
HDAC6 Signaling at Primary Cilia Promotes Proliferation and Restricts Differentiation of Glioma Cells

 , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
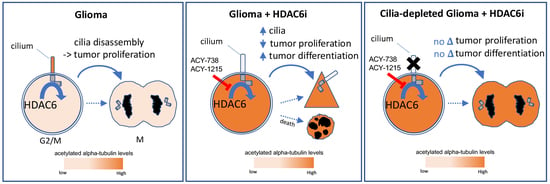
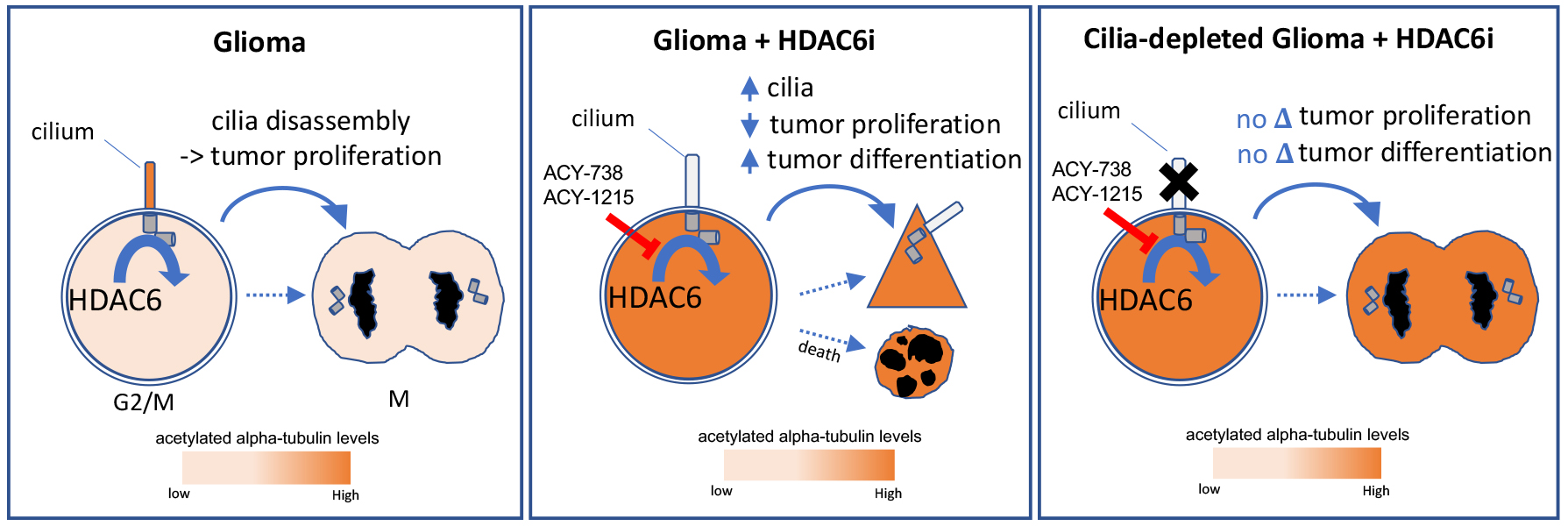{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. HDAC6 Inhibitors Inhibit the Proliferation of Murine and Human Glioma Cells
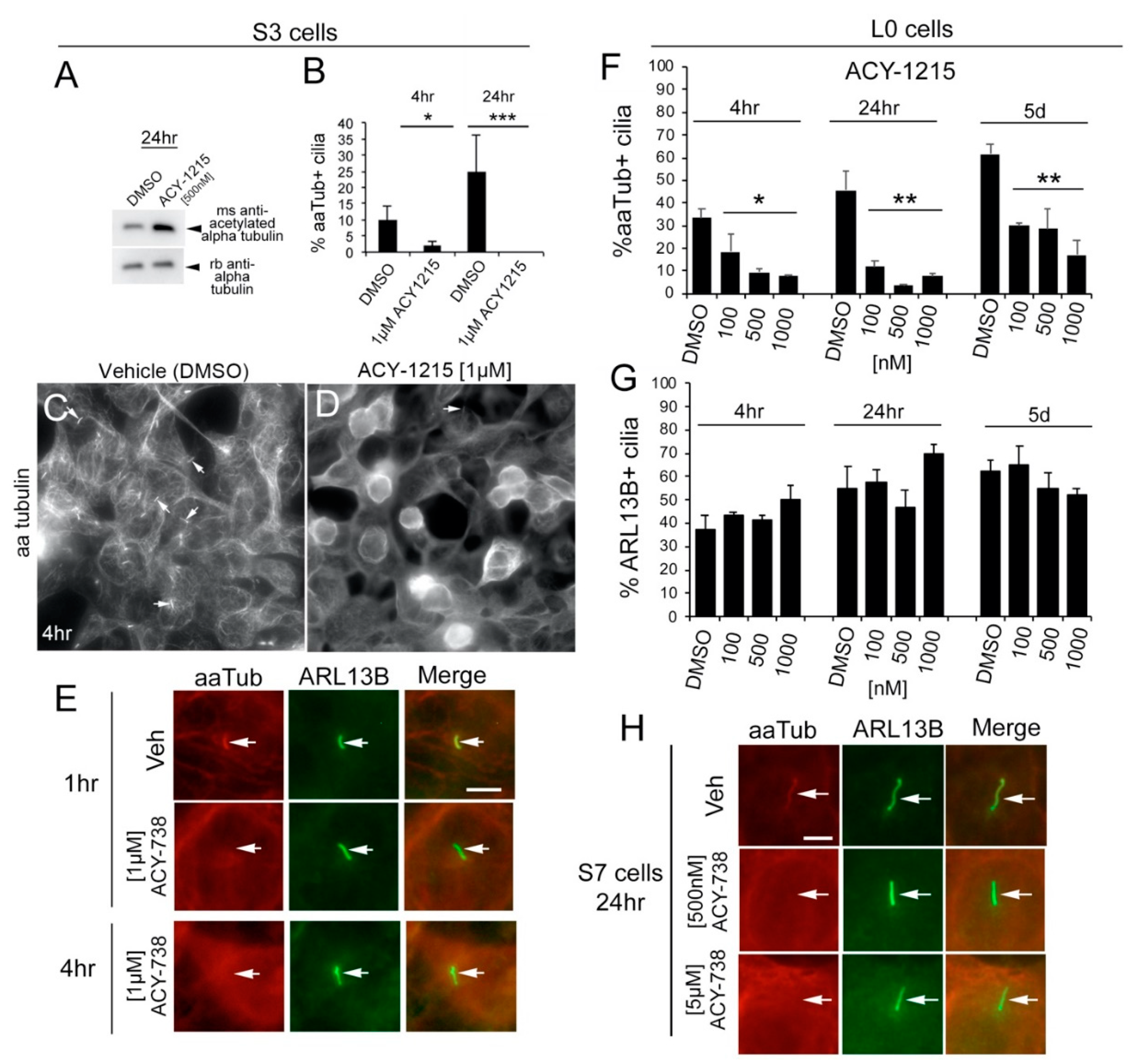
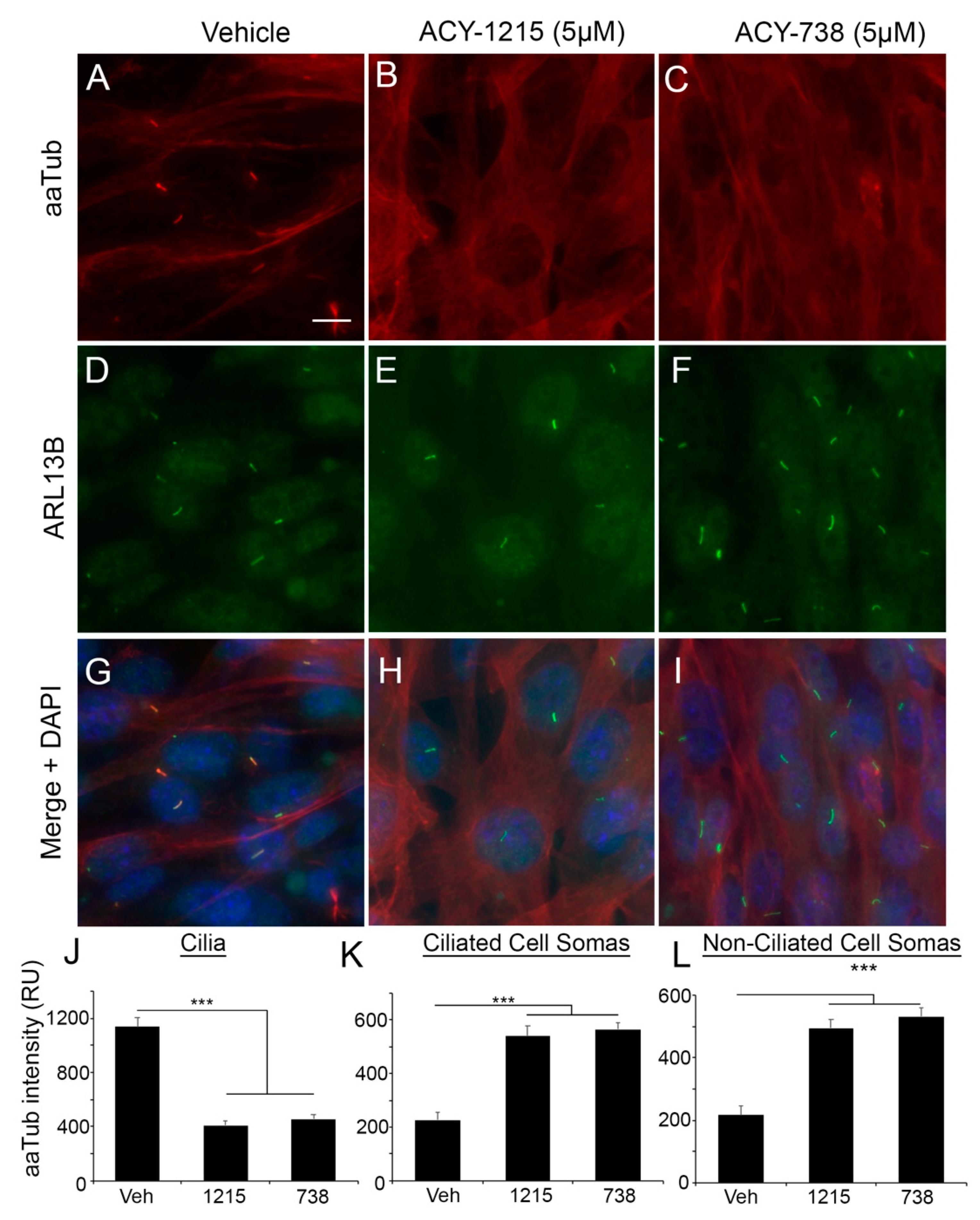
2.2. HDAC6 Inhibitors Rapidly and Differentially Alter Alpha-Tubulin Acetylation in Glioma Soma versus Primary Cilia
2.3. Normal Murine Astrocytes Show Ciliary Responses to HDAC6 Inhibition Similar to Glioma Cells
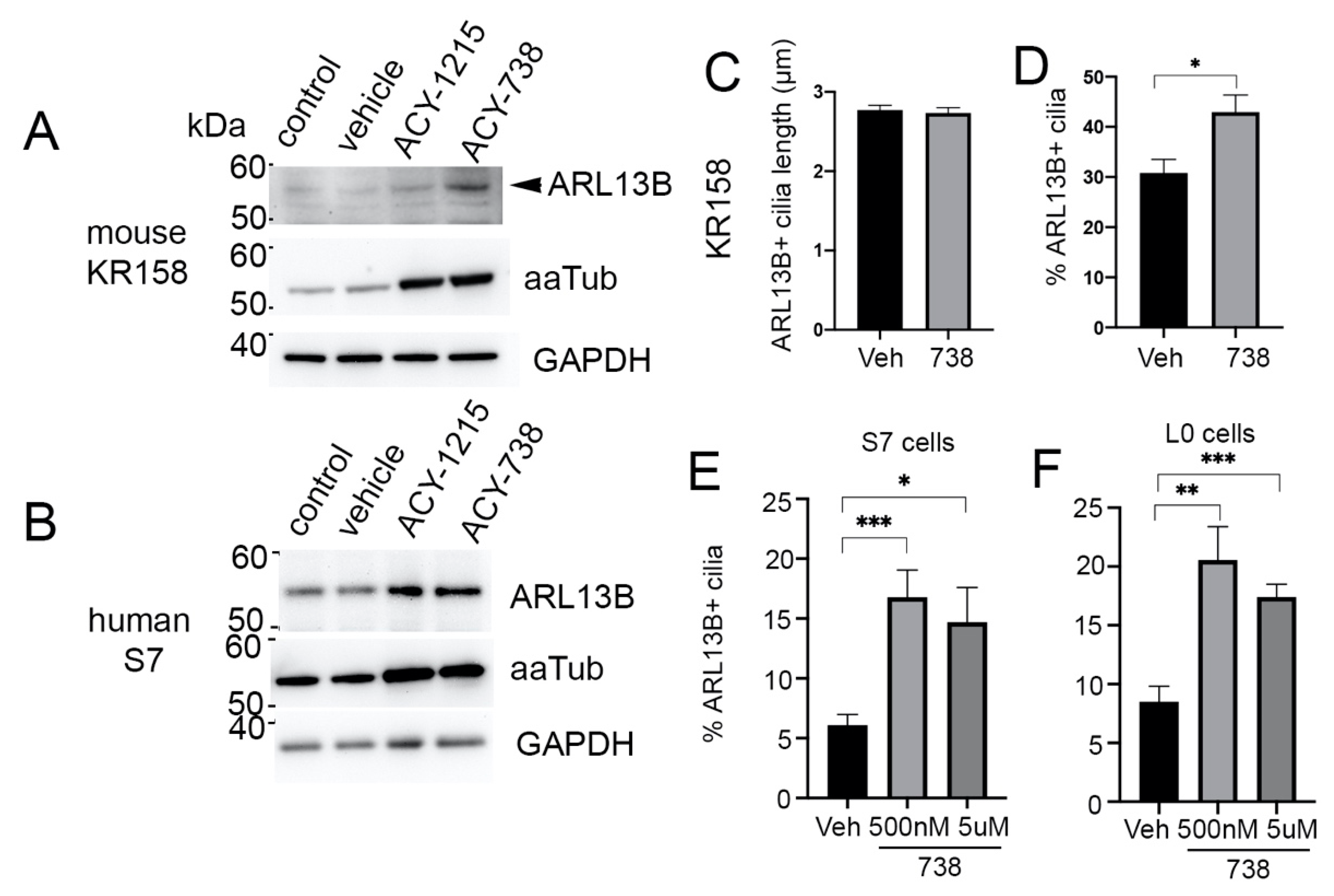
2.4. HDAC6 Inhibition Stimulates an Increase in ARL13B+ Ciliated Glioma Cells
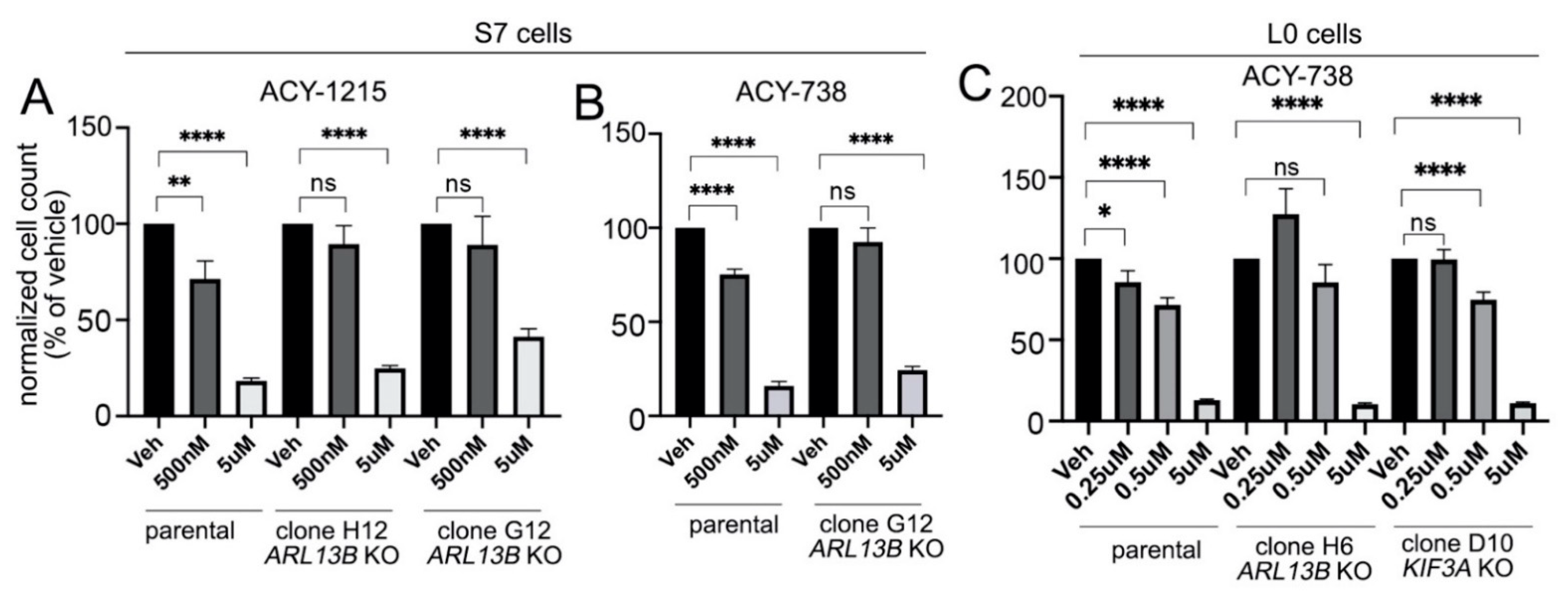
2.5. Preventing Cilia Formation Blocks the Antiproliferative Effect of HDAC6 Inhibitors on Glioma Cells at Low Drug Concentrations
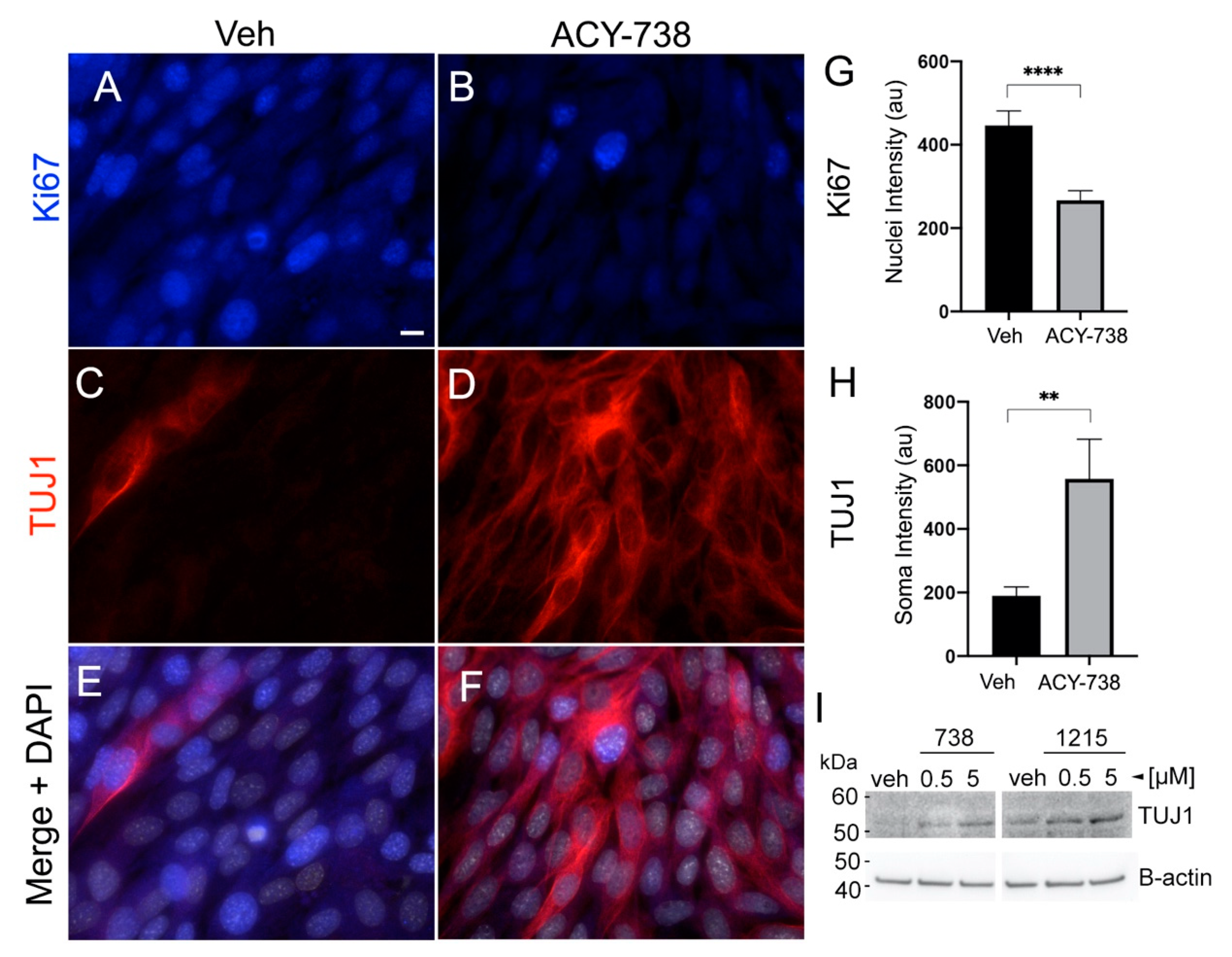
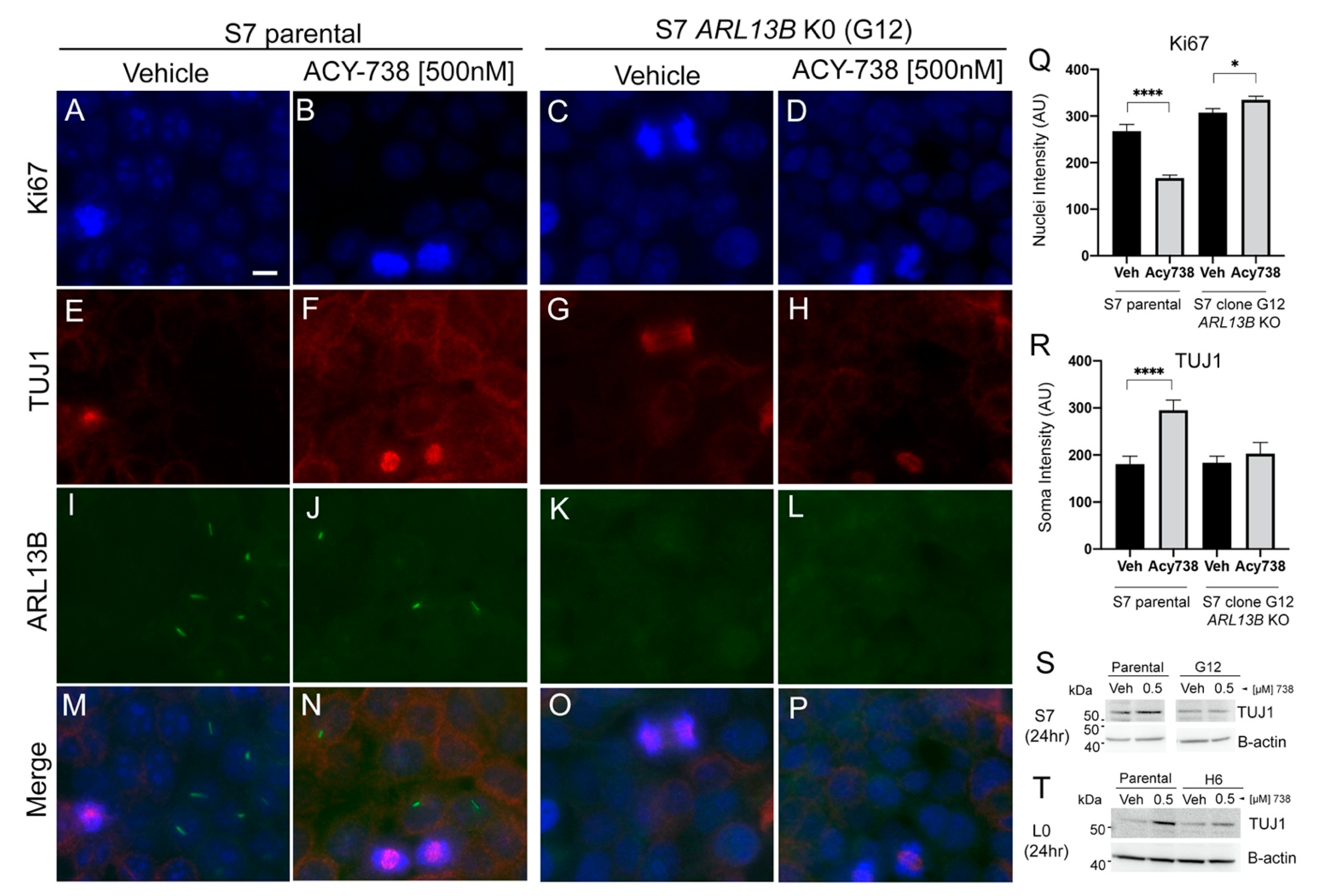
2.6. The Differentiation of Glioma Cells Induced by HDAC6 Inhibition Depends on Primary Cilia
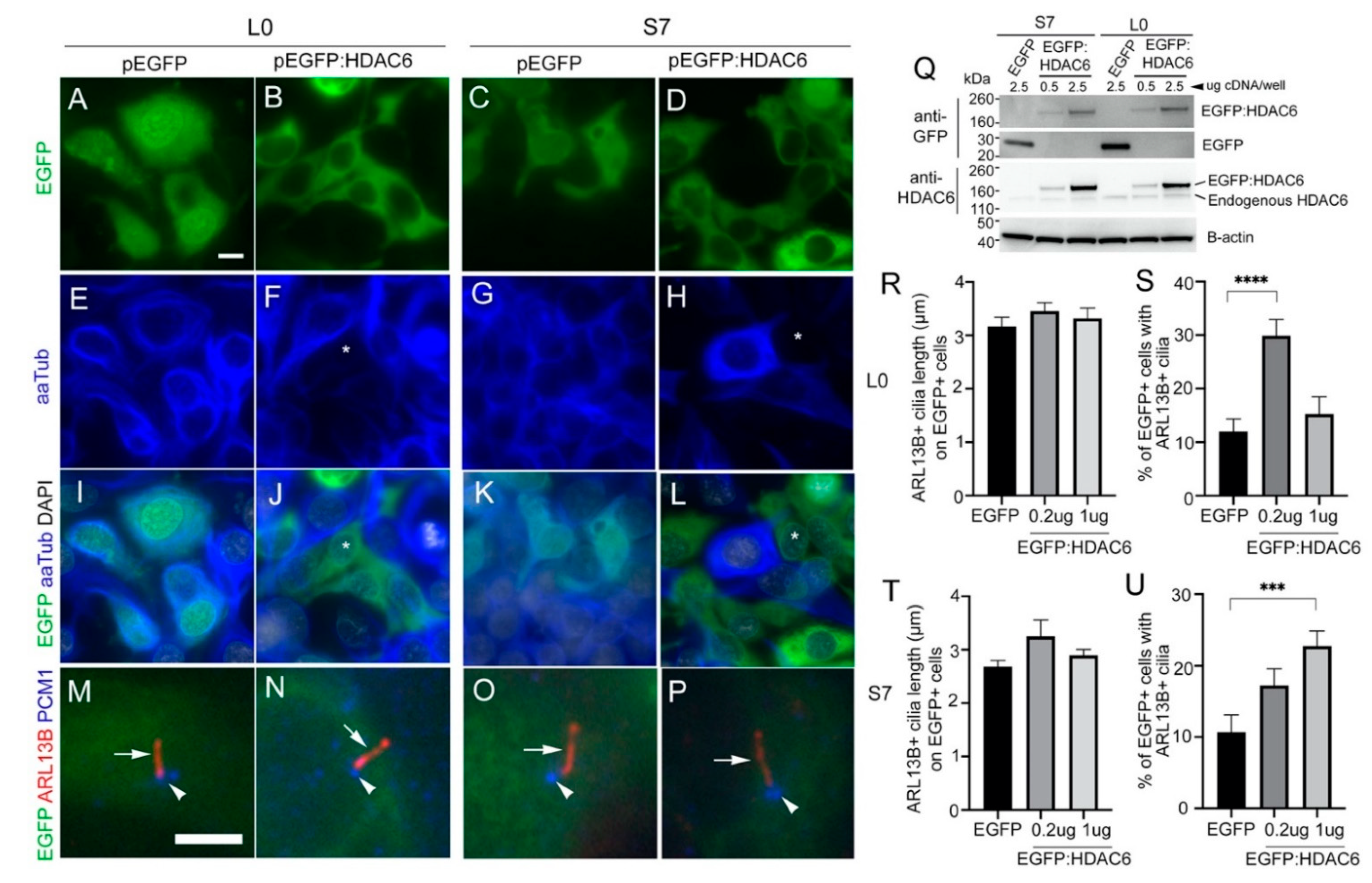
2.7. Overexpressing HDAC6 in Glioma Cells Reduces the Levels of Acetylated Alpha-Tubulin but Not Ciliary Length or Frequency of Ciliated Tumor Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Proliferation Assessment
4.3. Immunostaining
4.4. Western Blot (WB)
4.5. Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Auzmendi-Iriarte, J.; Saenz-Antonanzas, A.; Mikelez-Alonso, I.; Carrasco-Garcia, E.; Tellaetxe-Abete, M.; Lawrie, C.H.; Sampron, N.; Cortajarena, A.L.; Matheu, A. Characterization of a new small-molecule inhibitor of HDAC6 in glioblastoma. Cell Death Dis. 2020, 11, 417. