
FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers



 , , ,
, , , 

Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
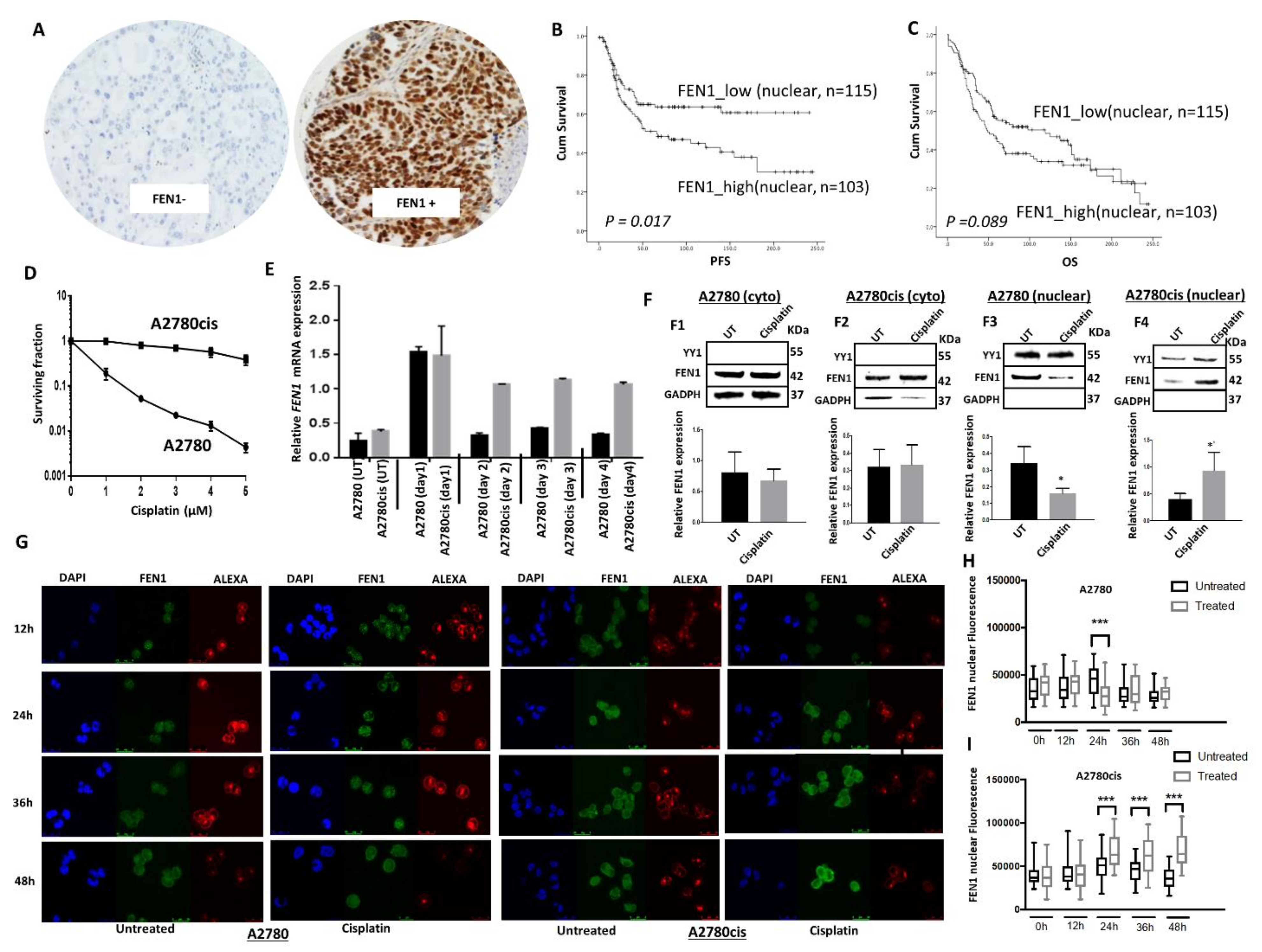
2.1. FEN1 Nuclear Overexpression Is Associated with Clinically Aggressive Epithelial Ovarian Cancers
