apically Localized Efflux Transporters P-Type Copper Transporting ATPases7A/B
3.2. Multidrug-Resistance-Associated Protein 2
3.3. Multi-Antimicrobial Extrusion Protein 1
4. Interventions Targeting Molecular Mechanisms CIAKI
4.1. Oxidative Stress
Monotropein (Nrf2/HO-1 Antioxidant Pathway)
4.2. Vascular Injury
The Renin Angiotensin System in Cisplatin-Induced Acute Kidney Injury
4.3. Cell Death
4.3.1. Necrosis
4.3.2. Apoptosis-Intrinsic/Mitochondrial Pathway
4.3.3. Apoptosis-Extrinsic Pathway
4.3.4. Endoplasmic Reticulum Stress-Induced Apoptosis
5. Cisplatin-Induced Acute Kidney Injury: Role of the Immune System
5.1. Toll-Like Receptors and Cisplatin-Induced Acute Kidney Injury
5.1.1. Toll-Like Receptor 2 is Protective in Cisplatin-Induced Acute Kidney Injury
5.1.2. Toll-Like Receptor 4 Has a Detrimental Role in Cisplatin-Induced Acute Kidney Injury
5.1.3. Toll-Like Receptor 9 is Protective in Cisplatin-Induced Acute Kidney Injury
5.2. Cytokines
5.2.1. Tumor Necrosis Factor Alpha
5.2.2. Nuclear Factor Kappa-Light-Chain Enhancer of Activated B Cells
5.2.3. Interleukin-10
5.2.4. Interleukin-33
5.2.5. Interleukin-6
5.3. Chemokines
5.3.1. CXCL16
5.3.2. CXCL1-CXCR2 Axis
6. Preclinic to Clinic Translation
7. CIAKI Clinical Trials
8. Potential Future Treatments
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gomez-Ruiz, S.; Maksimović-Ivanić, D.; Mijatović, S.; Kaluđerović, G.N. On the discovery, biological effects, and use of cisplatin and metallocenes in anticancer chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 140284. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, J.; Li, J.; Liu, F.; Wu, X.; Wang, Z. Cisplatin suppresses the growth and proliferation of breast and cervical cancer cell lines by inhibiting integrin β5-mediated glycolysis. Am. J. Cancer Res. 2016, 6, 1108–1117. [Google Scholar]
- Li, Z.; Zhang, P.; Ma, Q.; Wang, D.; Zhou, T. Cisplatin-based chemoradiotherapy with 5-fluorouracil or pemetrexed in patients with locally advanced, unresectable esophageal squamous cell carcinoma: A retrospective analysis. Mol. Clin. Oncol. 2017, 6, 743–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.; Palmer, D.; Lloyd, B.; Collins, S.; Barton, D.; Ansari, J.; James, N. A study of split-dose cisplatin-based neo-adjuvant chemotherapy in muscle-invasive bladder cancer. Oncol. Lett. 2012, 3, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Coward, J.I.G. Chemotherapy advances in small-cell lung cancer. J. Thorac. Dis. 2013, 5 (Suppl. 5), S565–S578. [Google Scholar] [CrossRef]
- Einhorn, L.H.; Donohue, J. Cis-Diamminedichloroplatinum, Vinblastine, and Bleomycin Combination Chemotherapy in Disseminated Testicular Cancer. Ann. Intern. Med. 1977, 87, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Livingston, J.A.; Patel, S.R.; Benjamin, R.S. Chemotherapy for Bone Sarcoma in Adults. J. Oncol. Pract. 2016, 12, 208–216. [Google Scholar] [CrossRef]
- Le, X.; Hanna, E.Y. Optimal regimen of cisplatin in squamous cell carcinoma of head and neck yet to be determined. Ann. Transl. Med. 2018, 6, 229. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, P.M.; Yu, F.; Kaldis, P.; Aleem, E.; Nowak, G.; Safirstein, R.L.; Megyesi, J. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J. Am. Soc. Nephrol. 2006, 17, 2434–2442. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Rybak, L.P.; Mukherjea, D.; Jajoo, S.; Ramkumar, V. Cisplatin ototoxicity and protection: Clinical and experimental studies. Tohoku J. Exp. Med. 2009, 219, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Rybak, L.P.; Whitworth, C.A.; Mukherjea, D.; Ramkumar, V. Mechanisms of cisplatin-induced ototoxicity and prevention. Hear. Res. 2007, 226, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, N.A.G.; Rodrigues, M.A.C.; Martins, N.M.; Dos Santos, A.C. Cisplatin-induced nephrotoxicity and targets of nephroprotection: An update. Arch. Toxicol. 2012, 86, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.J.; Hou, J.G.; Ma, Z.N.; Wang, Z.; Ren, S.; Wang, Y.P.; Liu, W.C.; Chen, C.; Li, W. Ginsenoside Rb3 provides protective effects against cisplatin-induced nephrotoxicity via regulation of AMPK-/mTOR-mediated autophagy and inhibition of apoptosis in vitro and in vivo. Cell Prolif. 2019, 52, e12627. [Google Scholar] [CrossRef]
- Hoek, J.; Bloemendal, K.M.; Van der Velden, L.A.A.; Van Diessen, J.N.; Van Werkhoven, E.; Klop, W.M.; Tesselaar, M.E. Nephrotoxicity as a Dose-Limiting Factor in a High-Dose Cisplatin-Based Chemoradiotherapy Regimen for Head and Neck Carcinomas. Cancers 2016, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, X.-J.; Hou, J.-G.; Wang, Z.; Han, Y.; Ren, S.; Hu, J.-N.; Chen, C.; Li, W. The protective effects of maltol on cisplatin-induced nephrotoxicity through the AMPK-mediated PI3K/Akt and p53 signaling pathways. Sci. Rep. 2018, 8, 15922. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, F.; Haghshenas, S.; Nematbakhsh, M.; Nasri, H.; Talebi, A.; Eshraghi-Jazi, F.; Pezeshki, Z.; Safari, T. The Role of Magnesium Supplementation in Cisplatin-induced Nephrotoxicity in a Rat Model: No Nephroprotectant Effect. Int. J. Prev. Med. 2012, 3, 637–643. [Google Scholar]
- Sastry, J.; Kellie, S.J. Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatric Hematol. Oncol. 2005, 22, 441–445. [Google Scholar] [CrossRef]
- Qin, Y.; Stokman, G.; Yan, K.; Ramaiahgari, S.; Verbeek, F.; De Graauw, M.; Van de Water, B.; Price, L.S. cAMP signalling protects proximal tubular epithelial cells from cisplatin-induced apoptosis via activation of Epac. Br. J. Pharmacol. 2012, 165, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Kellum, J.A.; Unruh, M.L.; Murugan, R. Acute kidney injury. BMJ Clin. Evid. 2011, 394, 1949–1964. [Google Scholar] [CrossRef]
- Lameire, N.; Van Biesen, W.; Vanholder, R. Acute kidney injury. Lancet 2008, 372, 1863–1865. [Google Scholar] [CrossRef]
- Hanif, M.O.; Bali, A.; Ramphul, K. Acute renal tubular necrosis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Rahman, M.; Shad, F.; Smith, M.C. Acute kidney injury: A guide to diagnosis and management. Am. Fam. Physician 2012, 86, 631–639. [Google Scholar]
- Dalal, R.; Bruss, Z.S.; Sehdev, J.S. Physiology, renal blood flow and filtration. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Oh, G.-S.; Kim, H.-J.; Shen, A.; Lee, S.B.; Khadka, D.; Pandit, A.; So, H.-S. Cisplatin-induced kidney dysfunction and perspectives on improving treatment strategies. Electrolytes Blood Press. 2014, 12, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins (Basel) 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [Green Version]
- Bennis, Y.; Savry, A.; Rocca, M.; Gauthier-Villano, L.; Pisano, P.; Pourroy, B. Cisplatin dose adjustment in patients with renal impairment, which recommendations should we follow? Int. J. Clin. Pharm. 2014, 36, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. Biomed. Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef]
- Rosner, M.H.; Perazella, M.A. Acute kidney injury in the patient with cancer. Kidney Res. Clin. Pract. 2019, 38, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, G.; Reeves, W.B. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramesh, G.; Norbury, C.; Reeves, W. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Li, X.; Lv, W.; Xu, Z. Inhibition of CXCL1-CXCR2 axis ameliorates cisplatin-induced acute kidney injury by mediating inflammatory response. Biomed. Pharmacother. 2020, 122, 109693. [Google Scholar] [CrossRef]
- Soni, H.; Kaminski, D.; Gangaraju, R.; Adebiyi, A. Cisplatin-induced oxidative stress stimulates renal Fas ligand shedding. Ren. Fail. 2018, 40, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Hou, X.; Wang, X.; Shi, Y.; Xu, L.; Zheng, X.; Liu, N.; Qiu, A.; Zhuang, S. 3-deazaneplanocin A protects against cisplatin-induced renal tubular cell apoptosis and acute kidney injury by restoration of E-cadherin expression. Cell Death Dis. 2019, 10, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potočnjak, I.; Marinić, J.; Batičić, L.; Šimić, L.; Broznić, D.; Domitrović, R. Aucubin administered by either oral or parenteral route protects against cisplatin-induced acute kidney injury in mice. Food Chem. Toxicol. 2020, 142, 111472. [Google Scholar] [CrossRef]
- Huang, S.-J.; Huang, J.; Yan, Y.-B.; Qiu, J.; Tan, R.-Q.; Liu, Y.; Tian, Q.; Guan, L.; Niu, S.-S.; Zhang, Y. The renoprotective effect of curcumin against cisplatin-induced acute kidney injury in mice: Involvement of miR-181a/PTEN axis. Ren. Fail. 2020, 42, 350–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El-Kader, M.; Taha, R.I. Comparative nephroprotective effects of curcumin and etoricoxib against cisplatin-induced acute kidney injury in rats. Acta Histochem. 2020, 122, 151534. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Zhu, K.; Li, C.; Wang, X.; Shen, J.; Yong, F.; Jia, H. Dexmedetomidine alleviates cisplatin-induced acute kidney injury by attenuating endoplasmic reticulum stress-induced apoptosis via the α2AR/PI3K/AKT pathway. Mol. Med. Rep. 2020, 21, 1597–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadir, A.; Sher, S.; Siddiqui, R.A.; Mirza, T. Nephroprotective role of eugenol against cisplatin-induced acute kidney injury in mice. Pak. J. Pharm. Sci. 2020, 33, 1281–1287. [Google Scholar]