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Lee, D.H.; Yeon, S.K.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Temozolomide-resistant glioblastoma depends on HDAC6 activity through regulation of DNA mismatch repair. Anticancer Res. 2019, 39, 6731–6741. [Google Scholar] [CrossRef]
- Li, S.; Liu, X.; Chen, X.; Zhang, L.; Wang, X. Histone deacetylase 6 promotes growth of glioblastoma through inhibition of SMAD2 signaling. Tumour Biol. 2015, 36, 9661–9665. [Google Scholar] [CrossRef]
- Liu, J.R.; Yu, C.W.; Hung, P.Y.; Hsin, L.W.; Chern, J.W. High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhanced antitumor immunity in glioblastoma. Biochem. Pharmacol. 2019, 163, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Urdiciain, A.; Erausquin, E.; Melendez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. Tubastatin A, an inhibitor of HDAC6, enhances temozolomideinduced apoptosis and reverses the malignant phenotype of glioblastoma cells. Int. J. Oncol. 2019, 54, 1797–1808. [Google Scholar] [PubMed]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef]
- Yang, W.B.; Hsu, C.C.; Hsu, T.I.; Liou, J.P.; Chang, K.Y.; Chen, P.Y.; Liu, J.J.; Yang, S.T.; Wang, J.Y.; Yeh, S.H.; et al. Increased activation of HDAC1/2/6 and Sp1 underlies therapeutic resistance and tumor growth in glioblastoma. Neuro. Oncol. 2020, 22, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Li, P. Growth suppression of glioma cells using HDAC6 inhibitor, Tubacin. Open Med. (Wars) 2018, 13, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.Y.; Xia, Y.; Hu, K.; Zeng, S.; Wu, L.; Liu, S.; Zhi, C.; Lai, M.; Chen, D.; Xie, L.; et al. Histone deacetylase 6 promotes growth of glioblastoma through the MKK7/JNK/c-Jun signaling pathway. J. Neurochem. 