2.2. Induction and Altered Sub-Cellular Localization of FEN1 Following Cisplatin Therapy
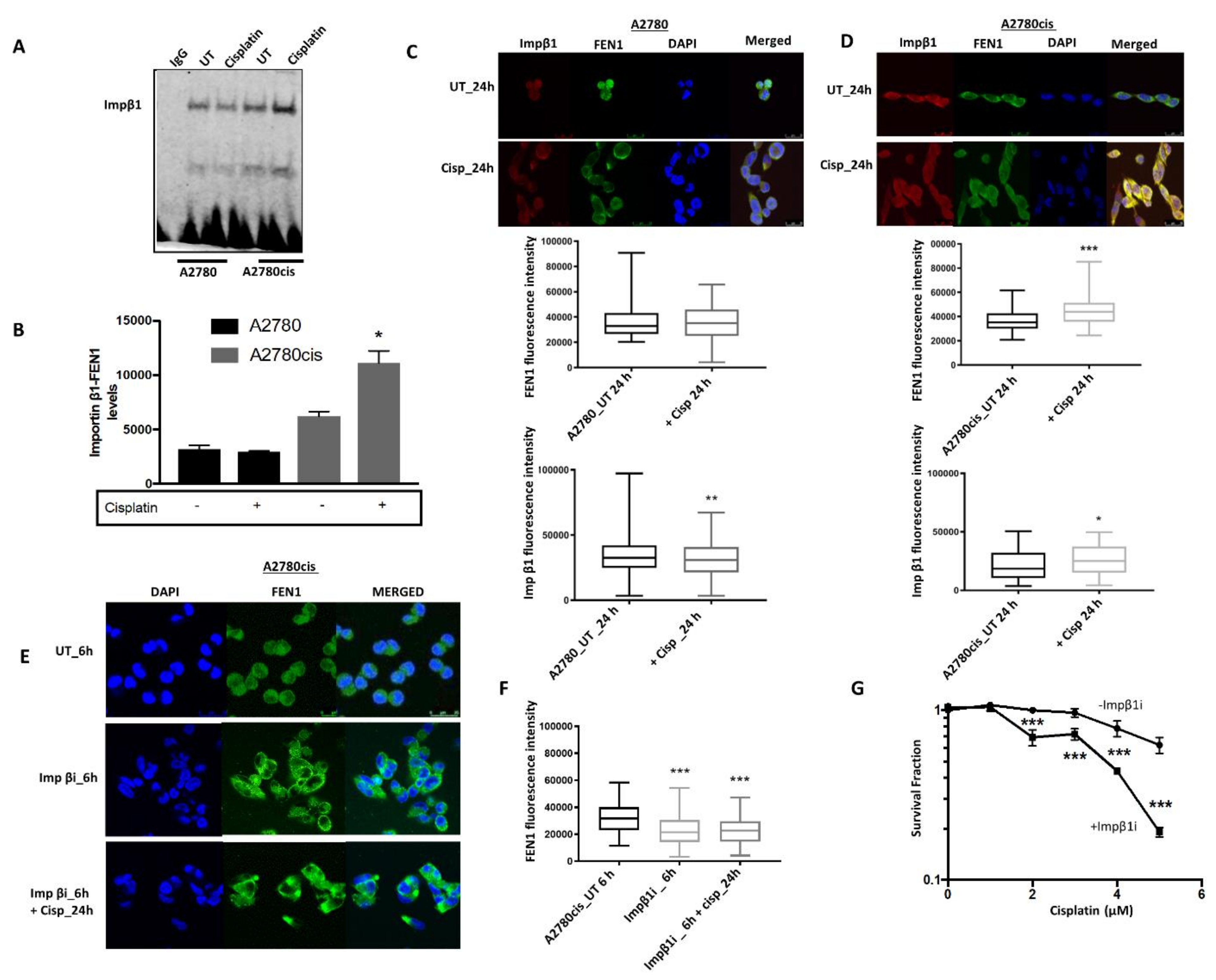
2.3. FEN1 Nuclear Localization