
Myc-Related Mitochondrial Activity as a Novel Target for Multiple Myeloma

 ,
, 
 , ,
, ,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Myeloma Cell Lines and Primary Multiple Myeloma Samples
2.2. MYC Gene Expression Analysis
2.3. RNA and DNA Extraction and Quantitative PCR (qPCR)
2.4. Optical Microscopy
2.5. Multiparametric Flow Cytometry Analysis
2.6. In Vitro and Ex Vivo Drug Assays
2.7. Western Blotting
2.8. Animal Model
2.9. Statistical Analysis
3. Results
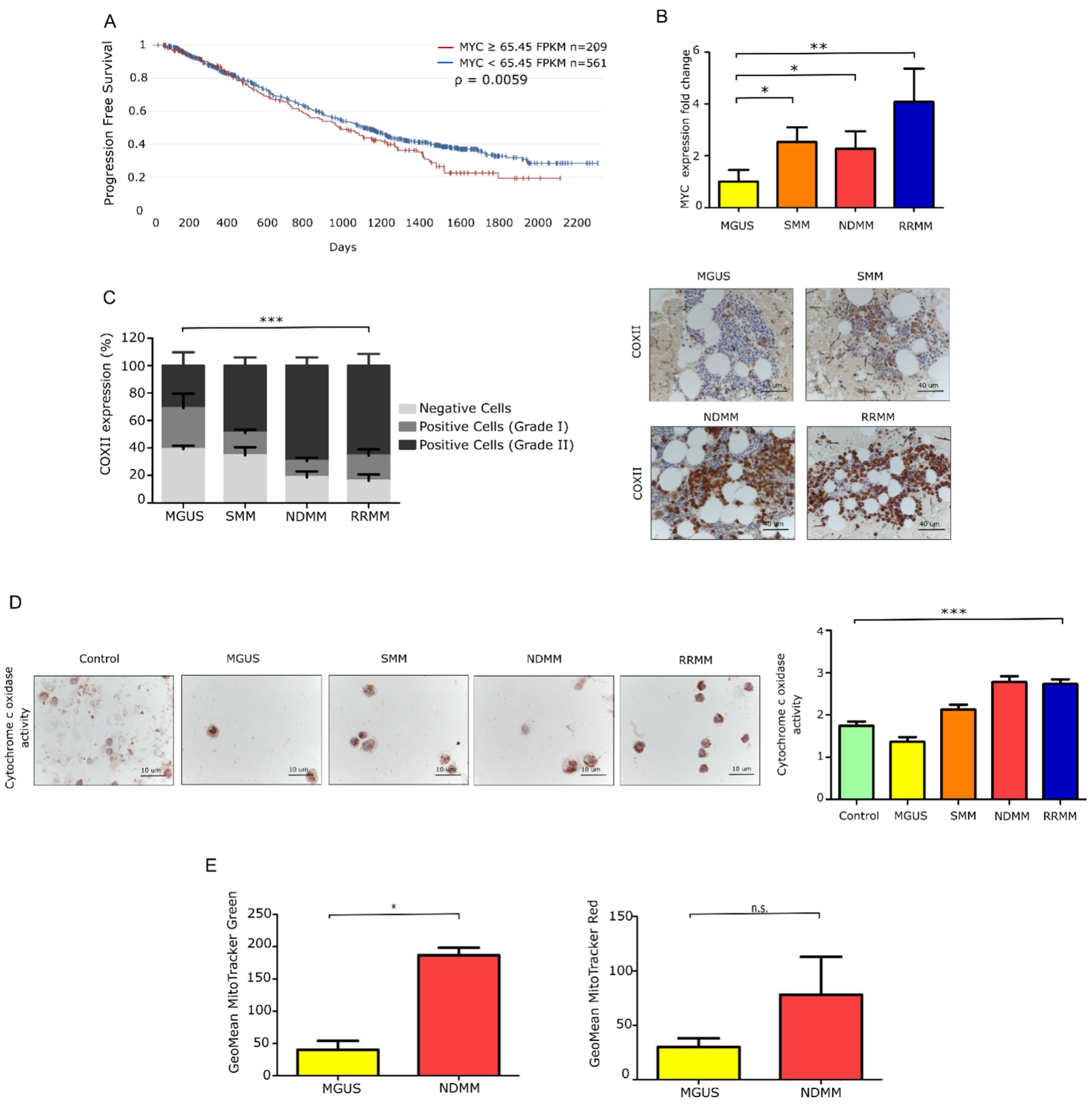
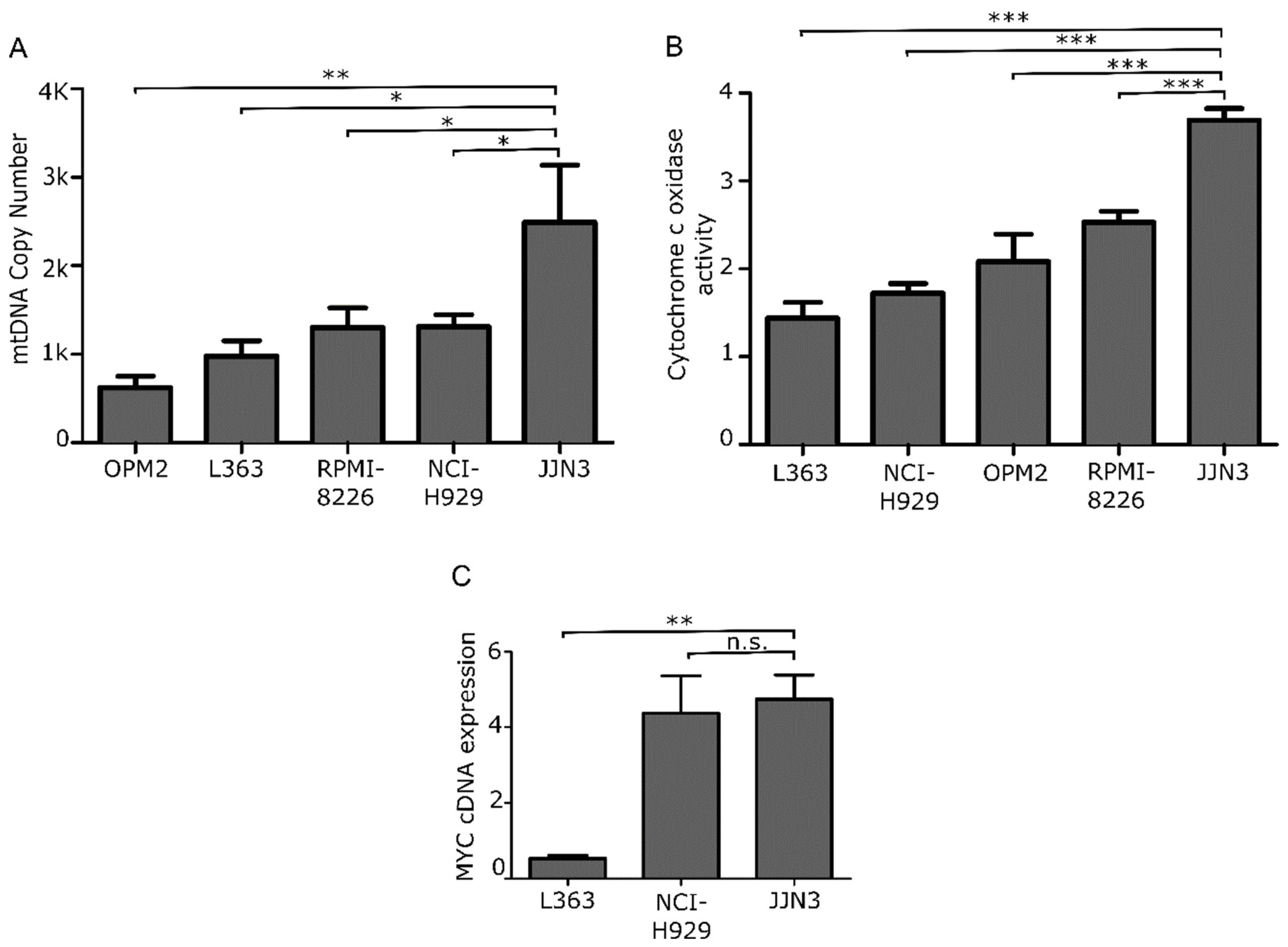
3.1. Patients with Newly Diagnosed and Relapsed MM Exhibit Elevated Myc Expression Features
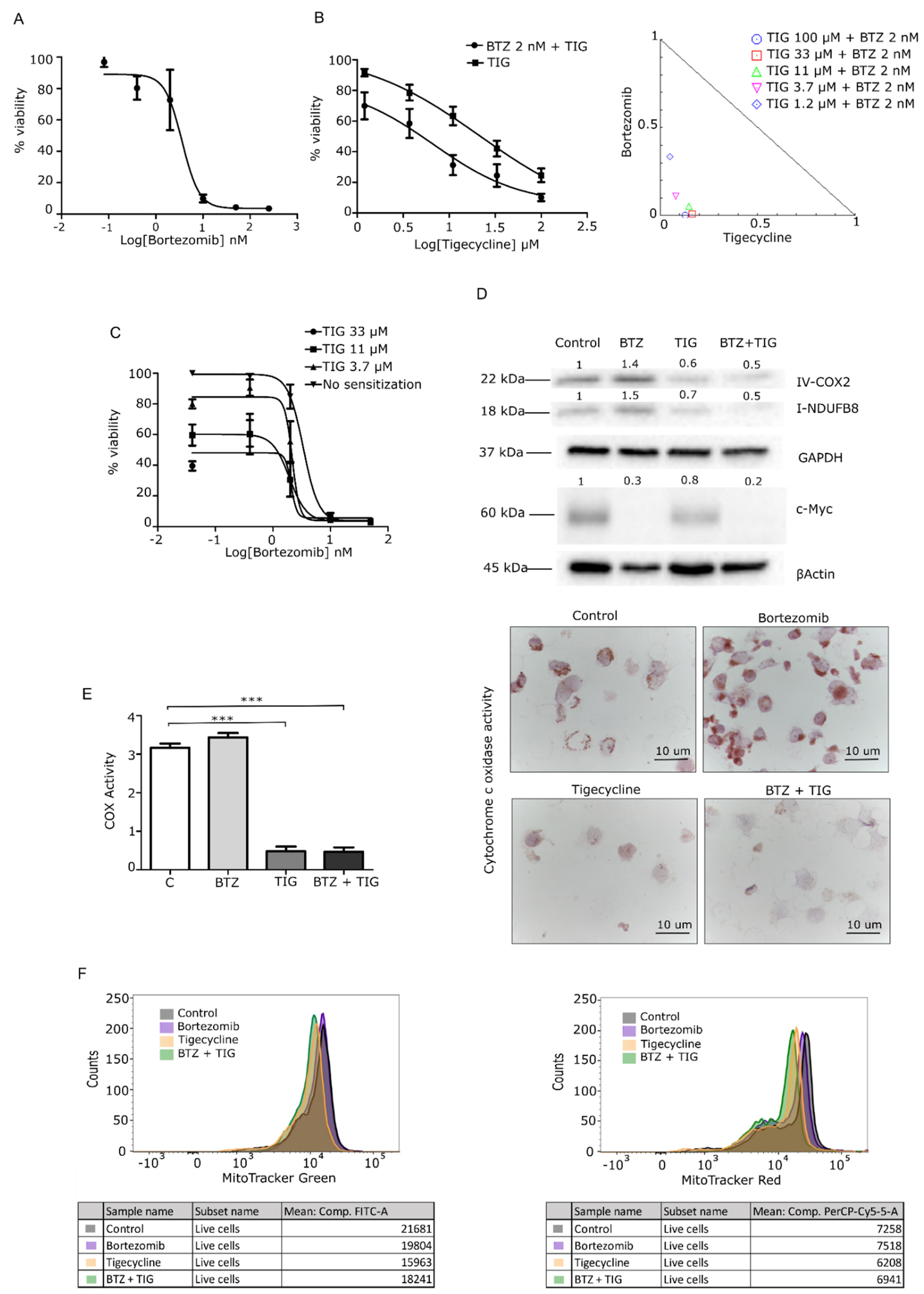
3.2. Mitochondrial Inhibition Is Cytotoxic to MM Cells In Vitro and Synergizes with Bortezomib
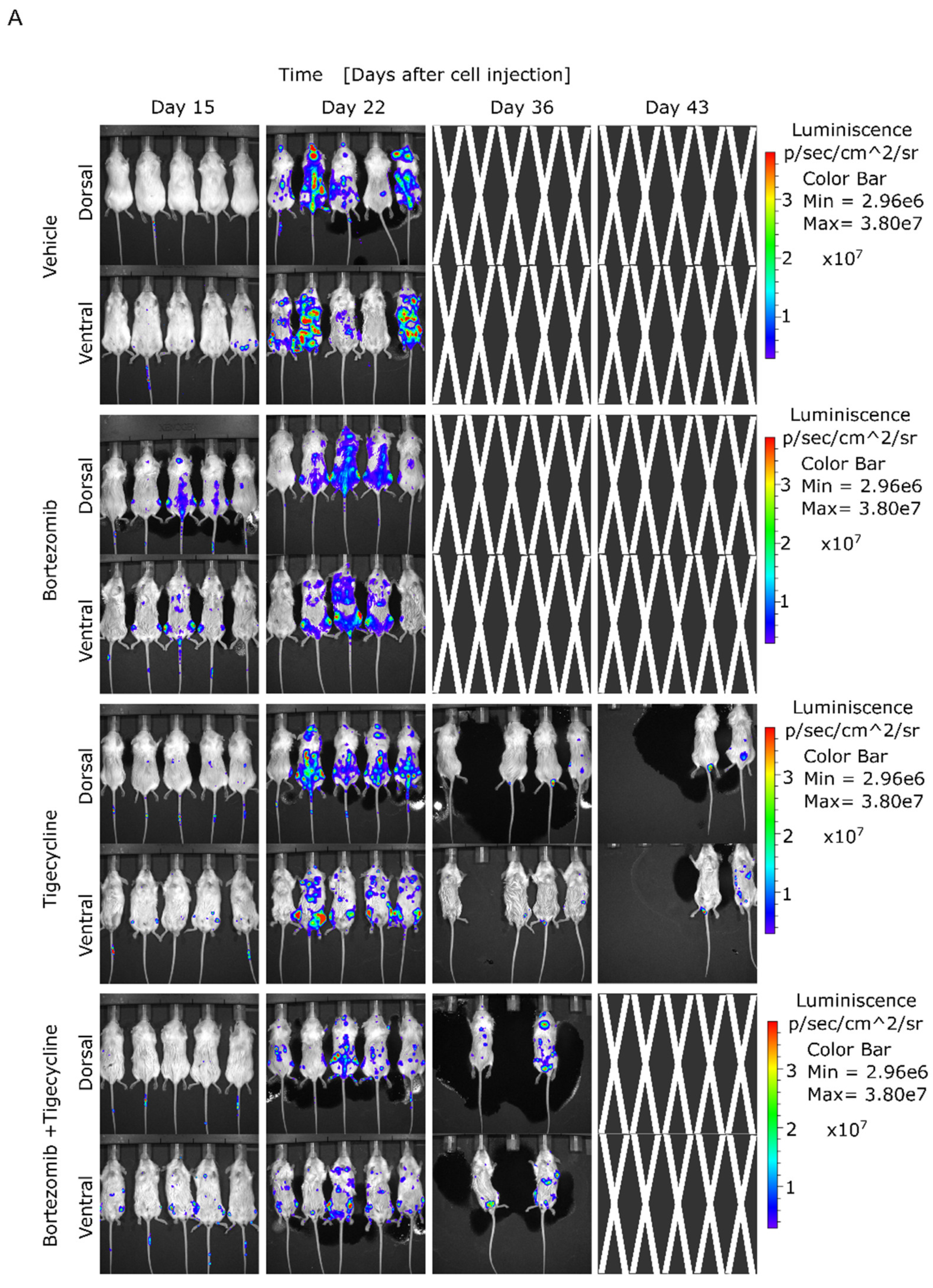
3.3. Tigecycline Is Active against Primary Myeloma Cells in Vitro and in a Murine MM Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rich, R.R.; Fleisher, T.A.; Shearer, W.T.; Schroeder, H.; Frew, A.J.; Weyand, C.M. Clinical Immunology. Principles and Practice, 5th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020, 34, 322–326. [Google Scholar] [CrossRef]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- De Barrios, O.; Meler, A.; Parra, M. MYC’s Fine Line Between B Cell Development and Malignancy. Cells 2020, 9, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloedjes, T.A.; De Wilde, G.; Guikema, J.E.J. Metabolic Effects of Recurrent Genetic Aberrations in Multiple Myeloma. Cancers 2021, 13, 396. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E. Myc is also the bad guy in myeloma. Leuk. Lymphoma 2016, 57, 2485–2486. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, N.; Ootsubo, K.; Wagatsuma, M.; Midorikawa, K.; Nagata, A.; Noto, S.; Yamada, K.; Takezako, N. Impact of C-Myc gene-related aberrations in newly diagnosed myeloma with bortezomib/dexamethasone therapy. Int. J. Hematol. 2014, 99, 288–295. [Google Scholar] [CrossRef]
- Seo, J.H.; Agarwal, E.; Chae, Y.C.; Lee, Y.G.; Garlick, D.S.; Storaci, A.M.; Ferrero, S.; Gaudioso, G.; Gianelli, U.; Vaira, V.; et al. Mitochondrial fission factor is a novel Myc-dependent regulator of mitochondrial permeability in cancer. EBioMedicine 2019, 48, 353–363. [Google Scholar] [CrossRef]
- Hsieh, A.L.; Dang, C.V. MYC, Metabolic Synthetic Lethality, and Cancer. Recent Results Cancer Res. 2016, 207, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Rizzieri, D.; Paul, B.; Kang, Y. Metabolic alterations and the potential for targeting metabolic pathways in the treatment of multiple myeloma. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, F.; Hockenbery, D. MYC and Mitochondrial Biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Yu, W.; Franqui-Machin, R.; Bates, M.L.; Nadiminti, K.; Cao, H.; Amendt, B.A.; Jethava, Y.; Frech, I.; Zhan, F.; et al. Alteration of mitochondrial biogenesis promotes disease progression in multiple myeloma. Oncotarget 2017, 8, 111213–111224. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.G.; Kim, Y.N.; Lee, J.H.; Szardenings, M.; Baek, H.J.; Kook, H.; Kim, H.R.; Shin, M.G. Clinicopathological Implications of Mitochondrial Genome Alterations in Pediatric Acute Myeloid Leukemia. Ann. Lab. Med. 2016, 36, 101–110. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. “ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis”—A critical commentary. Free Radic. Biol. Med. 2008, 45, 1217–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Fang, B.; Liu, Y.; Yan, S.; Cao, D.; Mei, H.; Wang, Q.; Hu, Y.; Guo, T. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci. 2020, 256, 117971. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Dalton, W.S. Targeting the mitochondria: An exciting new approach to myeloma therapy. Commentary re: N. J. Bahlis et al., Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin. Cancer Res. 2002, 8, 3643–3645. [Google Scholar] [PubMed]
- Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. Int. J. Mol. Sci. 2019, 20, 3577. [Google Scholar] [CrossRef] [Green Version]
- Andreu, A.L.; Martinez, R.; Marti, R.; Garcia-Arumi, E. Quantification of mitochondrial DNA copy number: Pre-analytical factors. Mitochondrion 2009, 9, 242–246. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath 2013, 3, 71–85. [Google Scholar] [PubMed]
- Bae, J.; Song, W.; Smith, R.; Daley, J.; Tai, Y.T.; Anderson, K.C.; Munshi, N.C. A novel immunogenic CS1-specific peptide inducing antigen-specific cytotoxic T lymphocytes targeting multiple myeloma. Br. J. Haematol. 2012, 157, 687–701. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Multiple Myeloma Genomics Initiative. Available online: https://research.themmrf.org/ (accessed on 22 April 2020).
