
Gene Amplification-Associated Overexpression of the Selenoprotein tRNA Enzyme TRIT1 Confers Sensitivity to Arsenic Trioxide in Small-Cell Lung Cancer

 , , , ,
, , , ,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Cell Lines
2.2. Human Biological Samples
2.3. Fluorescence In Situ Hybridization (FISH)
2.4. Multiplex Ligation-Dependent Probe Amplification (MLPA)
2.5. TRIT1 Short-Hairpin RNA
2.6. Western Blot
2.7. N6-Isopentenyladenosine (I6A) Quantification
2.8. Murine Models
2.9. Drug-Dose Response Assay
2.10. RNA Sequencing
2.11. Statistical Analysis
3. Results
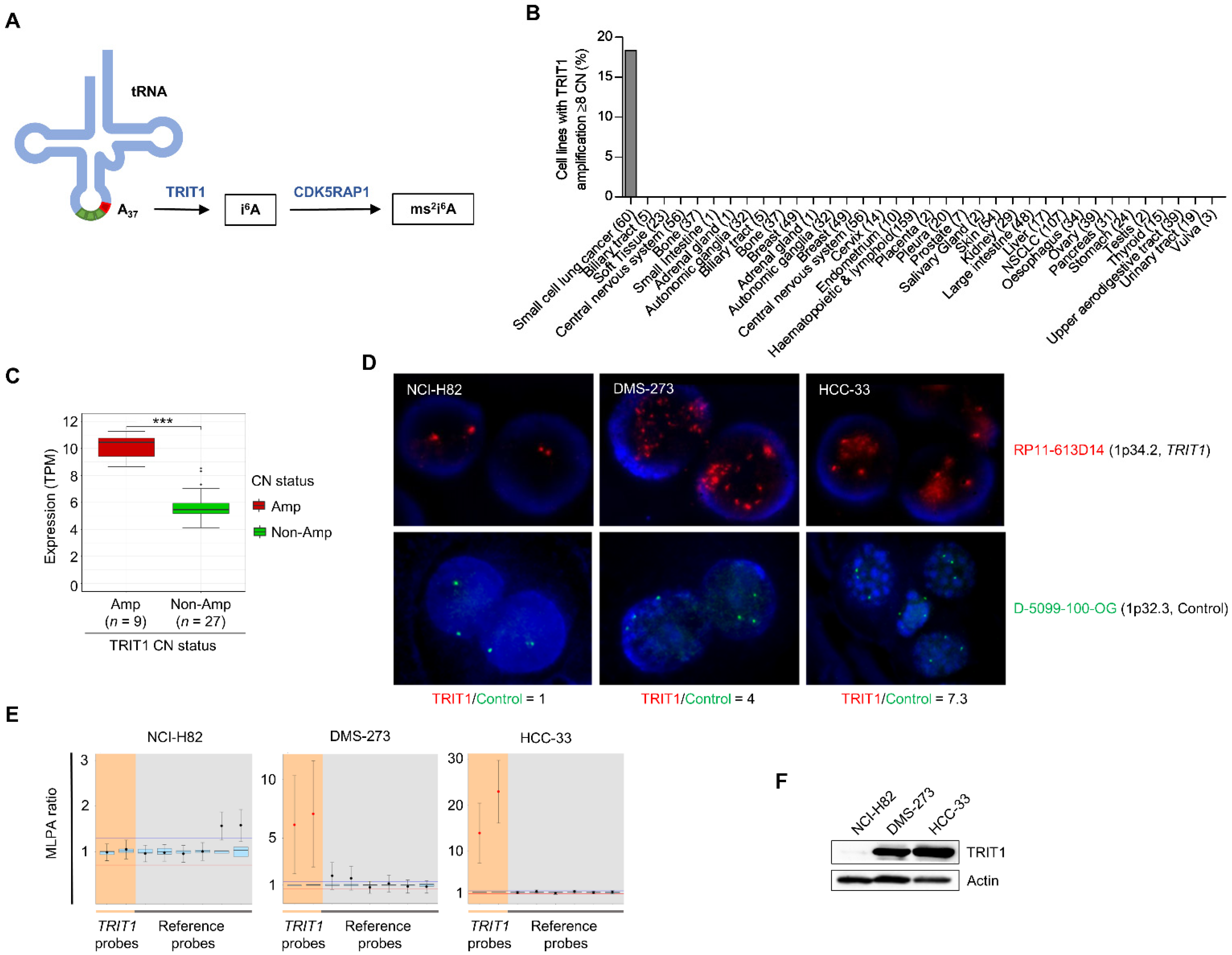
3.1. Detection of TRIT1 Gene Amplification-Associated Overexpression in Small-Cell Lung Cancer Cell Lines
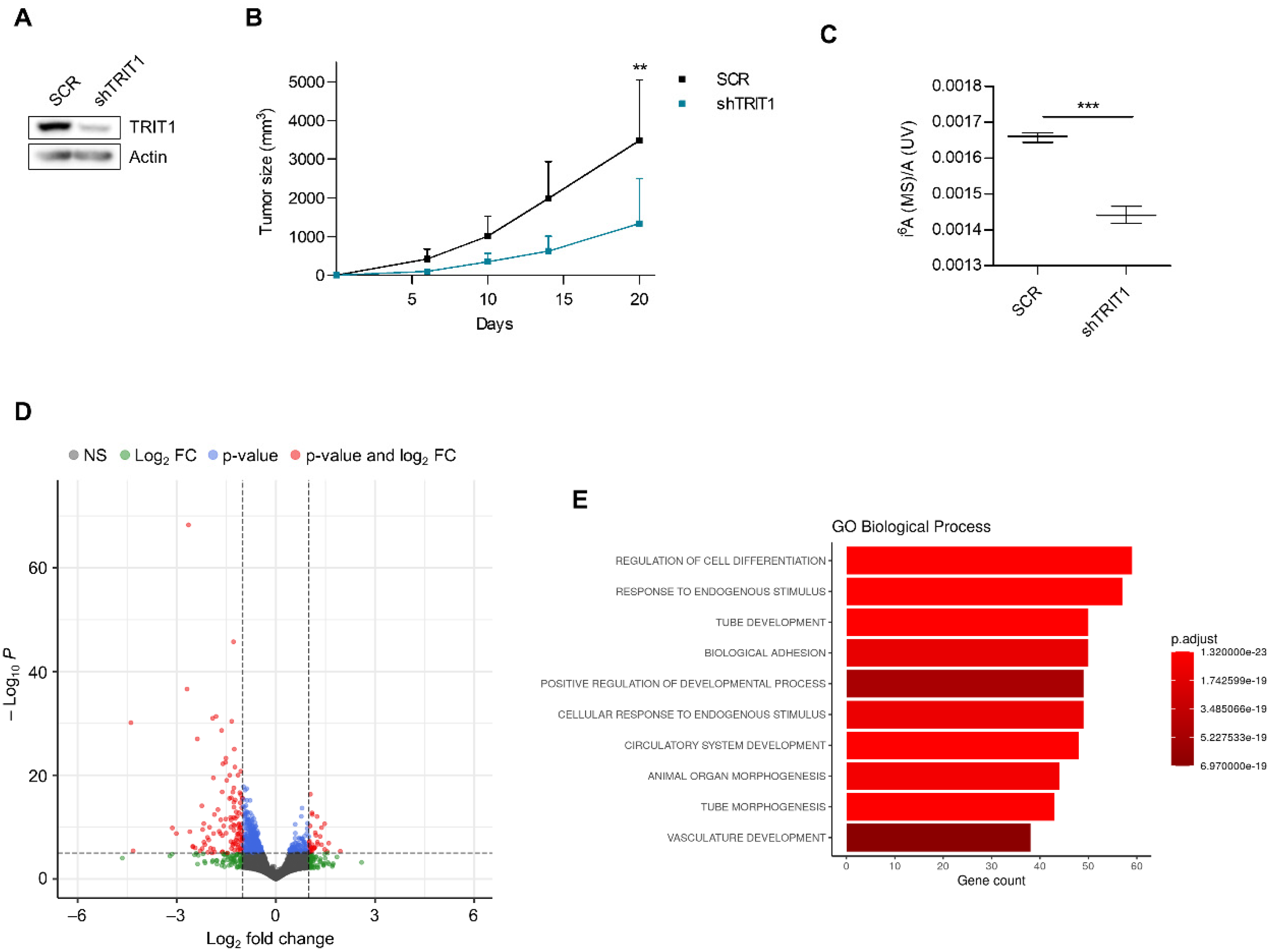
3.2. Cellular and Molecular Effects of TRIT1 Depletion in Gene Amplified Small-Cell Lung Cancer Cells
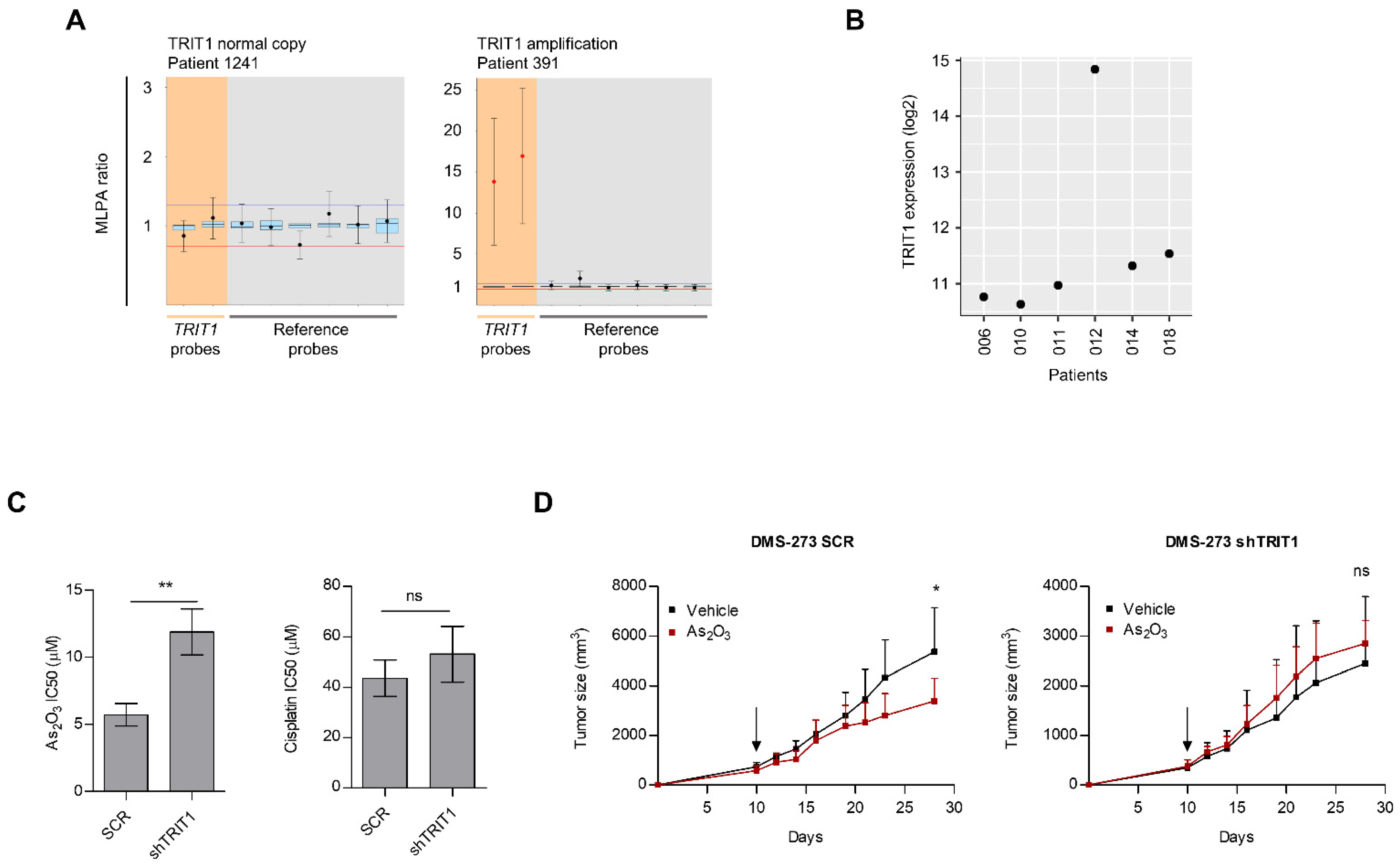
3.3. Occurrence of TRIT1 Gene Amplification in Small-Cell Lung Cancer Patients and In Vitro and In Vivo Sensitivity to Arsenic Trioxide
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esteller, M.; Pandolfi, P.P. The epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Rosselló-Tortella, M.; Ferrer, G.; Esteller, M. Epitranscriptomics in hematopoiesis and hematological malignancies. Blood Cancer Discov. 2020, 1, 26–31. [Google Scholar] [CrossRef]
- Duechler, M.; Leszczynska, G.; Sochacka, E.; Nawrot, B. Nucleoside modifications in the regulation of gene expression: Focus on tRNA. Cell. Mol. Life Sci. 2016, 73, 3075–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018, 28, 395–404. [Google Scholar] [CrossRef]
- Popis, M.C.; Blanco, S.; Frye, M. Posttranscriptional methylation of transfer and ribosomal RNA in stress response pathways, cell differentiation, and cancer. Curr. Opin. Oncol. 2016, 28, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Janin, M.; Coll-SanMartin, L.; Esteller, M. Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol. Cancer 2020, 19, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosselló-Tortella, M.; Llinàs-Arias, P.; Sakaguchi, Y.; Miyauchi, K.; Davalos, V.; Setien, F.; Careja-Cervantes, M.E.; Piñeyro, D.; Martínez-Gómez, J.; Guil, S.; et al. Epigenetic loss of the transfer RNA-modifying enzyme TYW2 induces ribosome frameshifts in colon cancer. Proc. Natl. Acad. Sci. USA 2020, 25, 20785–20793. [Google Scholar] [CrossRef]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fradejas, N.; Carlson, B.A.; Rijntjes, E.; Becker, N.P.; Tobe, R.; Schweizer, U. Mammalian Trit1 is a tRNA([Ser]Sec)-isopentenyl transferase required for full selenoprotein expression. Biochem. J. 2013, 450, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamichhane, T.N.; Mattijssen, S.; Maraia, R.J. Human cells have a limited set of tRNA anticodon loop substrates of the tRNA isopentenyltransferase TRIT1 tumor suppressor. Mol. Cell. Biol. 2013, 33, 4900–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, U.; Bohleber, S.; Fradejas-Villar, N. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biol. 2017, 14, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arragain, S.; Handelman, S.K.; Forouhar, F.; Wei, F.Y.; Tomizawa, K.; Hunt, J.F.; Douki, T.; Fontecave, M.; Mulliez, E.; Atta, M. Identification of eukaryotic and prokaryotic methylthiotransferase for biosynthesis of 2-methylthio-N6-threonylcarbamoyladenosine in tRNA. J. Biol. Chem. 2010, 285, 28425–28433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.Y.; Suzuki, T.; Watanabe, S.; Kimura, S.; Kaitsuka, T.; Fujimura, A.; Matsui, H.; Atta, M.; Michiue, H.; Fontecave, M.; et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Investig. 2011, 121, 3598–3608. [Google Scholar] [CrossRef] [Green Version]
- Reiter, V.; Matschkal, D.M.; Wagner, M.; Globisch, D.; Kneuttinger, A.C.; Müller, M.; Carell, T. The CDK5 repressor CDK5RAP1 is a methylthiotransferase acting on nuclear and mitochondrial RNA. Nucleic Acids Res. 2012, 40, 6235–6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakruddin, M.; Wei, F.Y.; Emura, S.; Matsuda, S.; Yasukawa, T.; Kang, D.; Tomizawa, K. Cdk5rap1-mediated 2-methylthio-N6-isopentenyladenosine modification is absent from nuclear-derived RNA species. Nucleic Acids Res. 2017, 45, 11954–11961. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, T.N.; Blewett, N.H.; Crawford, A.K.; Cherkasova, V.A.; Iben, J.R.; Begley, T.J.; Farabaugh, P.J.; Maraia, R.J. Lack of tRNA modification isopentenyl-A37 alters mRNA decoding and causes metabolic deficiencies in fission yeast. Mol. Cell. Biol. 2013, 33, 2918–2929. [Google Scholar] [CrossRef] [Green Version]
- Vindry, C.; Ohlmann, T.; Chavatte, L. Translation regulation of mammalian selenoproteins. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2480–2492. [Google Scholar] [CrossRef]
- Short, S.P.; Williams, C.S. Selenoproteins in tumorigenesis and cancer progression. Adv. Cancer Res. 2017, 136, 49–83. [Google Scholar]
- Marciel, M.P.; Hoffmann, P.R. Selenoproteins and metastasis. Adv. Cancer Res. 2017, 136, 85–108. [Google Scholar]
- Benko, A.L.; Vaduva, G.; Martin, N.C.; Hopper, A.K. Competition between a sterol biosynthetic enzyme and tRNA modification in addition to changes in the protein synthesis machinery causes altered nonsense suppression. Proc. Natl. Acad. Sci. USA 2000, 97, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A landscape of pharmacogenomic interactions in cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Chan, C.T.; Gu, C.; Lim, K.S.; Chionh, Y.H.; McBee, M.E.; Russell, B.S.; Babu, I.R.; Begley, T.J.; Dedon, P.C. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat. Protoc. 2014, 9, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, R.; Kohno, T.; Totoki, Y.; Shibata, T.; Tsuchihara, K.; Mimaki, S.; Tsuta, K.; Narita, Y.; Nishikawa, R.; Noguchi, M.; et al. Expression and clinical significance of genes frequently mutated in small cell lung cancers defined by whole exome/RNA sequencing. Carcinogenesis 2015, 36, 616–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakawa, R.; Takenaka, M.; Kohno, T.; Shimada, Y.; Totoki, Y.; Shibata, T.; Tsuta, K.; Nishikawa, R.; Noguchi, M.; Sato-Otsubo, A.; et al. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer 2013, 52, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Clinical Lung Cancer Genome Project (CLCGP); Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci. Transl. Med. 2013, 5, 209ra153. [Google Scholar]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Taniguchi, H.; Sen, T.; Rudin, C.M. Targeted therapies and biomarkers in small cell lung cancer. Front. Oncol. 2020, 10, 741. [Google Scholar] [CrossRef]