in Response to Cisplatin in Mediated by Importin β
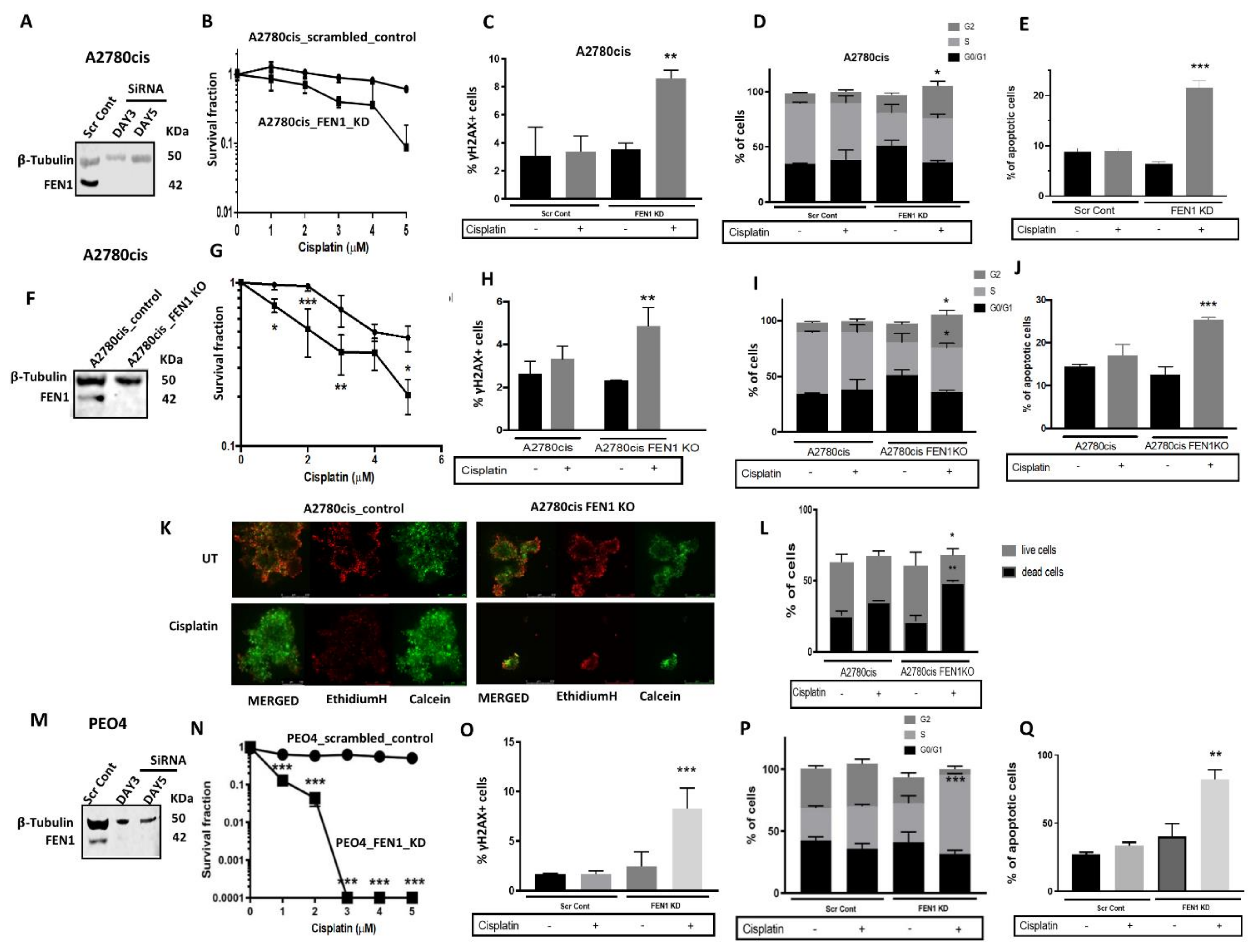
2.4. FEN1 Depletion or CRISPR Inactivation