- Hu, Z.; Zhang, H.; Yi, B.; Yang, S.; Liu, J.; Hu, J.; Wang, J.; Cao, K.; Zhang, W. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wei, W.; Huang, J.; Liu, X.; Ci, X. Isoorientin attenuates cisplatin-induced nephrotoxicity through the inhibition of oxidative stress and apoptosis via activating the SIRT1/SIRT6/Nrf-2 pathway. Front. Pharmacol. 2020, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, Y.; Li, B.; Ding, P.; Jin, D.; Hou, S.; Cai, X.; Sheng, X. The effect of monotropein on alleviating cisplatin-induced acute kidney injury by inhibiting oxidative damage, inflammation and apoptosis. Biomed. Pharmacother. 2020, 129, 110408. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.Z.; Wang, C.; Deng, C.; Zhong, X.; Yan, Y.; Luo, Y.; Lan, H.Y.; He, T.; Wang, L. Quercetin protects against cisplatin-induced acute kidney injury by inhibiting Mincle/Syk/NF-κB signaling maintained macrophage inflammation. Phytother. Res. 2020, 34, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, Z.; He, L.; Wang, Y. CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget 2016, 7, 31652. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Baliga, R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003, 63, 1687–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitazaki, S.; Honma, S.; Suto, M.; Kato, N.; Hiraiwa, K.; Yoshida, M.; Abe, S. Interleukin-6 plays a protective role in development of cisplatin-induced acute renal failure through upregulation of anti-oxidative stress factors. Life Sci. 2011, 88, 1142–1148. [Google Scholar] [CrossRef]
- Ravichandran, K.; Holditch, S.; Brown, C.N.; Wang, Q.; Ozkok, A.; Weiser-Evans, M.C.; Nemenoff, R.; Miyazaki, M.; Thiessen-Philbrook, H.; Parikh, C.R. IL-33 deficiency slows cancer growth but does not protect against cisplatin-induced AKI in mice with cancer. Am. J. Physiol. Ren. Physiol. 2018, 314, F356–F366. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, D.W.; Ravichandran, K.; Keys, D.O.; Akcay, A.; Nguyen, Q.; He, Z.; Jani, A.; Ljubanovic, D.; Edelstein, C.L. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 2013, 346, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, P.; Horváth, B.; Kechrid, M.; Tanchian, G.; Rajesh, M.; Naura, A.S.; Boulares, A.H.; Pacher, P. Poly (ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic. Biol. Med. 2011, 51, 1774–1788. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Chien, C.-C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; An, C.; Jin, X.; Hu, Z.; Safirstein, R.L.; Wang, Y. TAK1 deficiency attenuates cisplatin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 318, F209–F215. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Silva, M.; Cenedeze, M.A.; Perandini, L.A.; Felizardo, R.J.F.; Watanabe, I.K.M.; Agudelo, J.S.H.; Castoldi, A.; Gonçalves, G.M.; Origassa, C.S.T.; Semedo, P. TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 1725–1739. [Google Scholar] [CrossRef]
- Alikhan, M.A.; Summers, S.A.; Gan, P.Y.; Chan, A.J.; Khouri, M.B.; Ooi, J.D.; Ghali, J.R.; Odobasic, D.; Hickey, M.J.; Kitching, A.R. Endogenous toll-like receptor 9 regulates AKI by promoting regulatory T cell recruitment. J. Am. Soc. Nephrol. 2016, 27, 706–714. [Google Scholar] [CrossRef]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol Ren. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014, 34, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstadt, H.; Lanvers-Kaminsky, C.; Am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar] [CrossRef]
- Tanihara, Y.; Masuda, S.; Katsura, T.; Inui, K.-I. Protective effect of concomitant administration of imatinib on cisplatin-induced nephrotoxicity focusing on renal organic cation transporter OCT2. Biochem. Pharmacol. 2009, 78, 1263–1271. [Google Scholar] [CrossRef]
- Ismail, R.S.; El-Awady, M.S.; Hassan, M.H. Pantoprazole abrogated cisplatin-induced nephrotoxicity in mice via suppression of inflammation, apoptosis, and oxidative stress. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1–11. [Google Scholar] [CrossRef]
- Hu, S.; Leblanc, A.; Gibson, A.; Hong, K.; Kim, J.; Janke, L.; Li, L.; Vasilyeva, A.; Finkelstein, D.; Sprowl, J. Identification of OAT1/OAT3 as contributors to cisplatin toxicity. Clin. Transl. Sci. 2017, 10, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Morsy, M.A.; Heeba, G.H. Nebivolol ameliorates cisplatin-induced nephrotoxicity in rats. Basic Clin. Pharmacol. Toxicol. 2016, 118, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Yonezawa, A.; Hashimoto, S.; Katsura, T.; Inui, K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem. Pharm. 2010, 80, 1762–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Buckley, B.; McCandlish, E.; Goedken, M.J.; Syed, S.; Pelis, R.; Manautou, J.E.; Aleksunes, L.M. Transgenic expression of the human MRP2 transporter reduces cisplatin accumulation and nephrotoxicity in Mrp2-null mice. Am. J. Pathol. 2014, 184, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nieskens, T.T.; Peters, J.G.; Dabaghie, D.; Korte, D.; Jansen, K.; Van Asbeck, A.H.; Tavraz, N.N.; Friedrich, T.; Russel, F.G.; Masereeuw, R. Expression of organic anion transporter 1 or 3 in human kidney proximal tubule cells reduces cisplatin sensitivity. Drug Metab. Dispos. 2018, 46, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, K.D.; Wanchoo, R.; Sakhiya, V.; Ross, D.W.; Fishbane, S. Adverse renal effects of novel molecular oncologic targeted therapies: A narrative review. Kidney Int. Rep. 2017, 2, 108–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcolino, M.; Boersma, E.; Clementino, N.; Macedo, A.; Marx-Neto, A.; Silva, M.; van Gelder, T.; Akkerhuis, K.; Ribeiro, A. Imatinib treatment duration is related to decreased estimated glomerular filtration rate in chronic myeloid leukemia patients. Ann. Oncol. 2011, 22, 2073–2079. [Google Scholar] [CrossRef]
- Huang, D.; Wang, C.; Duan, Y.; Meng, Q.; Liu, Z.; Huo, X.; Sun, H.; Ma, X.; Liu, K. Targeting Oct2 and P53: Formononetin prevents cisplatin-induced acute kidney injury. Toxicol. Appl. Pharmacol. 2017, 326, 15–24. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Lin, X.; Okuda, T.; Holzer, A.; Howell, S. The Copper Transporter CTR1 Regulates Cisplatin Uptake in Saccharomyces cerevisiae. Mol. Pharmacol. 2002, 62, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.; Levin, K.; Zhu, Y.; Segers, B.; Balamuth, N.; Womer, R.; Bagatell, R.; Balis, F. Pantoprazole, an Inhibitor of the Organic Cation Transporter 2, Does Not Ameliorate Cisplatin-Related Ototoxicity or Nephrotoxicity in Children and Adolescents with Newly Diagnosed Osteosarcoma Treated with Methotrexate, Doxorubicin, and Cisplatin. Oncologist 2018, 23, 762. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Sørensen, B.H.; Sauter, D.P.; Lambert, I.H. Role of volume-regulated and calcium-activated anion channels in cell volume homeostasis, cancer and drug resistance. Channels 2015, 9, 380–396. [Google Scholar] [CrossRef] [Green Version]
- Osei-Owusu, J.; Yang, J.; Del Carmen Vitery, M.; Qiu, Z. Molecular biology and physiology of volume-regulated anion channel (VRAC). In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2018; Volume 81, pp. 177–203. [Google Scholar]
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalan, M.; Bagchi, T.; Misra, A. The Cell. In Challenges in Delivery of Therapeutic Genomics and Proteomics; Elsevier: Amsterdam, The Netherlands, 2011; pp. 1–43. [Google Scholar]
- Yonny, M.E.; García, E.M.; Lopez, A.; Arroquy, J.I.; Nazareno, M.A. Measurement of malondialdehyde as oxidative stress biomarker in goat plasma by HPLC-DAD. Microchem. J. 2016, 129, 281–285. [Google Scholar] [CrossRef]
- Galgamuwa, R.; Hardy, K.; Dahlstrom, J.E.; Blackburn, A.C.; Wium, E.; Rooke, M.; Cappello, J.Y.; Tummala, P.; Patel, H.R.; Chuah, A. Dichloroacetate prevents cisplatin-induced nephrotoxicity without compromising cisplatin anticancer properties. J. Am. Soc. Nephrol. 2016, 27, 3331–3344. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, T.; Sato, W.; Ishikawa, K.; Terao, Y.; Takahashi, K.; Noda, Y.; Yuzawa, Y.; Nagamatsu, T. Significance of downregulation of renal organic cation transporter (SLC47A1) in cisplatin-induced proximal tubular injury. Oncotargets Ther. 2015, 8, 1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.-S.; Terada, K.; Furukawa, T.; Yang, X.-L.; Gao, H.; Miura, N.; Sugiyama, T. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar] [PubMed]
- Samimi, G.; Varki, N.M.; Wilczynski, S.; Safaei, R.; Alberts, D.S.; Howell, S.B. Increase in expression of the copper transporter ATP7A during platinum drug-based treatment is associated with poor survival in ovarian cancer patients. Clin. Cancer Res. 2003, 9, 5853–5859. [Google Scholar] [PubMed]
- Kalayda, G.V.; Wagner, C.H.; Buß, I.; Reedijk, J.; Jaehde, U. Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer 2008, 8, 175. [Google Scholar] [CrossRef] [Green Version]
- Harrach, S.; Ciarimboli, G. Role of transporters in the distribution of platinum-based drugs. Front. Pharmacol. 2015, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, A.; Oda, K.; Mori, N.; Murakami, T. Modulated function of multidrug resistance-associated proteins in cisplatin-induced acute renal failure rats. Die Pharm. Int. J. Pharm. Sci. 2017, 72, 209–213. [Google Scholar]