- Ross, J.M. Visualization of mitochondrial respiratory function using cytochrome c oxidase/succinate dehydrogenase (COX/SDH) double-labeling histochemistry. J. Vis. Exp. 2011, 57, e3266. [Google Scholar] [CrossRef] [Green Version]
- Samelman, T.R.; Shiry, L.J.; Cameron, D.F. Endurance training increases the expression of mitochondrial and nuclear encoded cytochrome c oxidase subunits and heat shock proteins in rat skeletal muscle. Eur. J. Appl. Physiol. 2000, 83, 22–27. [Google Scholar] [CrossRef]
- Srinivasan, S.; Avadhani, N.G. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, M.A.; Paton-Hough, J.M.; Evans, H.R.; Walker, R.E.; Harris, W.; Ratnabalan, D.; Snowden, J.A.; Chantry, A.D. NOD/SCID-GAMMA mice are an ideal strain to assess the efficacy of therapeutic agents used in the treatment of myeloma bone disease. PLoS ONE 2015, 10, e0119546. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkucinska-Wieckowska, A.; Pronicki, M.; Wieckowski, M.R. Histoenzymatic methods for visualization of the activity of individual mitochondrial respiratory chain complexes in the muscle biopsies from patients with mitochondrial defects. Methods Mol. Biol. 2015, 1241, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Norberg, E.; Lako, A.; Chen, P.H.; Stanley, I.A.; Zhou, F.; Ficarro, S.B.; Chapuy, B.; Chen, L.; Rodig, S.; Shin, D.; et al. Differential contribution of the mitochondrial translation pathway to the survival of diffuse large B-cell lymphoma subsets. Cell Death Differ. 