- Poirier, J.T.; George, J.; Owonikoko, T.K.; Berns, A.; Brambilla, E.; Byers, L.A.; Carbone, D.; Chen, H.J.; Christensen, C.L.; Dive, C.; et al. New approaches to SCLC therapy: From the laboratory to the clinic. J. Thorac. Oncol. 2020, 15, 520–540. [Google Scholar] [CrossRef] [Green Version]
- Hoonjan, M.; Jadhav, V.; Bhatt, P. Arsenic trioxide: Insights into its evolution to an anticancer agent. J. Biol. Inorg. Chem. 2018, 23, 313–329. [Google Scholar] [CrossRef]
- Wahiduzzaman, M.; Ota, A.; Hosokawa, Y. Novel mechanistic insights into the anti-cancer mode of arsenic trioxide. Curr. Cancer Drug Targets 2020, 20, 115–129. [Google Scholar] [CrossRef]
- De Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Huang, W.; Zeng, Y.C. A candidate for lung cancer treatment: Arsenic trioxide. Clin. Transl. Oncol. 2019, 21, 1115–1126. [Google Scholar] [CrossRef]
- Sobh, A.; Loguinov, A.; Yazici, G.N.; Zeidan, R.S.; Tagmount, A.; Hejazi, N.S.; Hubbard, A.E.; Zhang, L.; Vulpe, C.D. Functional profiling identifies determinants of arsenic trioxide cellular toxicity. Toxicol. Sci. 2019, 169, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, S.; Nelson, R.; Self, W.T. Arsenic trioxide and auranofin inhibit selenoprotein synthesis: Implications for chemotherapy for acute promyelocytic leukaemia. Br. J. Pharmacol. 2008, 154, 940–948. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Rosario, S.E.; Self, W.T. Targeting selenium metabolism and selenoproteins: Novel avenues for drug discovery. Metallomics 2010, 2, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Tenjin, Y.; Matsuura, K.; Kudoh, S.; Usuki, S.; Yamada, T.; Matsuo, A.; Sato, Y.; Saito, H.; Fujino, K.; Wakimoto, J.; et al. Distinct transcriptional programs of SOX2 in different types of small cell lung cancers. Lab. Investig. 