- Hanigan, M.H.; Devarajan, P. Cisplatin nephrotoxicity: Molecular mechanisms. Cancer Ther. 2003, 1, 47. [Google Scholar] [PubMed]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K.-I. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1–3 and multidrug and toxin extrusion family). J. Pharmacol. Exp. Ther. 2006, 319, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guo, D.; Dong, Z.; Zhang, W.; Zhang, L.; Huang, S.-M.; Polli, J.E.; Shu, Y. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs). Toxicol. Appl. Pharmacol. 2013, 273, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubera, I.; Duranton, C.; Melis, N.; Cougnon, M.; Mograbi, B.; Tauc, M. Role of CFTR in oxidative stress and suicidal death of renal cells during cisplatin-induced nephrotoxicity. Cell Death Dis. 2013, 4, e817. [Google Scholar] [CrossRef] [PubMed]
- Jesse, C.R.; Bortolatto, C.F.; Wilhelm, E.A.; Roman, S.S.; Prigol, M.; Nogueira, C.W. The peroxisome proliferator-activated receptor-γ agonist pioglitazone protects against cisplatin-induced renal damage in mice. J. Appl. Toxicol. 2014, 34, 25–32. [Google Scholar] [CrossRef]
- Ma, Q.; Xu, Y.; Tang, L.; Yang, X.; Chen, Z.; Wei, Y.; Shao, X.; Shao, X.; Xin, Z.; Cai, B. Astragalus polysaccharide attenuates cisplatin-induced acute kidney injury by suppressing oxidative damage and mitochondrial dysfunction. Biomed. Res. Int. 2020, 2020, 2851349. [Google Scholar] [CrossRef]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Tew, K.D.; He, L.; King, J.B.; Hanigan, M.H. Role of glutathione S-transferase Pi in cisplatin-induced nephrotoxicity. Biomed. Pharmacother. 2009, 63, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-F.; Yang, C.-M.; Su, C.-M.; Hu, M.-L. Vitamin C protects against cisplatin-induced nephrotoxicity and damage without reducing its effectiveness in C57BL/6 mice xenografted with Lewis lung carcinoma. Nutr. Cancer 2014, 66, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Darwish, M.A.; Abo-Youssef, A.M.; Khalaf, M.M.; Abo-Saif, A.A.; Saleh, I.G.; Abdelghany, T.M. Vitamin E mitigates cisplatin-induced nephrotoxicity due to reversal of oxidative/nitrosative stress, suppression of inflammation and reduction of total renal platinum accumulation. J. Biochem. Mol. Toxicol. 2017, 31, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Shi, Y.; Chen, J.; Song, N.; Cai, J.; Fang, Y.; Yu, X.; Ji, J.; Ding, X. Necrostatin-1 attenuates cisplatin-induced nephrotoxicity through suppression of apoptosis and oxidative stress and retains klotho expression. Front. Pharmacol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Chen, X.; Wei, W.; Li, Y.; Huang, J.; Ci, X. Hesperetin relieves cisplatin-induced acute kidney injury by mitigating oxidative stress, inflammation and apoptosis. Chem. Biol. Interact. 2019, 308, 269–278. [Google Scholar] [CrossRef]
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef]
- Zhao, K.; Wen, L. DMF attenuates cisplatin-induced kidney injury via activating Nrf2 signaling pathway and inhibiting NF-kB signaling pathway. Eur. Rev. Med. Pharm. Sci. 2018, 22, 8924–8931. [Google Scholar]
- Ansari, M.A. Sinapic acid modulates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Biomed. Pharmacother. 2017, 93, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yehuda Greenwald, M.; Ben-Sasson, S.; Bianco-Peled, H.; Kohen, R. Skin redox balance maintenance: The need for an Nrf2-activator delivery system. Cosmetics 2016, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Hu, S.; He, Y.; Zhang, J.; Zeng, X.; Gong, F.; Liang, L.N. The protective effects of Zhen-Wu-Tang against cisplatin-induced acute kidney injury in rats. PloS ONE 2017, 12, e0179137. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Koike, N.; Murakami, T.; Suzuki, K. Dimethyl fumarate ameliorates cisplatin-induced renal tubulointerstitial lesions. J. Toxicol. Pathol. 2019, 32, 79–89. [Google Scholar] [CrossRef]
- Winston, J.A.; Safirstein, R. Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. Am. J. Physiol. 1985, 249, F490–F496. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, M.; Mou, J.; Zhao, Z.; Yang, J.; Zhu, F.; Pei, G.; Zhu, H.; Wang, Y.; Xu, G. Pretreatment of Huaiqihuang extractum protects against cisplatin-induced nephrotoxicity. Sci. Rep. 2018, 8, 1–10. [Google Scholar]
- Dibona, G.F.; Kopp, U.C. Neural control of renal function. Physiol. Rev. 1997, 77, 75–197. [Google Scholar] [CrossRef]
- Walsh, M.P. Regulation of vascular smooth muscle tone. Can. J. Physiol. Pharmacol. 1994, 72, 919–936. [Google Scholar] [CrossRef]
- Salomonsson, M.; Arendshorst, W.J. Calcium recruitment in renal vasculature: NE effects on blood flow and cytosolic calcium concentration. Am. J. Physiol. Ren. Physiol. 1999, 276, F700–F710. [Google Scholar] [CrossRef] [PubMed]
- Coats, A.; Jain, S. Protective effects of nebivolol from oxidative stress to prevent hypertension-related target organ damage. J. Hum. Hypertens. 2017, 31, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; West, N.E.; Pillai, R.; Taggart, D.P.; Channon, K.M. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension 2002, 39, 1088–1094. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahman, A.M.; Al Suleimani, Y.; Shalaby, A.; Ashique, M.; Manoj, P.; Al-Saadi, H.; Ali, B.H. Effect of levosimendan, a calcium sensitizer, on cisplatin-induced nephrotoxicity in rats. Toxicol. Rep. 2019, 6, 232–238. [Google Scholar] [CrossRef]
- Parissis, J.T.; Andreadou, I.; Bistola, V.; Paraskevaidis, I.; Filippatos, G.; Kremastinos, D.T. Novel biologic mechanisms of levosimendan and its effect on the failing heart. Expert Opin. Investig. Drugs 2008, 17, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.M.; Nolan, C.; Ryan, M.P.; Basinger, M.A.; Jones, M.M.; Hande, K.R. The role of the renin-angiotensin system in cisplatin nephrotoxicity. Ren. Fail. 1995, 17, 665–674. [Google Scholar] [CrossRef]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2017. [Google Scholar]
- Dhande, I.; Ma, W.; Hussain, T. Angiotensin AT 2 receptor stimulation is anti-inflammatory in lipopolysaccharide-activated THP-1 macrophages via increased interleukin-10 production. Hypertens. Res. 2015, 38, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Mori, H.; Masaki, T.; Nishiyama, A. Angiotensin II blockade and renal protection. Curr. Pharm. Des. 2013, 19, 3033–3042. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hein, T.W.; Wang, W.; Kuo, L. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ. Res. 2003, 92, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Bonnet, F.; Candido, R.; Nesteroff, S.P.; Burns, W.C.; Kawachi, H.; Shimizu, F.; Carey, R.M.; De Gasparo, M.; Cooper, M.E. Angiotensin type 2 receptor antagonism confers renal protection in a rat model of progressive renal injury. J. Am. Soc. Nephrol. 2002, 13, 1773–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhande, I.; Ali, Q.; Hussain, T. Proximal tubule angiotensin AT2 receptors mediate an anti-inflammatory response via interleukin-10: Role in renoprotection in obese rats. Hypertension 2013, 61, 1218–1226. [Google Scholar] [CrossRef] [Green Version]
- Hosoda, A.; Matsumoto, Y.; Toriyama, Y.; Tsuji, T.; Yoshida, Y.; Masamichi, S.; Kohno, T. Telmisartan Exacerbates Cisplatin-Induced Nephrotoxicity in a Mouse Model. Biol. Pharm. Bull. 2020, 43, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Suchal, K.; Gamad, N.; Dinda, A.K.; Arya, D.S.; Bhatia, J. Telmisartan ameliorates cisplatin-induced nephrotoxicity by inhibiting MAPK mediated inflammation and apoptosis. Eur. J. Pharm. 2015, 748, 54–60. [Google Scholar] [CrossRef]
- Okui, S.; Yamamoto, H.; Li, W.; Gamachi, N.; Fujita, Y.; Kashiwamura, S.-I.; Miura, D.; Takai, S.; Miyazaki, M.; Urade, M. Cisplatin-induced acute renal failure in mice is mediated by chymase-activated angiotensin-aldosterone system and interleukin-18. Eur. J. Pharm. 2012, 685, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Rudemiller, N.P.; Patel, M.B.; Wei, Q.; Karlovich, N.S.; Jeffs, A.D.; Wu, M.; Sparks, M.A.; Privratsky, J.R.; Herrera, M. Competing actions of type 1 angiotensin II receptors expressed on T lymphocytes and kidney epithelium during cisplatin-induced AKI. J. Am. Soc. Nephrol. 2016, 27, 2257–2264. [Google Scholar] [CrossRef] [Green Version]
- Sherif, I.O.; Al-Mutabagani, L.A.; Alnakhli, A.M.; Sobh, M.A.; Mohammed, H.E. Renoprotective effects of angiotensin receptor blocker and stem cells in acute kidney injury: Involvement of inflammatory and apoptotic markers. Exp. Biol. Med. 2015, 240, 1572–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharmashankar, K.; Widlansky, M.E. Vascular Endothelial Function and Hypertension: Insights and Directions. Curr. Hypertens. Rep. 2010, 12, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaradakhi, T.; Apostolopoulos, V.; Zulli, A. Angiotensin (1-7) and Alamandine: Similarities and differences. Pharmacol. Res. 2016, 111, 820–826. [Google Scholar] [CrossRef] [Green Version]
- Velkoska, E.; Patel, S.K.; Burrell, L.M. Angiotensin converting enzyme 2 and diminazene: Role in cardiovascular and blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2016, 25, 384–395. [Google Scholar] [CrossRef]
- Rabelo, L.A.; Todiras, M.; Nunes-Souza, V.; Qadri, F.; Szijarto, I.A.; Gollasch, M.; Penninger, J.M.; Bader, M.; Santos, R.A.; Alenina, N. Genetic Deletion of ACE2 Induces Vascular Dysfunction in C57BL/6 Mice: Role of Nitric Oxide Imbalance and Oxidative Stress. PloS ONE 2016, 11, e0150255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, V.; Gjymishka, A.; Jarajapu, Y.P.; Qi, Y.; Afzal, A.; Rigatto, K.; Ferreira, A.J.; Fraga-Silva, R.A.; Kearns, P.; Douglas, J.Y.; et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am. J. Respir. Crit. Care Med. 2013, 187, 648–657. [Google Scholar] [CrossRef]