2020, 152, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, I.; Beus, M.; Stochaj, U.; Le, P.U.; Zorc, B.; Rajic, Z.; Petrecca, K.; Maysinger, D. Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule. Cell Death Discov. 2018, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Buyandelger, B.; Bar, E.; Hung, K.; Chen, R.; Chiang, Y.; Liou, J.; Huang, H.; Wang, J. Histone deacetylase inhibitor MPT0B291 suppresses glioma growth in vitro and in vivo partially through acetylation of p53. Int. J. Biol. Sci. 2020, 16, 3184–3199. [Google Scholar] [CrossRef] [PubMed]
- Jochems, J.; Boulden, J.; Lee, B.G.; Blendy, J.A.; Jarpe, M.; Mazitschek, R.; Van Duzer, J.H.; Jones, S.; Berton, O. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 2014, 39, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Huo, X.; Jarpe, M.B.; Kavelaars, A.; Heijnen, C.J. Pharmacological inhibition of HDAC6 reverses cognitive impairment and tau pathology as a result of cisplatin treatment. Acta Neuropathol. Commun. 2018, 6, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.Y.; Zhang, C.; Zhang, Y.; Chen, L.; Chen, B.D.; Li, Q.Z.; Zhang, X.J.; Li, W.P. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. 2017, 391, 89–99. [Google Scholar] [CrossRef]
- Marampon, F.; Megiorni, F.; Camero, S.; Crescioli, C.; McDowell, H.P.; Sferra, R.; Vetuschi, A.; Pompili, S.; Ventura, L.; De Felice, F.; et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017, 397, 1–11. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef]