2017, 24, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Zhang, Y.; Wang, W.; Wu, J.; Yang, Q.; Xu, W.; Jiang, S.; Han, Y.; Yu, K.; Zhang, S. Inhibition of autophagy enhances the antitumour activity of tigecycline in multiple myeloma. J. Cell. Mol. Med. 2018, 22, 5955–5963. [Google Scholar] [CrossRef] [PubMed]
- Roca-Portoles, A.; Rodriguez-Blanco, G.; Sumpton, D.; Cloix, C.; Mullin, M.; Mackay, G.M.; O’Neill, K.; Lemgruber, L.; Luo, X.; Tait, S.W.G. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Ravà, M.; D’Andrea, A.; Nicoli, P.; Gritti, I.; Donati, G.; Doni, M.; Giorgio, M.; Olivero, D.; Amati, B. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Ronca, R.; Ghedini, G.C.; Maccarinelli, F.; Sacco, A.; Locatelli, S.L.; Foglio, E.; Taranto, S.; Grillo, E.; Matarazzo, S.; Castelli, R.; et al. FGF Trapping Inhibits Multiple Myeloma Growth through c-Myc Degradation-Induced Mitochondrial Oxidative Stress. Cancer Res. 2020, 80, 2340–2354. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.R.; Tonon, G.; Huang, Y.; Zhang, Y.; Sinha, R.; Feng, B.; Stewart, J.P.; Zhan, F.; Khatry, D.; Protopopova, M.; et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell 2006, 9, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.; Gritti, I.; Nicoli, P.; Giorgio, M.; Doni, M.; Conti, A.; Bianchi, V.; Casoli, L.; Sabò, A.; Mironov, A.; et al. The mitochondrial translation machinery as a therapeutic target in Myc-driven lymphomas. Oncotarget 2016, 7, 72415–72430. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Yan, Y.; Li, Z.; Qian, L.; Gong, Z. The Antibiotic Drug Tigecycline: A Focus on its Promising Anticancer Properties. Front. Pharmacol. 2016, 7, 473. [Google Scholar] [CrossRef] [Green Version]
- Niesvizky, R.; Jayabalan, D.S.; Christos, P.J.; Furst, J.R.; Naib, T.; Ely, S.; Jalbrzikowski, J.; Pearse, R.N.; Zafar, F.; Pekle, K.; et al. BiRD (Biaxin [clarithromycin]/Revlimid [lenalidomide]/dexamethasone) combination therapy results in high complete- and overall-response rates in treatment-naive symptomatic multiple myeloma. Blood 2008, 111, 1101–1109. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MM Patients Clinical Variables | MGUS (N = 15) | SMM (N = 19) | NDMM (N = 27) a | RRMM (N = 22) | |
|---|---|---|---|---|---|
| Median age (range), years | 65 (48–82) | 76 (54–85) | 68 (50–86) | 65.5 (38–79) | |
| Sex, (%) | Male | 40 | 36.8 | 59.3 | 50 |
| Female | 60 | 63.2 | 40.7 | 50 | |
| PC BM, average % (range) | 7.87 (3–17) | 22 (1.7–55) | 39.2 (1–84) | 42.4 (4–90) | |
| PPC MFC %, average (range) | 1 (0.3–5) | 5.3 (0.7–24) | 9.2 (1.4–38.7) | 15.4 (0.03–40) | |
| Type of Ig heavy chain (serum) | nondetected | 0 | 0 | 3.8 | 0 |
| IgG | 46.7 | 61.1 | 69.2 | 68.2 | |
| IgA | 53.3 | 38.9 | 26.9 | 18.2 | |
| IgM | 0 | 0 | 0 | 13.6 | |
| IgD | 0 | 0 | 0 | 0 | |
| biclonal | 0 | 0 | 0 | 14.8 | |