Reverses Platinum Resistance in Ovarian Cancer Cells
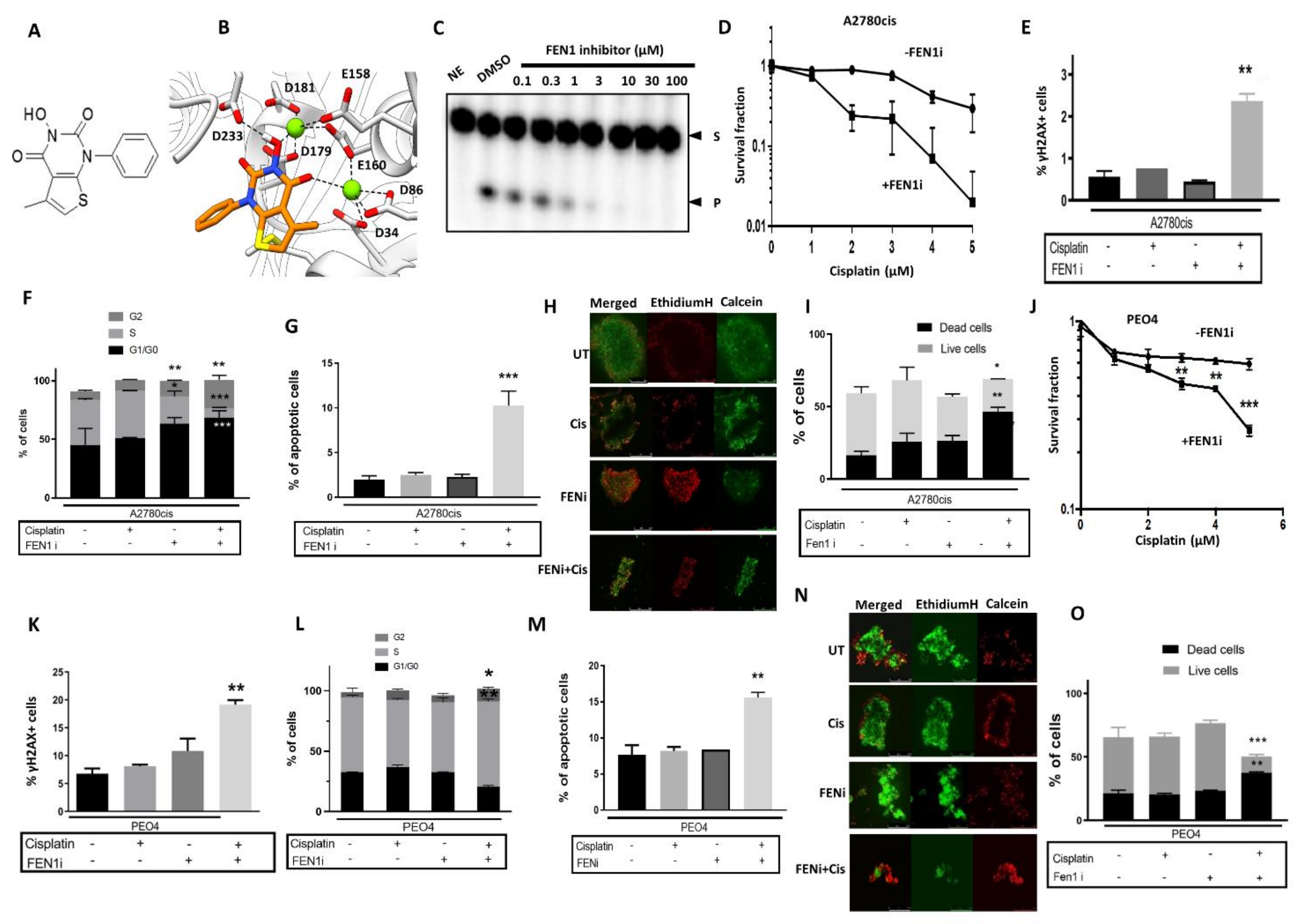
2.5. FEN1 Small Molecule Inhibitor Potentiates