- Wang, L.; Dynlacht, B.D. The regulation of cilium assembly and disassembly in development and disease. Development 2018, 145, dev151407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemi, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, J.; Yang, Y.; Li, D.; Liu, M.; Zhou, J. Deacetylation of alpha-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci. Rep. 2015, 5, 12917. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Radtke, B.N.; Bogert, P.S.; Huang, B.Q.; Gajdos, G.B.; LaRusso, N.F. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013, 73, 2259–2270. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Guo, F.; Cheng, W.; Zhang, J.; Huang, J.; Wang, R.; Ma, Z.; Xu, K. HDAC6 inhibition suppresses chondrosarcoma by restoring the expression of primary cilia. Oncol. Rep. 2017, 38, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Sarkisian, M.R.; Siebzehnrubl, D.; Hoang-Minh, L.; Deleyrolle, L.; Silver, D.J.; Siebzehnrubl, F.A.; Guadiana, S.M.; Srivinasan, G.; Semple-Rowland, S.; Harrison, J.K.; et al. Detection of primary cilia in human glioblastoma. J. Neurooncol. 2014, 117, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Hoang-Minh, L.B.; Deleyrolle, L.P.; Siebzehnrub, D.; Ugartemendia, G.; Futch, H.; Griffith, B.; Breunig, J.J.; De Leon, G.; Mitchell, D.A.; Semple-Rowland, S.; et al. Disruption of KIF3A in patient-derived glioblastoma cells: Effects on ciliogenesis, hedgehog sensitivity, and tumorigenesis. Oncotarget 2016, 7, 7029–7043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, J.J.; Fritzler, M.J.; Rattner, J.B. Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer 2009, 9, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.L.; Chapple, J.P.; Knight, M.M. Primary cilia disassembly down-regulates mechanosensitive hedgehog signalling: A feedback mechanism controlling ADAMTS-5 expression in chondrocytes. Osteoarthr. Cartil. 