- Sampaio, W.O.; Souza dos Santos, R.A.; Faria-Silva, R.; Da Mata Machado, L.T.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007, 49, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Wright, H.M. Nebivolol: A highly selective β1-adrenergic receptor blocker that causes vasodilation by increasing nitric oxide. Cardiovasc. Ther. 2008, 26, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Varagic, J.; Ahmad, S.; Voncannon, J.L.; Moniwa, N.; Simington, S.W., Jr.; Brosnihan, B.K.; Gallagher, P.E.; Habibi, J.; Sowers, J.R.; Ferrario, C.M. Nebivolol reduces cardiac angiotensin II, associated oxidative stress and fibrosis but not arterial pressure in salt-loaded spontaneously hypertensive rats. J. Hypertens. 2012, 30, 1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, S.; Sauter, G.; Preyer, M.; Poerner, T.; Kempf, V.; Risler, T.; Brehm, B. Influence of nebivolol and metoprolol on inflammatory mediators in human coronary endothelial or smooth muscle cells. Effects on neointima formation after balloon denudation in carotid arteries of rats treated with nebivolol. Cell. Physiol. Biochem. 2007, 19, 129–136. [Google Scholar] [CrossRef]
- Szturz, P.; Wouters, K.; Kiyota, N.; Tahara, M.; Prabhash, K.; Noronha, V.; Adelstein, D.; Van Gestel, D.; Vermorken, J.B. Low-dose vs. high-dose cisplatin: Lessons learned from 59 chemoradiotherapy trials in head and neck cancer. Front. Oncol. 2019, 9, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef]
- Gillespie, S.K.; Zhang, X.D.; Hersey, P. Ingenol 3-angelate induces dual modes of cell death and differentially regulates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in melanoma cells. Mol. Cancer 2004, 3, 1651–1658. [Google Scholar]
- Saito, Y.; Nishio, K.; Ogawa, Y.; Kimata, J.; Kinumi, T.; Yoshida, Y.; Noguchi, N.; Niki, E. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free Radic. Res. 2006, 40, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Martínez, S.M.; Piedrafita, F.J.; Cannata-Andía, J.B.; López-Novoa, J.M.; López-Hernández, F.J. Necrotic concentrations of cisplatin activate the apoptotic machinery but inhibit effector caspases and interfere with the execution of apoptosis. Toxicol. Sci. 2011, 122, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. JASN 2015, 26, 2647–2658. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb Perspect Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12, 1463–1467. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, S.; Gao, H.; Wang, P.; Zhang, Y.; Zhang, A.; Jia, Z.; Huang, S. Ursodeoxycholic acid protects against cisplatin-induced acute kidney injury and mitochondrial dysfunction through acting on ALDH1L2. Free Radic. Biol. Med. 2020, 152, 821–837. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yuan, Y.; Liu, X.; Li, Q.; An, X.; Huang, Z.; Wu, L.; Zhang, B.; Zhang, A.; Xing, C. Adenosine kinase inhibition protects against cisplatin-induced nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2019, 317, F107–F115. [Google Scholar] [CrossRef]
- Shimizu, S.; Eguchi, Y.; Kamiike, W.; Funahashi, Y.; Mignon, A.; Lacronique, V.; Matsuda, H.; Tsujimoto, Y. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc. Natl. Acad. Sci. USA 1998, 95, 1455–1459. [Google Scholar] [CrossRef] [Green Version]
- Gertz, M.; Nguyen, G.T.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Fränzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D. A molecular mechanism for direct sirtuin activation by resveratrol. PloS ONE 2012, 7, e49761. [Google Scholar] [CrossRef] [Green Version]
- Hao, Q.; Xiao, X.; Zhen, J.; Feng, J.; Song, C.; Jiang, B.; Hu, Z. Resveratrol attenuates acute kidney injury by inhibiting death receptor-mediated apoptotic pathways in a cisplatin-induced rat model. Mol. Med. Rep. 2016, 14, 3683–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Xie, Q.; Qi, L.; Wang, C.; Xu, N.; Liu, W.; Yu, Y.; Li, S.; Xu, Y. Bcl-2 overexpression reduces cisplatin cytotoxicity by decreasing ER-mitochondrial Ca2+ signaling in SKOV3 cells. Oncol. Rep. 2018, 39, 985–992. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Wang, J.; Du, Q.; Yu, J.; Zhang, L.; Dong, Z. Regulation of PUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Oncogene 2006, 25, 4056–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. JASN 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Wang, X.; Parrish, A.R. Loss of α(E)-catenin promotes Fas mediated apoptosis in tubular epithelial cells. Apoptosis 2015, 20, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. Embo J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef]
- Tsuruya, K.; Ninomiya, T.; Tokumoto, M.; Hirakawa, M.; Masutani, K.; Taniguchi, M.; Fukuda, K.; Kanai, H.; Kishihara, K.; Hirakata, H.; et al. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003, 63, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Berrocal, J.; Nevado, J.; Ramírez-Camacho, R.; Sanz, R.; González-García, J.; Sánchez-Rodríguez, C.; Cantos, B.; España, P.; Verdaguer, J.; Trinidad Cabezas, A. The anticancer drug cisplatin induces an intrinsic apoptotic pathway inside the inner ear. Br. J. Pharmacol. 2007, 152, 1012–1020. [Google Scholar] [CrossRef]
- Chirino, Y.I.; Pedraza-Chaverri, J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. Am. J. Physiol. Ren. Physiol. 2005, 289, F166–F174. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Dai, H.; Xiong, Z.; Song, Q.; Zou, Z.; Li, M.; Nie, J.; Bai, X.; Chen, Z. Loss of DEPTOR in renal tubules protects against cisplatin-induced acute kidney injury. Cell Death Dis. 2018, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Endoplasmic reticulum stress as a progression factor for kidney injury. Curr. Opin. Pharmacol. 2010, 10, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Peyrou, M.; Hanna, P.E.; Cribb, A.E. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol. Sci. 2007, 99, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Zhen, X.; Zhu, F.; Hu, Z.; Lei, W.; Li, S.; Zha, Y.; Nie, J. Hyperhomocysteinemia exacerbates cisplatin-induced acute kidney injury. Int. J. Biol. Sci. 2017, 13, 219. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Kaufman, R.J. Chapter Twenty Identification and Characterization of Endoplasmic Reticulum Stress-Induced Apoptosis In Vivo. Methods Enzymol. 2008, 442, 395–419. [Google Scholar] [PubMed] [Green Version]
- Liu, H.; Baliga, R. Endoplasmic reticulum stress–associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J. Am. Soc. Nephrol. 2005, 16, 1985–1992. [Google Scholar] [CrossRef]
- Huang, Z.; Guo, F.; Xia, Z.; Liang, Y.; Lei, S.; Tan, Z.; Ma, L.; Fu, P. Activation of GPR120 by TUG891 ameliorated cisplatin-induced acute kidney injury via repressing ER stress and apoptosis. Biomed. Pharmacother. 2020, 126, 110056. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, G.; Zou, P.; Li, X.; Hao, Q.; Jiang, B.; Yang, X.; Hu, Z. Epigallocatechin-3-gallate protects against cisplatin-induced nephrotoxicity by inhibiting endoplasmic reticulum stress-induced apoptosis. Exp. Biol. Med. 2015, 240, 1513–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Guo, F.; Huang, Z.; Feng, Y.; Xia, Z.; Liu, J.; Li, L.; Huang, R.; Lin, L.; Ma, L. 2-Methylquinazoline derivative 23BB as a highly selective histone deacetylase 6 inhibitor alleviated cisplatin-induced acute kidney injury. Biosci. Rep. 2020, 40, BSR20191538. [Google Scholar] [CrossRef]
- Kong, D.; Zhuo, L.; Gao, C.; Shi, S.; Wang, N.; Huang, Z.; Li, W.; Hao, L. Erythropoietin protects against cisplatin-induced nephrotoxicity by attenuating endoplasmic reticulum stress-induced apoptosis. J. Nephrol. 2013, 26, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Liu, G.; Hu, Z.; Li, X.; Yang, X.; Jiang, B.; Li, X. Grape seed proanthocyanidin extract protects from cisplatin-induced nephrotoxicity by inhibiting endoplasmic reticulum stress-induced apoptosis. Mol. Med. Rep. 2014, 9, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Guo, F.; Huang, Z.; Xia, Z.; Liu, J.; Tao, S.; Li, L.; Feng, Y.; Du, X.; Ma, L. Pharmacological and genetic inhibition of fatty acid-binding protein 4 alleviated cisplatin-induced acute kidney injury. J. Cell. Mol. Med. 2019, 23, 6260–6270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuichi, K.; Kaneko, S.; Wada, T. Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin. Exp. Nephrol. 2009, 13, 9–14. [Google Scholar] [CrossRef]
- Liu, X.-Q.; Jin, J.; Li, Z.; Jiang, L.; Dong, Y.-H.; Cai, Y.-T.; Wu, M.-F.; Wang, J.-N.; Ma, T.-T.; Wen, J.-G. Rutaecarpine Derivative Cpd-6c Alleviates Acute Kidney Injury by Targeting PDE4B, a Key Enzyme Mediating inflammation in Cisplatin Nephropathy. Biochem. Pharmacol. 2020, 180, 114132. [Google Scholar] [CrossRef]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbrell, D.A.; Beutler, B. The evolution and genetics of innate immunity. Nat. Rev. Genet. 2001, 2, 256–267. [Google Scholar] [CrossRef]
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Godfroy, J.I., III; Roostan, M.; Moroz, Y.S.; Korendovych, I.V.; Yin, H. Isolated Toll-like receptor transmembrane domains are capable of oligomerization. PloS ONE 2012, 7, e48875. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.K.; Mullen, G.E.; Leifer, C.A.; Mazzoni, A.; Davies, D.R.; Segal, D.M. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 2003, 24, 528–533. [Google Scholar] [CrossRef]