Cisplatin Cytotoxicity in Ovarian Cancer Cells
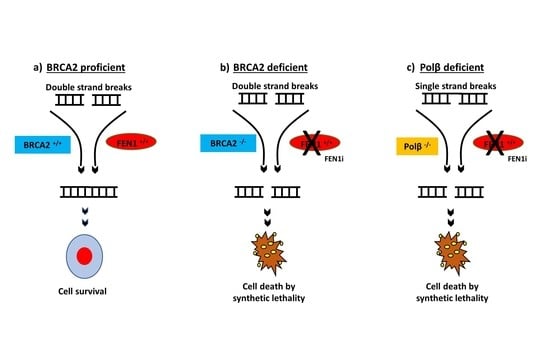
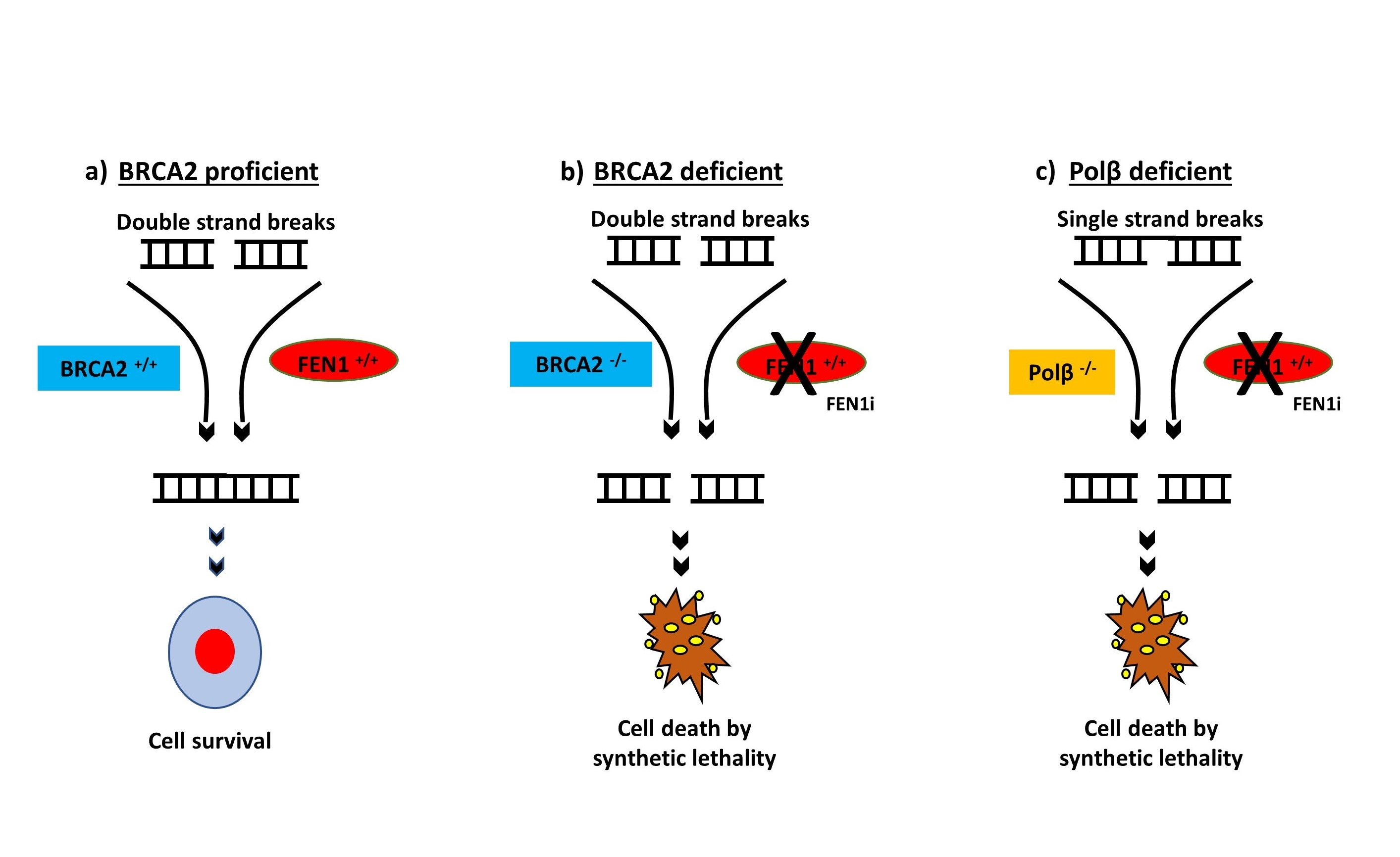