2014, 22, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Toh, M.T.; Narasimhan, V.; Thamilselvam, S.K.; Choksi, S.P.; Roy, S. A function for the Joubert syndrome protein Arl13b in ciliary membrane extension and ciliary length regulation. Dev. Biol. 2015, 397, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Revenkova, E.; Liu, Q.; Gusella, G.L.; Iomini, C. The Joubert syndrome protein ARL13B binds tubulin to maintain uniform distribution of proteins along the ciliary membrane. J. Cell Sci. 2018, 131, jcs212324. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.K.; Le, M.M.; Smith, T.S.; Hoang-Minh, L.B.; Atkinson, E.W.; Ugartemendia, G.; Semple-Rowland, S.; Coleman, J.E.; Sarkisian, M.R. Neonatal seizures induced by pentylenetetrazol or kainic acid disrupt primary cilia growth on developing mouse cortical neurons. Exp. Neurol. 2016, 282, 119–127. [Google Scholar] [CrossRef]
- Sterpka, A.; Chen, X. Neuronal and astrocytic primary cilia in the mature brain. Pharmacol. Res. 2018, 137, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.K.; Han, Y.G.; Shah, J.K.; Obernier, K.; Guinto, C.D.; Alvarez-Buylla, A. Primary cilia are required in a unique subpopulation of neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, 12438–12443. [Google Scholar] [CrossRef] [Green Version]
- Hoang-Minh, L.B.; Deleyrolle, L.P.; Nakamura, N.; Parker, A.K.; Martuscello, R.T.; Reynolds, B.A.; Sarkisian, M.R. PCM1 depletion inhibits glioblastoma cell ciliogenesis and increases cell death and sensitivity to temozolomide. Transl. Oncol. 