- Butcher, S.K.; O’Carroll, C.E.; Wells, C.A.; Carmody, R.J. Toll-like receptors drive specific patterns of tolerance and training on restimulation of macrophages. Front. Immunol. 2018, 9, 933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Ghosh, S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 2004, 303, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Zou, W.; Sun, Q.; Grandvaux, N.; Julkunen, I.; Hemmi, H.; Yamamoto, M.; Akira, S.; Yeh, W.-C.; Lin, R. Activation of TBK1 and IKKε kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol. 2004, 78, 10636–10649. [Google Scholar]
- Patel, S. Danger-associated molecular patterns (DAMPs): The derivatives and triggers of inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Kurup, S.P.; Tarleton, R.L. Perpetual expression of PAMPs necessary for optimal immune control and clearance of a persistent pathogen. Nat. Commun. 2013, 4, 2616. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, A.; Midwood, K. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, A.; Herrup, E.A.; Warren, H.S.; Trigilio, J.; Shin, H.-S.; Valentine, C.; Hellman, J. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J. Immunol. 2007, 178, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Mäkelä, S.M.; Strengell, M.; Pietilä, T.E.; Österlund, P.; Julkunen, I. Multiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cells. J. Leukoc. Biol. 2009, 85, 664–672. [Google Scholar] [CrossRef]
- Grassin-Delyle, S.; Abrial, C.; Salvator, H.; Brollo, M.; Naline, E.; Devillier, P. The role of toll-like receptors in the production of cytokines by human lung macrophages. J. Innate Immun. 2020, 12, 63–73. [Google Scholar] [CrossRef]
- Zhang, Y.; He, X.; Yu, F.; Xiang, Z.; Li, J.; Thorpe, K.L.; Yu, Z. Characteristic and functional analysis of toll-like receptors (TLRs) in the lophotrocozoan, Crassostrea gigas, reveals ancient origin of TLR-mediated innate immunity. PloS ONE 2013, 8, e76464. [Google Scholar]
- Mulla, M.J.; Myrtolli, K.; Tadesse, S.; Stanwood, N.L.; Gariepy, A.; Guller, S.; Norwitz, E.R.; Abrahams, V.M. Cutting-Edge Report: TLR 10 Plays a Role in Mediating Bacterial Peptidoglycan-Induced Trophoblast Apoptosis. Am. J. Reprod. Immunol. 2013, 69, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Shamsul, H.M.; Hasebe, A.; Iyori, M.; Ohtani, M.; Kiura, K.; Zhang, D.; Totsuka, Y.; Shibata, K.i. The Toll-like receptor 2 (TLR2) ligand FSL-1 is internalized via the clathrin-dependent endocytic pathway triggered by CD14 and CD36 but not by TLR2. Immunology 2010, 130, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zanoni, I.; Cullen, T.W.; Goodman, A.L.; Kagan, J.C. Mechanisms of Toll-like receptor 4 endocytosis reveal a common immune-evasion strategy used by pathogenic and commensal bacteria. Immunity 2015, 43, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Wörnle, M.; Schmid, H.; Banas, B.; Merkle, M.; Henger, A.; Roeder, M.; Blattner, S.; Bock, E.; Kretzler, M.; Gröne, H.-J. Novel role of toll-like receptor 3 in hepatitis C-associated glomerulonephritis. Am. J. Pathol. 2006, 168, 370–385. [Google Scholar] [CrossRef] [Green Version]
- Kropf, P.; Freudenberg, M.A.; Modolell, M.; Price, H.P.; Herath, S.; Antoniazi, S.; Galanos, C.; Smith, D.F.; Müller, I. Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect. Immun. 2004, 72, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Holscher, T.D.; King, A.J.; Hall, F.W.; Kiosses, W.B.; Tobias, P.S.; Mackman, N.; McKay, D.B. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and-independent pathways. J. Immunol. 2007, 178, 6252–6258. [Google Scholar] [CrossRef] [Green Version]
- Conti, F.; Spinelli, F.R.; Truglia, S.; Miranda, F.; Alessandri, C.; Ceccarelli, F.; Bombardieri, M.; Giannakakis, K.; Valesini, G. Kidney expression of toll like receptors in lupus nephritis: Quantification and clinicopathological correlations. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef]
- Ciferska, H.; Horak, P.; Konttinen, Y.T.; Krejci, K.; Tichy, T.; Hermanova, Z.; Zadrazil, J. Expression of nucleic acid binding Toll-like receptors in control, lupus and transplanted kidneys–a preliminary pilot study. Lupus 2008, 17, 580–585. [Google Scholar] [CrossRef]
- Lin, M.; Yiu, W.H.; Wu, H.J.; Chan, L.Y.; Leung, J.C.; Au, W.S.; Chan, K.W.; Lai, K.N.; Tang, S.C. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 86–102. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, T.; Sharma, M.; Yew, K.-H.; Sharma, R.; Duncan, R.S.; Saleem, M.A.; McCarthy, E.T.; Kats, A.; Cudmore, P.A.; Alon, U.S. LPS and PAN-induced podocyte injury in an in vitro model of minimal change disease: Changes in TLR profile. J. Cell Commun. Signal. 2013, 7, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, M.; Ishimoto, T.; Lee, P.Y.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Wymer, D.T.; Yamabe, H.; Mathieson, P.W.; Saleem, M.A. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-κ B-dependent pathway. Nephrol. Dial. Transplant. 2012, 27, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zou, L.; Feng, Y.; Xu, G.; Gong, Y.; Zhao, G.; Ouyang, W.; Thurman, J.M.; Chao, W. Complement factor B production in renal tubular cells and its role in sodium transporter expression during polymicrobial sepsis. Crit. Care Med. 2016, 44, e289. [Google Scholar] [CrossRef] [Green Version]
- Bäckhed, F.; Söderhäll, M.; Ekman, P.; Normark, S.; Richter-Dahlfors, A. Induction of innate immune responses by Escherichia coli and purified lipopolysaccharide correlate with organ-and cell-specific expression of Toll-like receptors within the human urinary tract. Cell. Microbiol. 2001, 3, 153–158. [Google Scholar] [CrossRef]
- Jonassen, J.A.; Kohjimoto, Y.; Scheid, C.R.; Schmidt, M. Oxalate toxicity in renal cells. Urol. Res. 2005, 33, 329–339. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, J.-S.; Han, N.J.; Lee, M.J.; Park, S.-K. Toll-like receptor 4/spleen tyrosine kinase complex in high glucose signal transduction of proximal tubular epithelial cells. Cell. Physiol. Biochem. 2015, 35, 2309–2319. [Google Scholar] [CrossRef]
- Han, S.J.; Li, H.; Kim, M.; Shlomchik, M.J.; Lee, H.T. Kidney proximal tubular TLR9 exacerbates ischemic acute kidney injury. J. Immunol. 2018, 201, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, P.; Hang, L.; Wullt, B.; Irjala, H.; Svanborg, C. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect. Immun. 2004, 72, 3179–3186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimitraki, E.; Tzardi, M.; Bertsias, G.; Sotsiou, E.; Boumpas, D.T. Glomerular expression of toll-like receptor-9 in lupus nephritis but not in normal kidneys: Implications for the amplification of the inflammatory response. Lupus 2009, 18, 831–835. [Google Scholar] [CrossRef]
- González-Guerrero, C.; Cannata-Ortiz, P.; Guerri, C.; Egido, J.; Ortiz, A.; Ramos, A.M. TLR4-mediated inflammation is a key pathogenic event leading to kidney damage and fibrosis in cyclosporine nephrotoxicity. Arch. Toxicol. 2017, 91, 1925–1939. [Google Scholar] [CrossRef] [PubMed]
- Lepenies, J.; Eardley, K.S.; Kienitz, T.; Hewison, M.; Ihl, T.; Stewart, P.M.; Cockwell, P.; Quinkler, M. Renal TLR4 mRNA expression correlates with inflammatory marker MCP-1 and profibrotic molecule TGF-β1 in patients with chronic kidney disease. Nephron Clin. Pract. 2011, 119, c97–c104. [Google Scholar] [CrossRef] [PubMed]
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.; Kirschning, C.J.; Akira, S.; Van der Poll, T.; Weening, J.J.; Florquin, S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Investig. 2005, 115, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Verzola, D.; Cappuccino, L.; D’amato, E.; Villaggio, B.; Gianiorio, F.; Mij, M.; Simonato, A.; Viazzi, F.; Salvidio, G.; Garibotto, G. Enhanced glomerular Toll-like receptor 4 expression and signaling in patients with type 2 diabetic nephropathy and microalbuminuria. Kidney Int. 2014, 86, 1229–1243. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Stokes, J.A.; Corr, M.; Yaksh, T.L. Toll-like receptor signaling regulates cisplatin-induced mechanical allodynia in mice. Cancer Chemother. Pharmacol. 2014, 73, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volarevic, V.; Markovic, B.S.; Jankovic, M.G.; Djokovic, B.; Jovicic, N.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N.; Lukic, M.L. Galectin 3 protects from cisplatin-induced acute kidney injury by promoting TLR-2-dependent activation of IDO1/Kynurenine pathway in renal DCs. Theranostics 2019, 9, 5976. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yan, L.; Zhu, Q.; Shao, F. Puerarin attenuates cisplatin-induced rat nephrotoxicity: The involvement of TLR4/NF-κB signaling pathway. PloS ONE 2017, 12, e0171612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenedeze, M.; Goncalves, G.; Feitoza, C.; Wang, P.; Damiao, M.; Bertocchi, A.; Pacheco-Silva, A.; Camara, N. The role of toll-like receptor 4 in cisplatin-induced renal injury. Transplant. Proc. 2007, 39, 409–411. [Google Scholar] [CrossRef]
- Ramesh, G.; Zhang, B.; Uematsu, S.; Akira, S.; Reeves, W.B. Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am. J. Physiol. Ren. Physiol. 2007, 293, F325–F332. [Google Scholar] [CrossRef] [PubMed]
- Babolmorad, G.; Latif, A.; Pollock, N.M.; Domingo, I.K.; Delyea, C.; Rieger, A.M.; Allison, W.T.; Bhavsar, A.P. Toll-like receptor 4 is activated by platinum and contributes to cisplatin-induced ototoxicity. bioRxiv 2020, e51280. [Google Scholar] [CrossRef]
- Dessing, M.C.; Kers, J.; Damman, J.; Leuvenink, H.G.; Van Goor, H.; Hillebrands, J.-L.; Hepkema, B.G.; Snieder, H.; Van den Born, J.; De Borst, M.H. Toll-like receptor family polymorphisms are associated with primary renal diseases but not with renal outcomes following kidney transplantation. PloS ONE 2015, 10, e0139769. [Google Scholar] [CrossRef]