2020, 100, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.J.; Yang, M.H.; Zheng, J.C.; Li, B.; Nie, W. Arsenic trioxide inhibits cancer stem-like cells via down-regulation of Gli1 in lung cancer. Am. J. Transl. Res. 2016, 8, 1133–1143. [Google Scholar] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Ott, P.A.; Elez, E.; Hiret, S.; Kim, D.W.; Morosky, A.; Saraf, S.; Piperdi, B.; Mehnert, J.M. Pembrolizumab in patients with extensive-stage small-cell lung cancer: Results from the phase Ib KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 3823–3829. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 2018, 33, 853–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, J.W.; Garassino, M.C.; Chen, Y.; Reinmuth, N.; Hotta, K.; Poltoratskiy, A.; Trukhin, D.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. LBA86 Durvalumab (D) ± tremelimumab (T) + platinum-etoposide (EP) in 1L ES-SCLC: Characterization of long-term clinical benefit and tumour mutational burden (TMB) in CASPIAN. Ann. Oncol. 2020, 31, S1212–S1213. [Google Scholar] [CrossRef]
- Liu, S.V.; Horn, L.; Mok, T.; Mansfield, A.; De Boer, R.; Losonczy, G.; Sugawara, S.; Dziadziuszko, R.; Krzakowski, M.; Smolin, A.; et al. 1781MO IMpower133: Characterisation of long-term survivors treated first-line with chemotherapy ± atezolizumab in extensive-stage small cell lung cancer. Ann. Oncol. 2020, 31, S1032–S1033. [Google Scholar] [CrossRef]
- Swoboda, R.K.; Somasundaram, R.; Caputo-Gross, L.; Marincola, F.M.; Robbins, P.; Herlyn, M.; Herlyn, D. Antimelanoma CTL recognizes peptides derived from an ORF transcribed from the antisense strand of the 3’ untranslated region of TRIT1. Mol. Ther. Oncolytics 2015, 1, 14009. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Total (n = 39) | TRIT1 Non-Amplified (n = 35) | TRIT1 Amplified (n = 4) | p Value * |
|---|---|---|---|---|
| Age years [median (range)] | 65 (49–84) | 65 (49–84) | 65 (55–71) | |
| Gender | ||||
| Female | 8 (20.5%) | 7 (20.0%) | 1 (25.0%) | 1.000 |
| Male | 31 (79.5%) | 28 (80.0%) | 3 (75.0%) | |
| Smoker | ||||
| Yes | 33 (84.6%) | 29 (82.9%) | 4 (100.0%) | 1.000 |