| Type of Ig light chain (serum) | nondetected | 0 | 0 | 0 | 0 |
| kappa | 25 | 44.4 | 74.1 | 50 | |
| lambda | 75 | 55.6 | 25.9 | 45.5 | |
| biclonal | 0 | 0 | 0 | 4.5 | |
| Serum M-spike, ≥3 g/dL (%) | 0 | 13.3 | 19 | 25 | |
| Urine M-spike, detected (%) | 14.3 | 18.2 | 60 | 53.3 | |
| Kappa, (%) | ≥19.4 mg/L | 77.8 | 40 | 57.9 | 50 |
| Lambda, (%) | ≥26.3 mg/L | 66.7 | 40 | 36.8 | 41.7 |
| Free kappa/lambda ratio, (%) | <0.26 mg/L | 22.2 | 13.3 | 21.1 | 30 |
| ≥0.26 <1.65 mg/L | 22.2 | 53.3 | 0 | 0 | |
| ≥1.65 mg/L | 55.6 | 33.3 | 78.9 | 70 | |
| Creatinine (%) | ≥1.3 mg/dL | 33.3 | 21.1 | 30.8 | 13.6 |
| Serum calcium (%) | ≥11 mg/dL | 0 | 5.3 | 4 | 0 |
| LDH (%) | ≥225 U/I | 14.3 | 21.1 | 12 | 23.8 |
| Albumin (%) | ≤2.8 g/dL | 0 | 5.3 | 15.4 | 4.5 |
| Immunoparesis, yes (%) | 18.2 | 35.7 | 84.2 | 87.5 | |
| Refractory, yes (%) | NA | NA | 31.8 | 50 | |
| Type of prior treatment (%) | with PI | NA | 0 | 0 | 59.1 |
| without PI | NA | 100 | 100 | 40.9 | |
| Type of following treatment (%) | with PI | NA | 31.6 | 44.4 | 68.4 |
| without PI | NA | 68.4 | 55.6 | 31.6 | |
| Best response categories *, (%) | VGPR | 0 | 5.2 | 3.7 | 0 |
| PR | 0 | 15.8 | 14.8 | 40 | |
| CR | 0 | 5.3 | 25.9 | 25 | |
| CR MRD+ | 0 | 0 | 14.8 | 5 | |
| CR MRD− | 0 | 0 | 11.1 | 5 | |
| SD | 0 | 0 | 3.7 | 25 | |
| NA | 100 | 73.7 | 25.9 | 0 | |
| Method | MGUS (N = 15) | SMM (N = 19) | NDMM (N = 27) | RRMM (N = 22) |
|---|---|---|---|---|
| Total samples, N | 21 | 21 | 42 | 26 |
| gene expression | 9 | 9 | 18 | 11 |
| IHC | 4 | 7 | 13 | 11 |