- Elloumi, N.; Fakhfakh, R.; Abida, O.; Ayadi, L.; Marzouk, S.; Hachicha, H.; Fourati, M.; Bahloul, Z.; Mhiri, M.; Kammoun, K. Relevant genetic polymorphisms and kidney expression of Toll-like receptor (TLR)-5 and TLR-9 in lupus nephritis. Clin. Exp. Immunol. 2017, 190, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Ducloux, D.; Deschamps, M.; Yannaraki, M.; Ferrand, C.; Bamoulid, J.; Saas, P.; Kazory, A.; Chalopin, J.-M.; Tiberghien, P. Relevance of Toll-like receptor-4 polymorphisms in renal transplantation. Kidney Int. 2005, 67, 2454–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Martins, M.; Sameiro-Faria, M.; Ribeiro, S.; Rocha-Pereira, P.; Nascimento, H.; Reis, F.; Miranda, V.; Quintanilha, A.; Belo, L.; Beirão, I. TLR4 and TLR9 polymorphisms effect on inflammatory response in end-stage renal disease patients. Eur. J. Inflamm. 2014, 12, 521–529. [Google Scholar] [CrossRef]
- Farhat, K.; Riekenberg, S.; Heine, H.; Debarry, J.; Lang, R.; Mages, J.; Buwitt-Beckmann, U.; Röschmann, K.; Jung, G.; Wiesmüller, K.H. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J. Leukoc. Biol. 2008, 83, 692–701. [Google Scholar] [CrossRef]
- Guan, Y.; Ranoa, D.R.E.; Jiang, S.; Mutha, S.K.; Li, X.; Baudry, J.; Tapping, R.I. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J. Immunol. 2010, 184, 5094–5103. [Google Scholar] [CrossRef]
- Matsushima, N.; Tanaka, T.; Enkhbayar, P.; Mikami, T.; Taga, M.; Yamada, K.; Kuroki, Y. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. Bmc Genom. 2007, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- De Oliviera Nascimento, L.; Massari, P.; Wetzler, L.M. The role of TLR2 in infection and immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Raby, A.-C.; González-Mateo, G.T.; Williams, A.; Topley, N.; Fraser, D.; López-Cabrera, M.; Labéta, M.O. Targeting Toll-like receptors with soluble Toll-like receptor 2 prevents peritoneal dialysis solution–induced fibrosis. Kidney Int. 2018, 94, 346–362. [Google Scholar] [CrossRef]
- Shen, Q.; Zhang, X.; Li, Q.; Zhang, J.; Lai, H.; Gan, H.; Du, X.; Li, M. TLR2 protects cisplatin-induced acute kidney injury associated with autophagy via PI3K/Akt signaling pathway. J. Cell. Biochem. 2019, 120, 4366–4374. [Google Scholar] [CrossRef]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef]
- Myeku, N.; Figueiredo-Pereira, M.E. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy association With sequestosome 1/p62. J. Biol. Chem. 2011, 286, 22426–22440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherra III, S.J.; Kulich, S.M.; Uechi, G.; Balasubramani, M.; Mountzouris, J.; Day, B.W.; Chu, C.T. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 2010, 190, 533–539. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chen, S.; Cao, Y.; Zhou, P.; Chen, Z.; Cheng, K. Discovery of novel small molecule TLR4 inhibitors as potent anti-inflammatory agents. Eur. J. Med. Chem. 2018, 154, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramesh, G.; Uematsu, S.; Akira, S.; Reeves, W.B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol. 2008, 19, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola virus glycoprotein induces an innate immune response in vivo via TLR4. Front. Microbiol. 2017, 8, 1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashidi, N.; Mirahmadian, M.; Jeddi-Tehrani, M.; Rezania, S.; Ghasemi, J.; Kazemnejad, S.; Mirzadegan, E.; Vafaei, S.; Kashanian, M.; Rasoulzadeh, Z. Lipopolysaccharide-and lipoteichoic acid-mediated pro-inflammatory cytokine production and modulation of TLR2, TLR4 and MyD88 expression in human endometrial cells. J. Reprod. Infertil. 2015, 16, 72. [Google Scholar]
- Thomas, P.G.; Carter, M.R.; Atochina, O.; Da’Dara, A.A.; Piskorska, D.; McGuire, E.; Harn, D.A. Maturation of dendritic cell 2 phenotype by a helminth glycan uses a Toll-like receptor 4-dependent mechanism. J. Immunol. 2003, 171, 5837–5841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.-M.; Park, G.; Lee, S.H.; Seo, C.-S.; Shin, H.-K.; Oh, D.-S. Assessing the recovery from prerenal and renal acute kidney injury after treatment with single herbal medicine via activity of the biomarkers HMGB1, NGAL and KIM-1 in kidney proximal tubular cells treated by cisplatin with different doses and exposure times. BMC Complementary Altern. Med. 2017, 17, 544. [Google Scholar]
- Ullah, M.; Liu, D.D.; Rai, S.; Concepcion, W.; Thakor, A.S. HSP70-Mediated NLRP3 Inflammasome Suppression Underlies Reversal of Acute Kidney Injury Following Extracellular Vesicle and Focused Ultrasound Combination Therapy. Int. J. Mol. Sci. 2020, 21, 4085. [Google Scholar] [CrossRef] [PubMed]
- Rachmawati, D.; Bontkes, H.J.; Verstege, M.I.; Muris, J.; Von Blomberg, B.M.E.; Scheper, R.J.; Van Hoogstraten, I.M. Transition metal sensing by Toll-like receptor-4: Next to nickel, cobalt and palladium are potent human dendritic cell stimulators. Contact Dermat. 2013, 68, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Fenhammar, J.; Rundgren, M.; Hultenby, K.; Forestier, J.; Taavo, M.; Kenne, E.; Weitzberg, E.; Eriksson, S.; Ozenci, V.; Wernerson, A. Renal effects of treatment with a TLR4 inhibitor in conscious septic sheep. Crit. Care 2014, 18, 488. [Google Scholar]
- Fenhammar, J.; Rundgren, M.; Forestier, J.; Kalman, S.; Eriksson, S.; Frithiof, R. Toll-like receptor 4 inhibitor TAK-242 attenuates acute kidney injury in endotoxemic sheep. Anesthesiol. J. Am. Soc. Anesthesiol. 2011, 114, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Ichikawa, T.; Ii, M.; Sunamoto, M.; Itoh, K.; Tamura, N.; Kitazaki, T. Discovery of novel and potent small-molecule inhibitors of NO and cytokine production as antisepsis agents: Synthesis and biological activity of alkyl 6-(N-substituted sulfamoyl) cyclohex-1-ene-1-carboxylate. J. Med. Chem. 2005, 48, 7457–7467. [Google Scholar] [CrossRef] [PubMed]
- Takashima, K.; Matsunaga, N.; Yoshimatsu, M.; Hazeki, K.; Kaisho, T.; Uekata, M.; Hazeki, O.; Akira, S.; Iizawa, Y.; Ii, M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br. J. Pharmacol. 2009, 157, 1250–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashani, B.; Zandi, Z.; Karimzadeh, M.R.; Bashash, D.; Nasrollahzadeh, A.; Ghaffari, S.H. Blockade of TLR4 using TAK-242 (resatorvid) enhances anti-cancer effects of chemotherapeutic agents: A novel synergistic approach for breast and ovarian cancers. Immunol. Res. 2019, 67, 505–516. [Google Scholar] [CrossRef]
- Pedersen, G.; Andresen, L.; Matthiessen, M.; Rask-Madsen, J.; Brynskov, J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin. Exp. Immunol. 2005, 141, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Abel, K.; Lantz, K.; Krieg, A.M.; McChesney, M.B.; Miller, C.J. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J. Virol. 2005, 79, 14355–14370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Peng, S.L. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2006, 54, 336–342. [Google Scholar] [CrossRef]
- Machida, H.; Ito, S.; Hirose, T.; Takeshita, F.; Oshiro, H.; Nakamura, T.; Mori, M.; Inayama, Y.; Yan, K.; Kobayashi, N. Expression of Toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis. Nephrol. Dial. Transplant. 2010, 25, 2430–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallavia, B.; Liu, F.; Lefrançais, E.; Cleary, S.J.; Kwaan, N.; Tian, J.J.; Magnen, M.; Sayah, D.M.; Soong, A.; Chen, J. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. Am. J. Respir. Cell Mol. Biol. 2020, 62, 364–372. [Google Scholar] [CrossRef]
- Busconi, L.; Bauer, J.W.; Tumang, J.R.; Laws, A.; Perkins-Mesires, K.; Tabor, A.S.; Lau, C.; Corley, R.B.; Rothstein, T.L.; Lund, F.E. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J. Immunol. 2007, 179, 7397–7405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, J.; Sun, W.; Chiosis, G.; Leifer, C.A. Heat shock protein gp96 regulates Toll-like receptor 9 proteolytic processing and confrimational stability. Biochem. Biophy. Res. Commun. 2012, 421, 780–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Chen, X. Expression of high-mobility group box 1 protein (HMGB1) and toll-like receptor 9 (TLR9) in retinas of diabetic rats. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 3115. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Kukita, K.; Kutomi, G.; Okuya, K.; Asanuma, H.; Tabeya, T.; Naishiro, Y.; Yamamoto, M.; Takahashi, H.; Torigoe, T. Heat shock protein 90 associates with Toll-like receptors 7/9 and mediates self-nucleic acid recognition in SLE. Eur. J. Immunol. 2015, 45, 2028–2041. [Google Scholar] [CrossRef] [PubMed]
- Urry, Z.; Xystrakis, E.; Richards, D.F.; McDonald, J.; Sattar, Z.; Cousins, D.J.; Corrigan, C.J.; Hickman, E.; Brown, Z.; Hawrylowicz, C.M. Ligation of TLR9 induced on human IL-10–secreting Tregs by 1α, 25-dihydroxyvitamin D3 abrogates regulatory function. J. Clin. Investig. 2009, 119, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.-W.; Yong, K.-C.; Lien, Y.-H.H. Pharmacologic recruitment of regulatory T cells as a therapy for ischemic acute kidney injury. Kidney Int. 2012, 81, 983–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humanes, B.; Camaño, S.; Lara, J.M.; Sabbisetti, V.; González-Nicolás, M.Á.; Bonventre, J.V.; Tejedor, A.; Lázaro, A. Cisplatin-induced renal inflammation is ameliorated by cilastatin nephroprotection. Nephrol. Dial. Transplant. 2017, 32, 1645–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szlosarek, P.W.; Balkwill, F.R. Tumour necrosis factor α: A potential target for the therapy of solid tumours. Lancet Oncol. 2003, 4, 565–573. [Google Scholar] [CrossRef]