| No | 3 (7.7%) | 3 (8.6%) | 0 (0.0%) | |
| Unknown | 3 (7.7%) | 3 (8.6%) | 0 (0.0%) | |
| Stage | ||||
| I | 10 (25.6%) | 7 (20.0%) | 3 (75.0%) | 0.098 |
| II | 4 (10.3%) | 4 (11.4%) | 0 (0.0%) | |
| III | 11 (28.2%) | 10 (28.6%) | 1 (25.0%) | |
| IV | 14 (35.9%) | 14 (40.0%) | 0 (0.0%) | |
| Type of clinical disease | ||||
| Localized | 18 (46.2%) | 15 (42.9%) | 3 (75.0%) | 0.318 |
| Extensive | 21 (53.8%) | 20 (57.1%) | 1 (25.0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coll-SanMartin, L.; Davalos, V.; Piñeyro, D.; Rosselló-Tortella, M.; Bueno-Costa, A.; Setien, F.; Villanueva, A.; Granada, I.; Ruiz-Xiviller, N.; Kotter, A.; et al. Gene Amplification-Associated Overexpression of the Selenoprotein tRNA Enzyme TRIT1 Confers Sensitivity to Arsenic Trioxide in Small-Cell Lung Cancer. Cancers 2021, 13, 1869. https://doi.org/10.3390/cancers13081869
Coll-SanMartin L, Davalos V, Piñeyro D, Rosselló-Tortella M, Bueno-Costa A, Setien F, Villanueva A, Granada I, Ruiz-Xiviller N, Kotter A, et al. Gene Amplification-Associated Overexpression of the Selenoprotein tRNA Enzyme TRIT1 Confers Sensitivity to Arsenic Trioxide in Small-Cell Lung Cancer. Cancers. 2021; 13(8):1869. https://doi.org/10.3390/cancers13081869
Chicago/Turabian StyleColl-SanMartin, Laia, Veronica Davalos, David Piñeyro, Margalida Rosselló-Tortella, Alberto Bueno-Costa, Fernando Setien, Alberto Villanueva, Isabel Granada, Neus Ruiz-Xiviller, Annika Kotter, and et al. 2021. "Gene Amplification-Associated Overexpression of the Selenoprotein tRNA Enzyme TRIT1 Confers Sensitivity to Arsenic Trioxide in Small-Cell Lung Cancer" Cancers 13, no. 8: 1869. https://doi.org/10.3390/cancers13081869
APA StyleColl-SanMartin, L., Davalos, V., Piñeyro, D., Rosselló-Tortella, M., Bueno-Costa, A., Setien, F., Villanueva, A., Granada, I., Ruiz-Xiviller, N., Kotter, A., Helm, M., Yokota, J., Kawabata-Iwakawa, R., Kohno, T., & Esteller, M. (2021). Gene Amplification-Associated Overexpression of the Selenoprotein tRNA Enzyme TRIT1 Confers Sensitivity to Arsenic Trioxide in Small-Cell Lung Cancer. Cancers, 13(8), 1869. https://doi.org/10.3390/cancers13081869