| HE | 4 | 5 | 5 | 4 |
| MFC | 4 | 0 | 6 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Ruiz, A.; Ruiz-Heredia, Y.; Morales, M.L.; Aguilar-Garrido, P.; García-Ortiz, A.; Valeri, A.; Bárcena, C.; García-Martin, R.M.; Garrido, V.; Moreno, L.; et al. Myc-Related Mitochondrial Activity as a Novel Target for Multiple Myeloma. Cancers 2021, 13, 1662. https://doi.org/10.3390/cancers13071662
Ortiz-Ruiz A, Ruiz-Heredia Y, Morales ML, Aguilar-Garrido P, García-Ortiz A, Valeri A, Bárcena C, García-Martin RM, Garrido V, Moreno L, et al. Myc-Related Mitochondrial Activity as a Novel Target for Multiple Myeloma. Cancers. 2021; 13(7):1662. https://doi.org/10.3390/cancers13071662
Chicago/Turabian StyleOrtiz-Ruiz, Alejandra, Yanira Ruiz-Heredia, María Luz Morales, Pedro Aguilar-Garrido, Almudena García-Ortiz, Antonio Valeri, Carmen Bárcena, Rosa María García-Martin, Vanesa Garrido, Laura Moreno, and et al. 2021. "Myc-Related Mitochondrial Activity as a Novel Target for Multiple Myeloma" Cancers 13, no. 7: 1662. https://doi.org/10.3390/cancers13071662
APA StyleOrtiz-Ruiz, A., Ruiz-Heredia, Y., Morales, M. L., Aguilar-Garrido, P., García-Ortiz, A., Valeri, A., Bárcena, C., García-Martin, R. M., Garrido, V., Moreno, L., Gimenez, A., Navarro-Aguadero, M. Á., Velasco-Estevez, M., Lospitao, E., Cedena, M. T., Barrio, S., Martínez-López, J., Linares, M., & Gallardo, M. (2021). Myc-Related Mitochondrial Activity as a Novel Target for Multiple Myeloma. Cancers, 13(7), 1662. https://doi.org/10.3390/cancers13071662