- Wu, X.; Wu, M.-Y.; Jiang, M.; Zhi, Q.; Bian, X.; Xu, M.-D.; Gong, F.-R.; Hou, J.; Tao, M.; Shou, L.-M. TNF-α sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? 1. Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-κB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 2016, 240, 105–113. [Google Scholar] [CrossRef]
- Zhou, J.; Fan, Y.; Zhong, J.; Huang, Z.; Huang, T.; Lin, S.; Chen, H. TAK1 mediates excessive autophagy via p38 and ERK in cisplatin-induced acute kidney injury. J. Cell. Mol. Med. 2018, 22, 2908–2921. [Google Scholar] [CrossRef] [Green Version]
- Mihaly, S.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.-J.; Lu, L.; Klein, C.L.; Dinarello, C.A. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 2007, 322, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Kohda, Y.; Chiao, H.; Wang, Y.; Hu, X.; Hewitt, S.M.; Miyaji, T.; Mcleroy, P.; Nibhanupudy, B.; Li, S. Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int. 2001, 60, 2118–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.H.; Oh, D.-J.; Dursun, B.; He, Z.; Hoke, T.S.; Faubel, S.; Edelstein, C.L. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J. Pharmacol. Exp. Ther. 2008, 324, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Tadagavadi, R.K.; Reeves, W.B. Endogenous IL-10 attenuates cisplatin nephrotoxicity: Role of dendritic cells. J. Immunol. 2010, 185, 4904–4911. [Google Scholar] [CrossRef] [Green Version]
- Jones, V.S.; Huang, R.-Y.; Chen, L.-P.; Chen, Z.-S.; Fu, L.; Huang, R.-P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Akcay, A.; Nguyen, Q.; He, Z.; Turkmen, K.; Lee, D.W.; Hernando, A.A.; Altmann, C.; Toker, A.; Pacic, A.; Ljubanovic, D.G. IL-33 exacerbates acute kidney injury. J. Am. Soc. Nephrol. 2011, 22, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Scott, H.; Jones, D.; Pui, C. The tumor lysis syndrome. N. Engl. J. Med. 2011, 364, 1844–1854. [Google Scholar]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro-and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennen, P.; Altmann, C.; Kaufman, J.; Klein, C.L.; Andres-Hernando, A.; Ahuja, N.H.; Edelstein, C.L.; Cadnapaphornchai, M.A.; Keniston, A.; Faubel, S. Urine interleukin-6 is an early biomarker of acute kidney injury in children undergoing cardiac surgery. Crit. Care 2010, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mitazaki, S.; Kato, N.; Suto, M.; Hiraiwa, K.; Abe, S. Interleukin-6 deficiency accelerates cisplatin-induced acute renal failure but not systemic injury. Toxicology 2009, 265, 115–121. [Google Scholar] [CrossRef]
- Suchi, K.; Fujiwara, H.; Okamura, S.; Okamura, H.; Umehara, S.; Todo, M.; Furutani, A.; Yoneda, M.; Shiozaki, A.; Kubota, T. Overexpression of Interleukin-6 suppresses cisplatin-induced cytotoxicity in esophageal squamous cell carcinoma cells. Anticancer Res. 2011, 31, 67–75. [Google Scholar] [PubMed]
- Reeves, W.B. Innate Immunity in Nephrotoxic Acute Kidney Injury. Trans. Am. Clin. Climatol. Assoc. 2019, 130, 33. [Google Scholar] [PubMed]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.B.; Chen, Y.; Gong, Y.X.; Gao, M.; Zhang, Y.; Wang, G.H.; Tang, R.N.; Liu, H.; Liu, B.C.; Ma, K.L. Activation of the CXCL16/CXCR6 pathway by inflammation contributes to atherosclerosis in patients with end-stage renal disease. Int. J. Med. Sci. 2016, 13, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehr, A.; Tacke, F. The roles of CXCL16 and CXCR6 in liver inflammation and fibrosis. Curr. Pathobiol. Rep. 2015, 3, 283–290. [Google Scholar] [CrossRef]
- Lehrke, M.; Millington, S.C.; Lefterova, M.; Cumaranatunge, R.G.; Szapary, P.; Wilensky, R.; Rader, D.J.; Lazar, M.A.; Reilly, M.P. CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J. Am. Coll. Cardiol. 2007, 49, 442–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Garcia, G.E.; Truong, L.D.; Li, P.; Zhang, P.; Johnson, R.J.; Wilson, C.B.; Feng, L. Inhibition of CXCL16 attenuates inflammatory and progressive phases of anti-glomerular basement membrane antibody-associated glomerulonephritis. Am. J. Pathol. 2007, 170, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Ma, Z.; Peng, H.; He, L.; Hu, Z.; Wang, Y. CXCL16 deficiency attenuates renal injury and fibrosis in salt-sensitive hypertension. Sci. Rep. 2016, 6, 28715. [Google Scholar] [CrossRef] [PubMed]
- Wehr, A.; Baeck, C.; Ulmer, F.; Gassler, N.; Hittatiya, K.; Luedde, T.; Neumann, U.P.; Trautwein, C.; Tacke, F. Pharmacological inhibition of the chemokine CXCL16 diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. PloS ONE 2014, 9, e112327. [Google Scholar] [CrossRef] [PubMed]
- Sawant, K.V.; Poluri, K.M.; Dutta, A.K.; Sepuru, K.M.; Troshkina, A.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci. Rep. 2016, 6, 33123. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, A.; Sawant, K.V.; Sarmiento, J.; Navarro, J.; Rajarathnam, K. Chemokine CXCL1 dimer is a potent agonist for the CXCR2 receptor. J. Biol. Chem. 2013, 288, 12244–12252. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, G.R.; Okusa, M.D. Role of leukocytes in the pathogenesis of acute kidney injury. Crit. Care 2012, 16, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, T.F. Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 2014, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Leenaars, C.H.; Kouwenaar, C.; Stafleu, F.R.; Bleich, A.; Ritskes-Hoitinga, M.; De Vries, R.B.; Meijboom, F.L. Animal to human translation: A systematic scoping review of reported concordance rates. J. Transl. Med. 2019, 17, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Skrypnyk, N.I.; Siskind, L.J.; Faubel, S.; De Caestecker, M.P. Bridging translation for acute kidney injury with better preclinical modeling of human disease. Am. J. Physiol. Ren. Physiol. 2016, 310, F972–F984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirrakhimov, A.E.; Ali, A.M.; Khan, M.; Barbaryan, A. Tumor lysis syndrome in solid tumors: An up to date review of the literature. Rare Tumors 2014, 6, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Bégin, A.-M.; Monfette, M.-L.; Boudrias-Dalle, É.; Lavallée, E.; Samouelian, V.; Soulières, D.; Chagnon, M.; Fournier, M.-A.; Letarte, N.; Adam, J.-P. Effect of mannitol on acute kidney injury induced by cisplatin. Supportive Care Cancer 2021, 29, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Makimoto, G.; Hotta, K.; Oze, I.; Ninomiya, K.; Ninomiya, T.; Kubo, T.; Ohashi, K.; Ichihara, E.; Rai, K.; Tabata, M. Randomized phase II study comparing mannitol with furosemide for the prevention of cisplatin-induced renal toxicity in advanced non-small cell lung cancer. OLCSG1406 Trial 2019, 72, 319–323. [Google Scholar] [CrossRef]
- Cvitkovic, E.; Spaulding, J.; Bethune, V.; Martin, J.; Whitmore, W.F. Improvement of Cis-dichlorodiammineplatinum (NSC 119875): Therapeutic index in an animal model. Cancer 1977, 39, 1357–1361. [Google Scholar] [CrossRef]
- McKibbin, T.; Cheng, L.L.; Kim, S.; Steuer, C.E.; Owonikoko, T.K.; Khuri, F.R.; Shin, D.M.; Saba, N.F. Mannitol to prevent cisplatin-induced nephrotoxicity in patients with squamous cell cancer of the head and neck (SCCHN) receiving concurrent therapy. Supportive Care Cancer 2016, 24, 1789–1793. [Google Scholar] [CrossRef]
- Phitakwatchara, W.; Reungweteattana, T.; Kananuraks, S.; Pathumarak, A.; Assanatham, M.; Nongnuch, A. Preloading Magnesium Attenuate Cisplatin-Induced Nephrotoxicity: SP241. Nephrol. Dial. Transplant. 2017, 32. [Google Scholar]
- Ghadrdan, E.; Sadighi, S.; Ebrahimpour, S.; Abdollahi, A.; Hadjibabaei, M.; Gholami, K.; Jahangard-Rafsanjani, Z. The effect of melatonin on cisplatin-induced nephrotoxicity: A pilot, randomized, double-blinded, placebo-controlled clinical trial. Eur. J. Integr. Med. 2020, 34, 101065. [Google Scholar] [CrossRef]
- Baek, S.H.; Kim, S.H.; Kim, J.W.; Kim, Y.J.; Lee, K.-W.; Na, K.Y. Effects of a DPP4 inhibitor on cisplatin-induced acute kidney injury: Study protocol for a randomized controlled trial. Trials 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hamamsy, M.; Kamal, N.; Bazan, N.S.; El Haddad, M. Evaluation of the effect of acetazolamide versus mannitol on cisplatin-induced nephrotoxicity, a pilot study. Int. J. Clin. Pharm. 2018, 40, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, F.; Sadighi, S.; Dashti-Khavidaki, S.; Shahi, F.; Mirzania, M.; Abdollahi, A.; Ghahremani, M.H. Effect of silymarin administration on cisplatin nephrotoxicity: Report from a pilot, randomized, double-blinded, placebo-controlled clinical trial. Phytother. Res. 2015, 29, 1046–1053. [Google Scholar] [CrossRef]
- Kemp, G.; Rose, P.; Lurain, J.; Berman, M.; Manetta, A.; Roullet, B.; Homesley, H.; Belpomme, D.; Glick, J. Amifostine pretreatment for protection against cyclophosphamide-induced and cisplatin-induced toxicities: Results of a randomized control trial in patients with advanced ovarian cancer. J. Clin. Oncol. 1996, 14, 2101–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nematbakhsh, M.; Pezeshki, Z.; Jazi, F.E.; Mazaheri, B.; Moeini, M.; Safari, T.; Azarkish, F.; Moslemi, F.; Maleki, M.; Rezaei, A. Cisplatin-induced nephrotoxicity; protective supplements and gender differences. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 295. [Google Scholar]