2016, 9, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroukov, W.; Rosch, A.; Schwan, C.; Jeney, A.; Romer, W.; Theunauer, R. Synchronizing protein traffic to the primary cilium. Front. Genet. 2019, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.J.; Fritzler, M.J.; Rattner, J.B. Ultrastructural characterization of primary cilia in pathologically characterized human glioblastoma multiforme (GBM) tumors. BMC Clin. Pathol. 2014, 14, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, S.; Baur, K.; Hellwig, A.; Holzl-Wenig, G.; Mandl, C.; Ciccolini, F. A flow cytometry-based approach for the isolation and characterization of neural stem cell primary cilia. Front. Cell. Neurosci. 2018, 12, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaconelli, J.; Xuan, L.; Karmacharya, R. HDAC6 modulates signaling pathways relevant to synaptic biology and neuronal differentiation in human stem-cell-derived neurons. Int. J. Mol. Sci. 2019, 20, 1605. [Google Scholar] [CrossRef] [Green Version]
- Breunig, J.J.; Sarkisian, M.R.; Arellano, J.I.; Morozov, Y.M.; Ayoub, A.E.; Sojitra, S.; Wang, B.; Flavell, R.A.; Rakic, P.; Town, T. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 13127–13132. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.G.; Spassky, N.; Romaguera-Ros, M.; Garcia-Verdugo, J.M.; Aguilar, A.; Schneider-Maunoury, S.; Alvarez-Buylla, A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 2008, 11, 277–284. [Google Scholar] [CrossRef]
- Mugge, L.; Mansour, T.R.; Crippen, M.; Alam, Y.; Schroeder, J. Depression and glioblastoma, complicated concomitant diseases: A systemic review of published literature. Neurosurg. Rev. 2020, 43, 497–511. [Google Scholar] [CrossRef]