- Beeler, B.; Delacruz, W.P.; Flynt, F.L.; Terrazzino, S.; Hullinger, N.; Aden, J.; Byrd, T.; King, M.M.; Nelson, D.A. Saline alone vs saline plus mannitol hydration for the prevention of acute cisplatin nephrotoxicity: A randomized trial. J. Clin. Oncol. 2018, 36, 242. [Google Scholar] [CrossRef]
- Ruggiero, A.; Rizzo, D.; Trombatore, G.; Maurizi, P.; Riccardi, R. The ability of mannitol to decrease cisplatin-induced nephrotoxicity in children: Real or not? Cancer Chemother. Pharmacol. 2016, 77, 19–26. [Google Scholar] [CrossRef]
- Komaki, K.; Kusaba, T.; Tanaka, M.; Kado, H.; Shiotsu, Y.; Matsui, M.; Shiozaki, A.; Nakano, H.; Ishikawa, T.; Fujiwara, H. Lower blood pressure and risk of cisplatin nephrotoxicity: A retrospective cohort study. BMC Cancer 2017, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Gammune, T.M.; Kubatka, P.; Caprnda, M.; Fedotova, J.; Radonak, J.; Kruzliak, P.; Zulli, A. Imipridone enhances vascular relaxation via FOXO1 pathway. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1816–1823. [Google Scholar] [CrossRef]
- Mercantepe, F.; Mercantepe, T.; Topcu, A.; Yılmaz, A.; Tumkaya, L. Protective effects of amifostine, curcumin, and melatonin against cisplatin-induced acute kidney injury. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 915–931. [Google Scholar] [CrossRef] [PubMed]







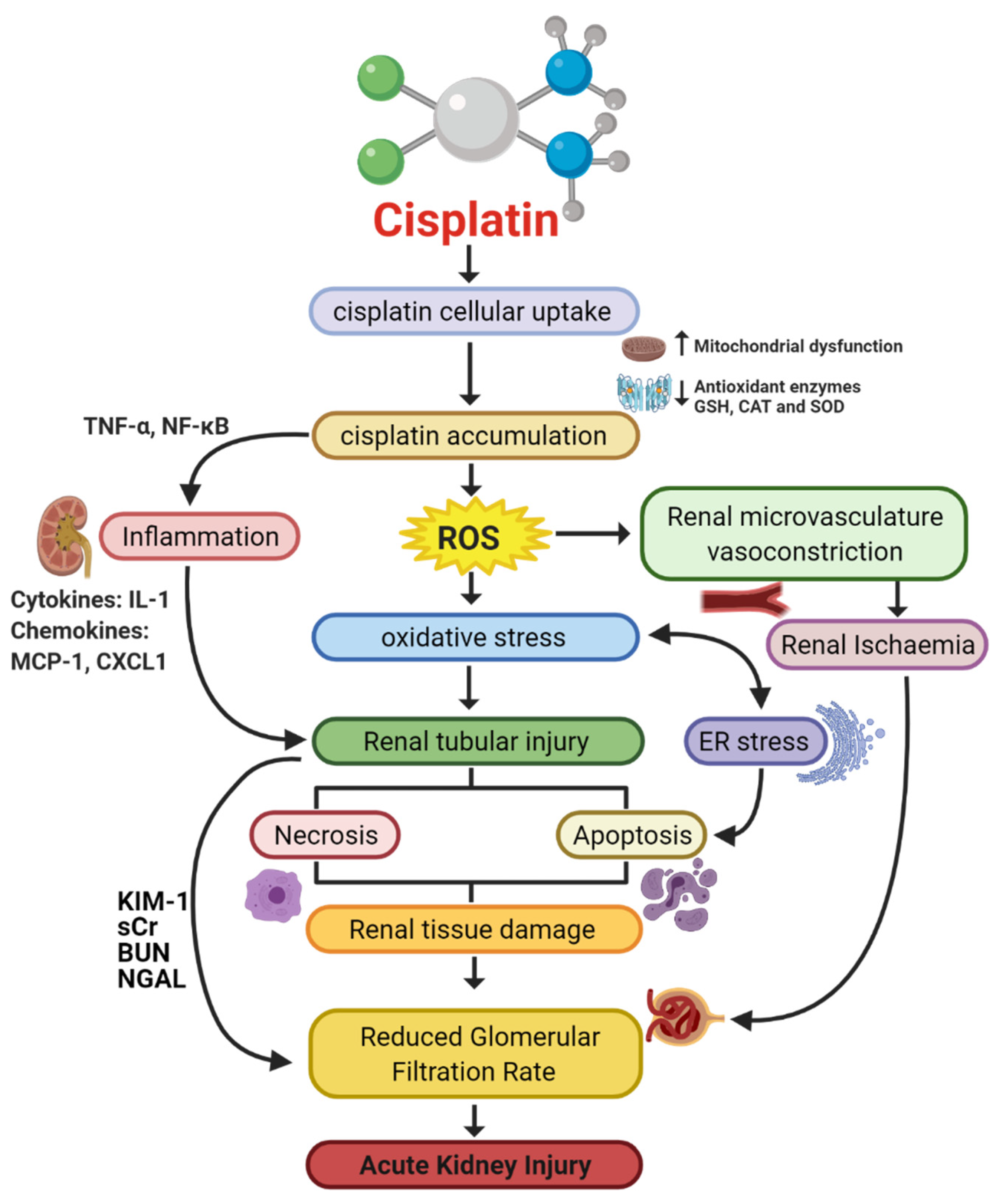{kind=link}
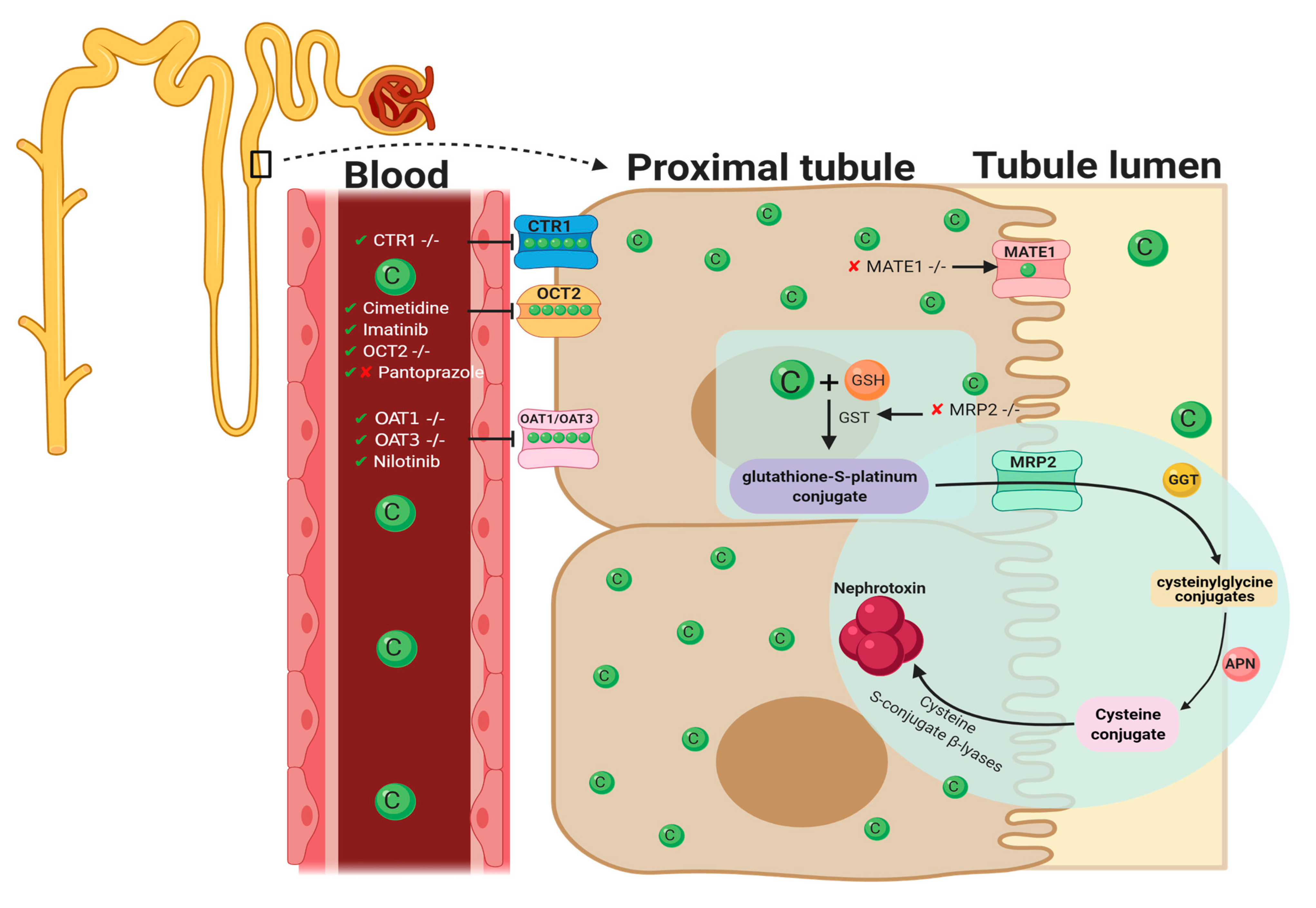{kind=link}
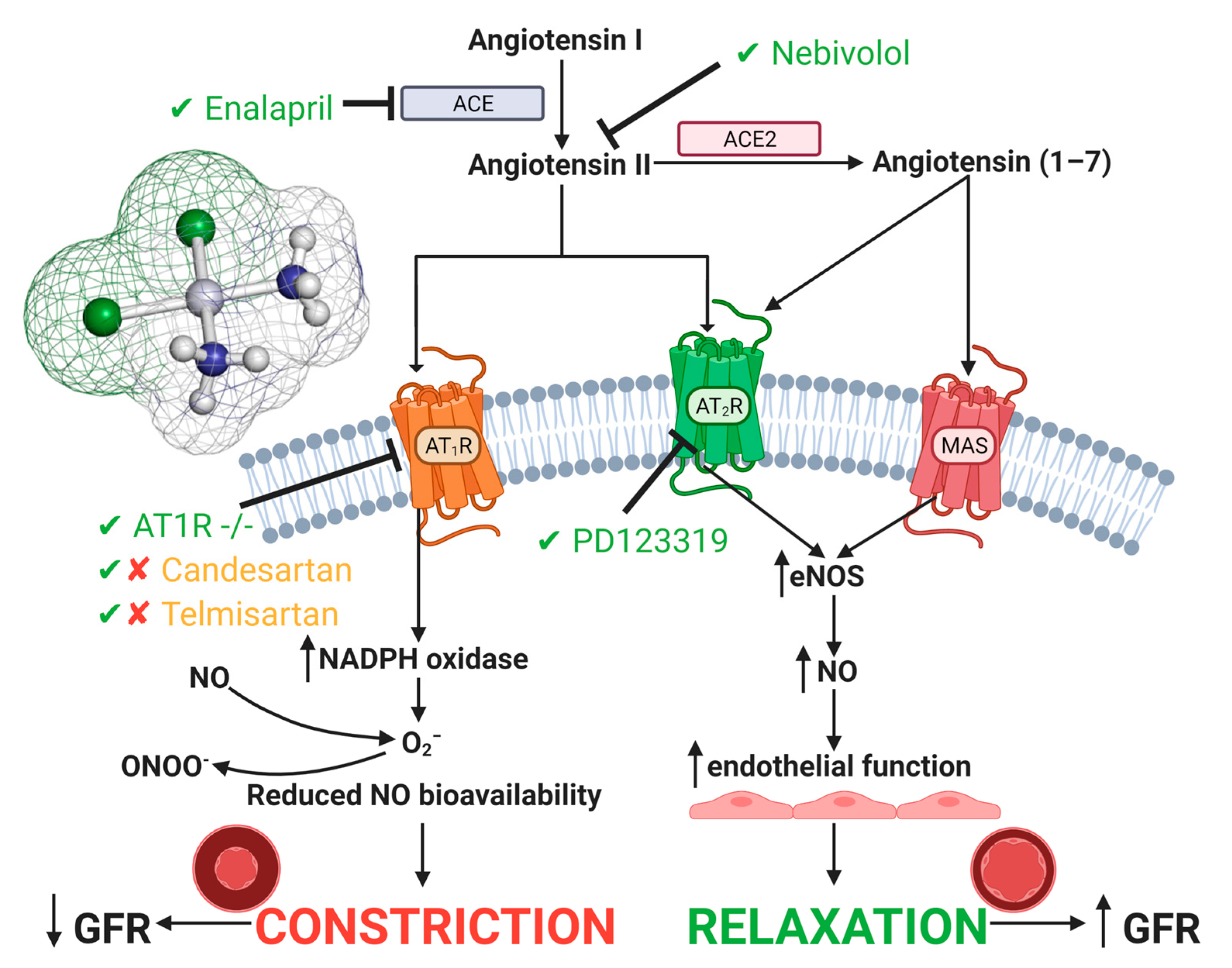{kind=link}
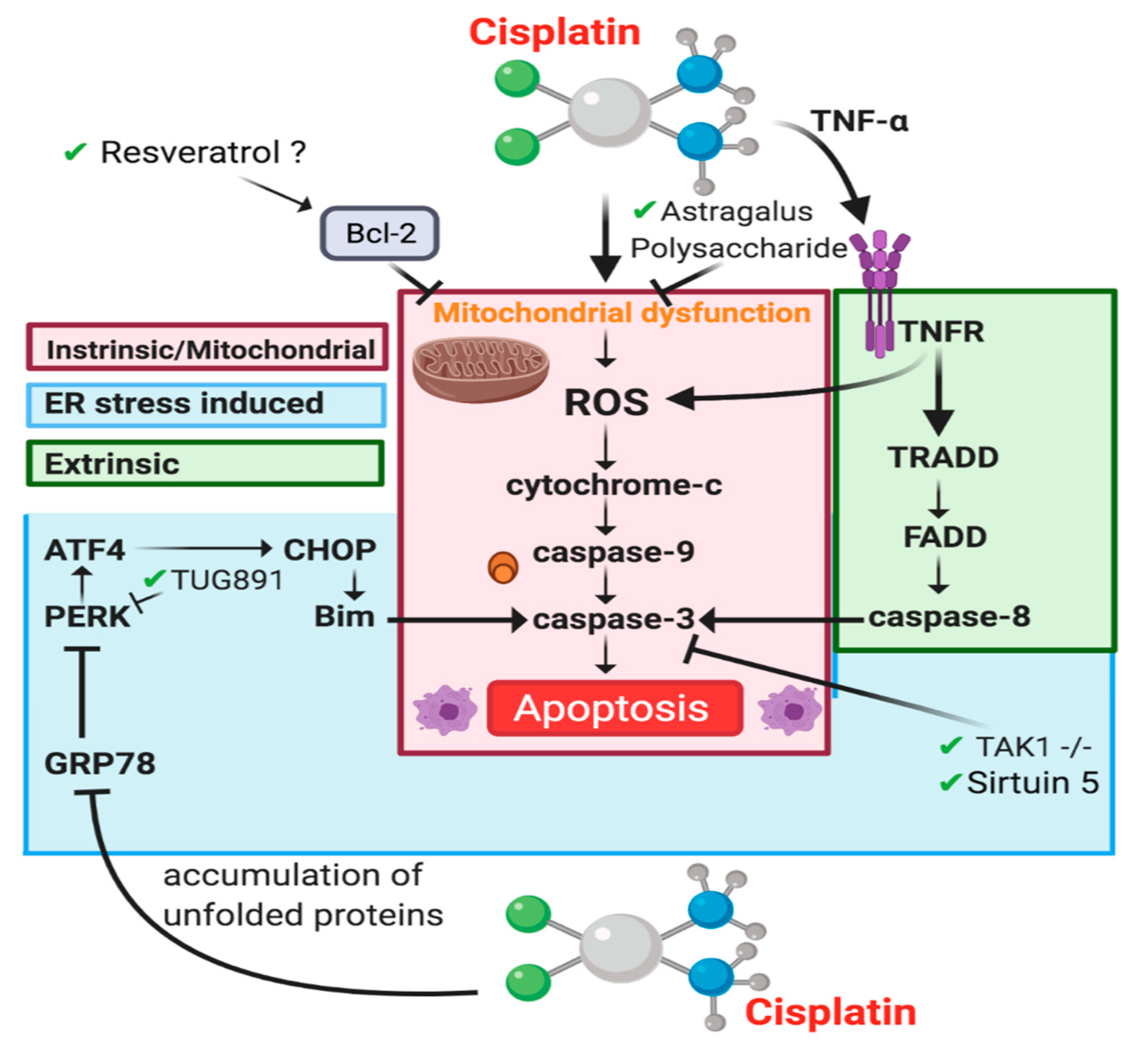{kind=link}
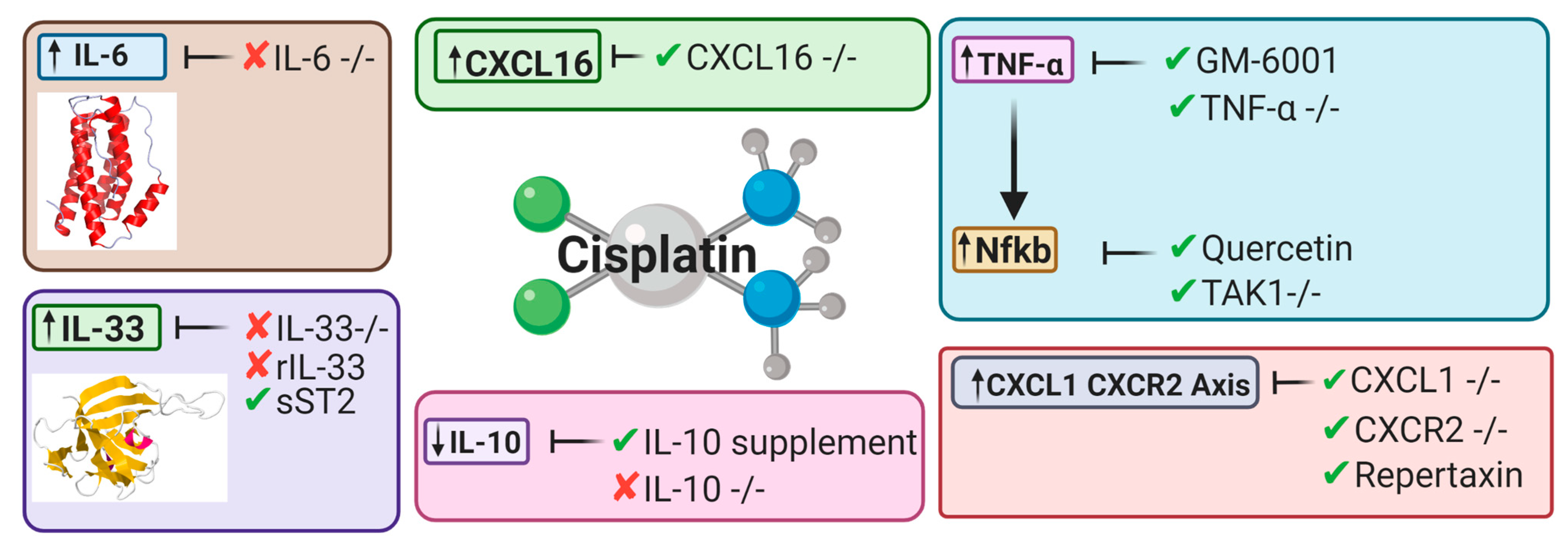{kind=link}
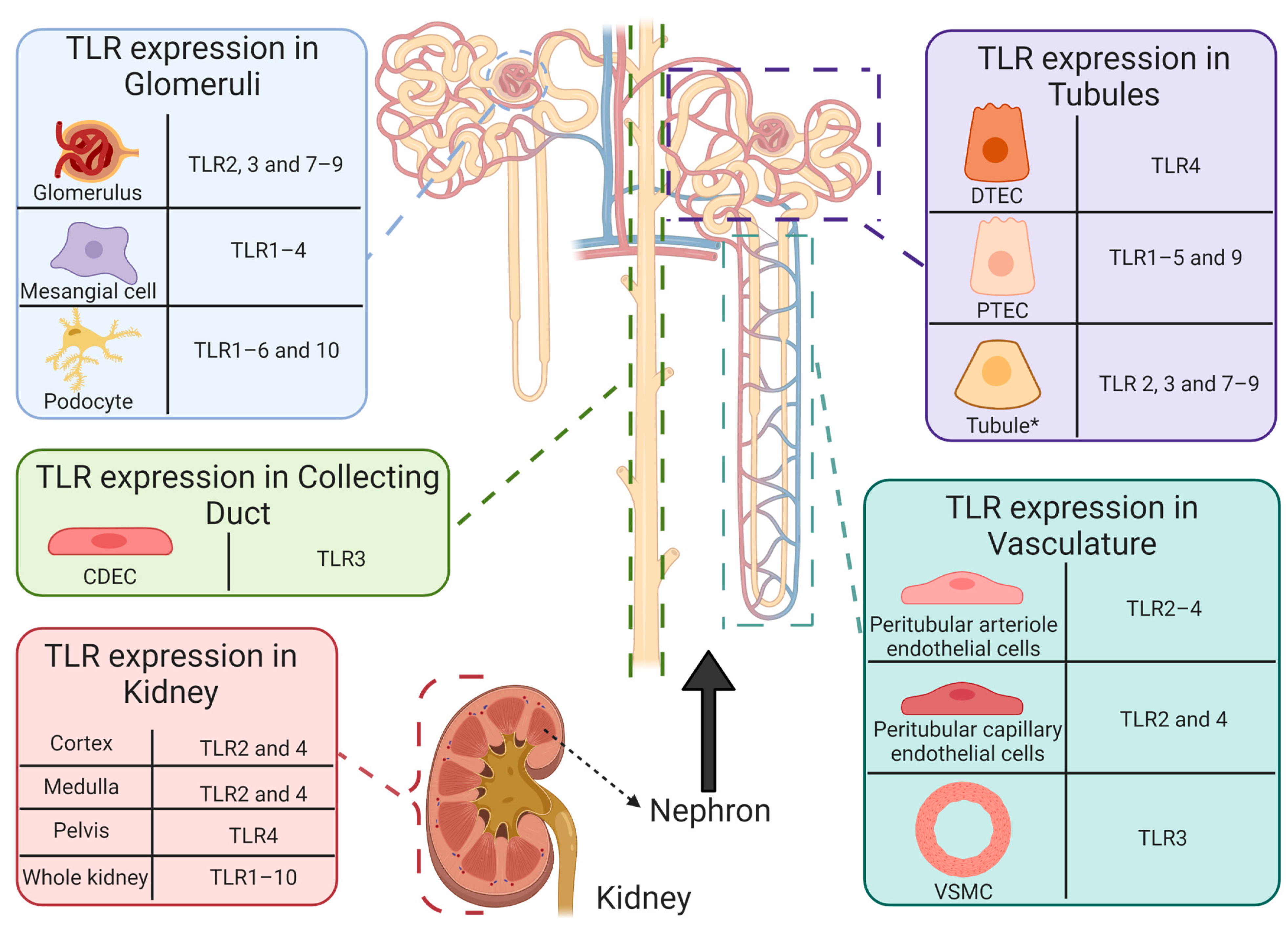{kind=link}
| Drug | Mechanism of Action | Findings | In Vitro | In Vivo | Reference |
|---|---|---|---|---|---|
| Aucubin | Anti-inflammatory | ↓ Markers of oxidative stress (HO-1 and 4-HNE) ↓ Apoptosis (caspase-3, caspase-9 and PARP) | – | BALB/c mice | [38] |
| Curcumin | Anti-inflammatory, Antioxidative, oxygen-free radical scavenging, antifibrotic, and anticancer activities | ↓ Tubular Injury ↓ BUN ↓ sCr (rats) | – | C57BL/6J mice/rats | [39,40] |
| Dexmedetomidine | Antiapoptotic via α2AR/PI3K/AKT pathway | ↑ Body weight and renal index ↓ Tubular epithelial cell apoptosis ↓ Expression of GRP78, CHOP and Caspase-12 | – | Sprague Dawley Rats | [41] |
| Etoricoxib | Anti-inflammatory | ↓ Inflammation (iNOS) ↓ Apoptosis (BAX) No changes to creatinine, BUN, GSH, and MDA | – | Rats | [40] |
| Eugenol | Antioxidant and anti-inflammatory properties | ↓ sCr and BUN ↓ PAS tubular injury score ↓ cytoplasmic vacuolization of proximal tubular cells | – | BALB/c mice | [42] |
| Ferrostatin-1 | Inhibits Ferroptotic cell death | ↓ sCr and BUN ↓ apoptosis (TUNEL stain) ↓ Tubular injury score (H&E) ↓ Lipid peroxidation | – | C57BL/6J mice | [43] |
| Isoorientin | Anti-inflammatory, antioxidant | ↓ ROS generation ↓ Apoptosis ↓ Inflammation | mTECs | Nrf2−/− | [44] |
| Monotropein | Antioxidant, anti-inflammatory and antiapoptotic | ↓ Tubular injury ↓ markers of oxidative stress ↓ markers of apoptosis ↓ BUN, no reduction in sCr | – | BALB/c mice | [45] |
| Paricalcitol | Synthetic vitamin D deficiency | ↓ MDA (HK-2 cells) ↓ Cell death (HK-2 cells) ↓ sCr and BUN (WT mouse) ↓ Tissue Injury (WT mouse) | HK-2 cells | WT mice | [43] |
| Quercetin | Anti-inflammatory | ↓ sCr and BUN ↓ mRNA expression of IL-1β, IL-6, TNF-α ↓ reduced tubular necrosis score ↓ activity of Syk/NF-κB | – | C57BL/6J mice | [46] |
| Genetic Deletion | Mechanism of Action | Results | Knockout Model | Reference |
|---|---|---|---|---|
| CXCL16 | Antiapoptosis and anti-inflammatory | ↓ Apoptosis of tubular cells ↓ Caspase-3 activation ↓ inhibition of macrophage and T cell infiltration | CXCL16 −/− mice C57BL/6J background WT | [47] |
| CYP2e1 | Antioxidant | ↓ ROS ↓ BUN ↓ sCr ↑ creatinine clearance | CYP2e1 −/− mice 129/sv background WT | [48] |
| IL-6 | Antioxidant | ↑ 4-HNE ↓ SOD1 ↓ SOD2 (no significance) ↑ ERK phosphorylation ↑ COX-2 | IL-6 −/− mice C57BL/6J background WT | [49] |
| IL-33 | Pro-inflammatory | ↑ BUN ↑ sCr ↑ NGAL No attenuation in ATN and tubular apoptosis scores ↓ tumor weight, volume, and growth ↑ Cisplatin efficacy | IL-33 −/− mice C57BL/6J background WT | [50] |
| NLRP3 | Unknown | No change to BUN, sCr, ATN score and tubular apoptosis score. | NLRP3 −/− mice C57BL/6J background WT | [51] |
| PARP-1 | Anti-inflammatory, antioxidant and antinitrative | ↓ BUN ↓ sCr ↓ PAS tubular injury score | PARP-1 −/− mice C57BL/6J background WT | [52] |
| T cell | Pro-inflammatory | ↑ cisplatin administration survival rate ↓ sCr ↓ tubular injury score ↓ TNF-α | nu/nu mice | [53] |
| TAK1 | Antiapoptotic, Anti-inflammatory | ↓ Apoptosis of tubular cells ↓ Caspase-3 activation ↓ reduced mRNA expression of IL-6, TNF-α, MCP-1 and MIP-2 ↓ JNK phosphorylation | PT-TAK1 −/− mice | [54] |
| TLR-2 | Inflammatory response | ↑ BUN ↑ sCr ↑ tissue injury score | TLR2 −/− | [55] |
| TLR4 | Anti-inflammatory response | ↓ BUN ↓ sCr ↓ tissue injury index ↑ IL-4 and IL-10 | TLR4 −/− | [55] |
| TLR-9 | Pro-inflammatory | No significant change to either serum urea or tubular injury score. | TLR-9 −/− | [56] |
| TNF-α | Potentially anti-inflammatory | ↓ BUN ↓ tubular necrosis score | TNF-α −/− | [33] |
| Toll-Like Receptor | Location | Primary Pathogen (s) |
|---|---|---|
| 1 | Extracellular | Gram-positive bacterium Fungus Mycobacterium |
| 2 | Extracellular | Fungus Gram-positive bacterium Mycobacterium |
| 3 | Intracellular | Double-stranded virus |
| 4 | Extracellular | Gram-negative bacterium |
| 5 | Extracellular | Flagellum |
| 6 | Extracellular | Gram-positive bacterium Fungus |
| 7 | Intracellular | Single-stranded virus |
| 8 | Intracellular | Virus |
| 9 | Intracellular | Bacterium |
| 10 | Extracellular | Gram-positive bacterium |
| Name of Trial | Status | Year | Results | Clinical Trial Identifier/ Reference |
|---|---|---|---|---|
| The Effect of Intravenous Mannitol Plus Saline on the Prevention of Cisplatin-induced Nephrotoxicity: A Randomized, Double-blind, Placebo Controlled Trial (MACIN) | Recruiting | 2020 | TBA | NCT04251689 |
| The effect of melatonin on cisplatin-induced nephrotoxicity: A pilot, randomized, double-blinded, placebo-controlled clinical trial | Completed | 2020 | Reduced KIM-1/creatinine and NGAL/creatinine ratios indicative of reduced AKI. patients treated with melatonin showed reduced AKI episodes compared to the placebo group. This however was non-significant suggested to be due to the small sample size. | [305] |
| Mesenchymal Stem Cells in Cisplatin-Induced Acute Renal Failure in Patients with Solid Organ Cancers | Withdrawn | 2011, updated 2018 | Study withdraw as patients failed to develop acute renal failure (Key criterion for the study) | NCT01275612 |
| Preloading Magnesium Attenuate Cisplatin-induced Nephrotoxicity | Completed | 2015, updated 2019 | —Beneficial in prevention of cisplatin-induced acute kidney disease No statistical significance in the prevention of CIAKI. | NCT02481518 [304] |
| Effects of DPP4 Inhibitor on Cisplatin-Induced Acute Kidney Injury | Unknown | NCT02250872 [306] | ||
| Preventing Nephrotoxicity and Ototoxicity from Osteosarcoma Therapy | Completed | 2013, updated 2020 | —serum creatinine/biomarkers of AKI (KIM-1/NGAL) were not improved by pantoprazole —The study concluded that pantoprazole was unable to ameliorate cisplatin-induced AKI.- | NCT01848457 [75] |
| Randomized phase II feasibility study of mannitol or furosemide hydration in moderate dose of cisplatin-based chemotherapy with short hydration for advanced non-small cell lung cancer | Completed | 2014, updated 2021 | No significant difference in renal toxicity compared to treatment without mannitol | UMIN000015293 [301] |
| Evaluation of the Effect of Acetazolamide, Mannitol and N-acetylcysteine on Cisplatin-Induced Nephrotoxicity | Completed | 2016, updated 2017 | TBA | NCT02760901 [307] |
| Effect of Silymarin Administration on Cisplatin Nephrotoxicity: Report from A Pilot, Randomized, Double-Blinded, Placebo-Controlled Clinical Trial | Completed | 2013, updated 2015 | NGAL/creatinine ratio was the same between silymarin and placebo groups Overall conclusion of the study is that silymarin was not effective against cisplatin-induced AKI. | [308] |
| Amifostine pretreatment for protection against cyclophosphamide-induced and cisplatin-induced toxicities: results of a randomized control trial in patients with advanced ovarian cancer | Completed | 1996 | Reduced number of patients requiring delaying or discontinue cisplatin treatment due to nephrotoxicity in Amifostine co treatment group compared to cisplatin alone. Amifostine reduced incidence of hypomagnesemia (key characteristic of cisplatin nephrotoxicity). | [309] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. https://doi.org/10.3390/cancers13071572
McSweeney KR, Gadanec LK, Qaradakhi T, Ali BA, Zulli A, Apostolopoulos V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers. 2021; 13(7):1572. https://doi.org/10.3390/cancers13071572
Chicago/Turabian StyleMcSweeney, Kristen Renee, Laura Kate Gadanec, Tawar Qaradakhi, Benazir Ashiana Ali, Anthony Zulli, and Vasso Apostolopoulos. 2021. "Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations" Cancers 13, no. 7: 1572. https://doi.org/10.3390/cancers13071572
APA StyleMcSweeney, K. R., Gadanec, L. K., Qaradakhi, T., Ali, B. A., Zulli, A., & Apostolopoulos, V. (2021). Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers, 13(7), 1572. https://doi.org/10.3390/cancers13071572