- Peixoto, E.; Jin, S.; Thelen, K.; Biswas, A.; Richard, S.; Morleo, M.; Mansini, A.; Holtorf, S.; Carbone, F.; Pastore, N.; et al. HDAC6-dependent ciliophagy is involved in ciliary loss and cholangiocarcinoma growth in human cells and murine models. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G1022–G1033. [Google Scholar] [CrossRef]
- Jenks, A.D.; Vyse, S.; Wong, J.P.; Kostaras, E.; Keller, D.; Burgoyne, T.; Shoemark, A.; Tsalikis, A.; de la Roche, M.; Michaelis, M.; et al. Primary cilia mediate diverse kinase inhibitor resistance mechanisms in cancer. Cell Rep. 2018, 23, 3042–3055. [Google Scholar] [CrossRef]
- Deleyrolle, L.P.; Harding, A.; Cato, K.; Siebzehnrubl, F.A.; Rahman, M.; Azari, H.; Olson, S.; Gabrielli, B.; Osborne, G.; Vescovi, A.; et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 2011, 134, 1331–1343. [Google Scholar] [CrossRef] [Green Version]
- Hothi, P.; Martins, T.J.; Chen, L.; Deleyrolle, L.; Yoon, J.G.; Reynolds, B.; Foltz, G. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget 2012, 3, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Gursel, D.B.; Connell-Albert, Y.S.; Tuskan, R.G.; Anastassiadis, T.; Walrath, J.C.; Hawes, J.J.; Amlin-Van Schaick, J.C.; Reilly, K.M. Control of proliferation in astrocytoma cells by the receptor tyrosine kinase/PI3K/AKT signaling axis and the use of PI-103 and TCN as potential anti-astrocytoma therapies. Neuro Oncol. 2011, 13, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Sambo, D.; Khoshbouei, H. Methamphetamine regulation of firing activity of dopamine neurons. J. Neurosci. 2016, 36, 10376–10391. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.R.; Lebowitz, J.J.; Guenther, D.T.; Refowich, A.J.; Hansen, C.; Maurer, A.P.; Khoshbouei, H. Methamphetamine regulation of activity and topology of ventral midbrain networks. PLoS ONE 2019, 14, e0222957. [Google Scholar] [CrossRef]
- Richardson, B.D.; Saha, K.; Krout, D.; Cabrera, E.; Felts, B.; Henry, L.K.; Swant, J.; Zou, M.F.; Newman, A.H.; Khoshbouei, H. Membrane potential shapes regulation of dopamine transporter trafficking at the plasma membrane. Nat. Comm. 2016, 7, 10423. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, P.; Hoang-Minh, L.B.; Tian, J.; Cheng, A.; Basrai, R.; Kalaria, N.; Lebowitz, J.J.; Khoshbouei, H.; Deleyrolle, L.P.; Sarkisian, M.R. HDAC6 Signaling at Primary Cilia Promotes Proliferation and Restricts Differentiation of Glioma Cells. Cancers 2021, 13, 1644. https://doi.org/10.3390/cancers13071644
Shi P, Hoang-Minh LB, Tian J, Cheng A, Basrai R, Kalaria N, Lebowitz JJ, Khoshbouei H, Deleyrolle LP, Sarkisian MR. HDAC6 Signaling at Primary Cilia Promotes Proliferation and Restricts Differentiation of Glioma Cells. Cancers. 2021; 13(7):1644. https://doi.org/10.3390/cancers13071644
Chicago/Turabian StyleShi, Ping, Lan B. Hoang-Minh, Jia Tian, Alice Cheng, Reemsha Basrai, Neil Kalaria, Joseph J. Lebowitz, Habibeh Khoshbouei, Loic P. Deleyrolle, and Matthew R. Sarkisian. 2021. "HDAC6 Signaling at Primary Cilia Promotes Proliferation and Restricts Differentiation of Glioma Cells" Cancers 13, no. 7: 1644. https://doi.org/10.3390/cancers13071644
APA StyleShi, P., Hoang-Minh, L. B., Tian, J., Cheng, A., Basrai, R., Kalaria, N., Lebowitz, J. J., Khoshbouei, H., Deleyrolle, L. P., & Sarkisian, M. R. (2021). HDAC6 Signaling at Primary Cilia Promotes Proliferation and Restricts Differentiation of Glioma Cells. Cancers, 13(7), 1644. https://doi.org/10.3390/cancers13071644