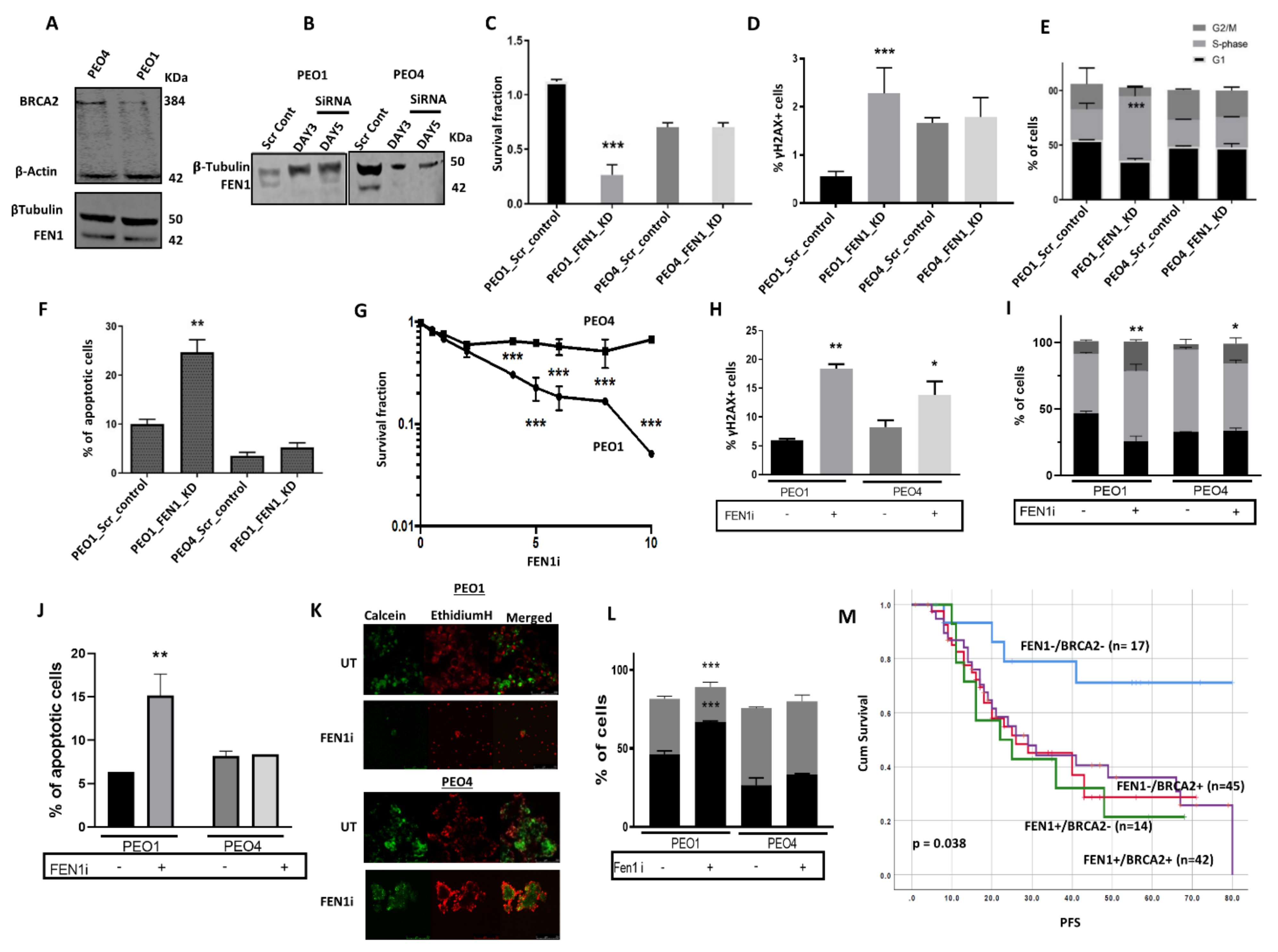
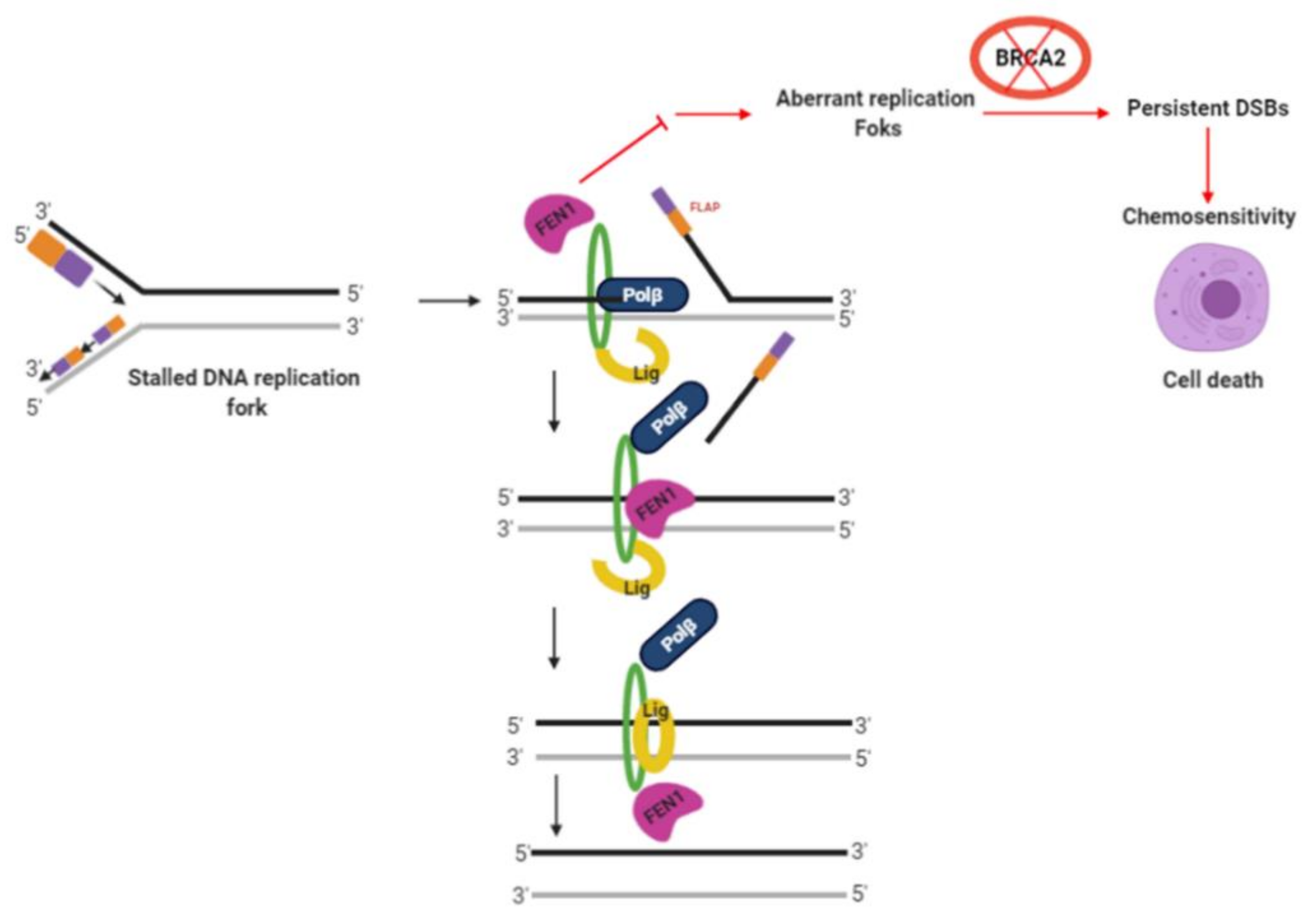
2.6. FEN1 Depletion or Inhibition Is Synthetically Lethal with BRCA2 Deficiency
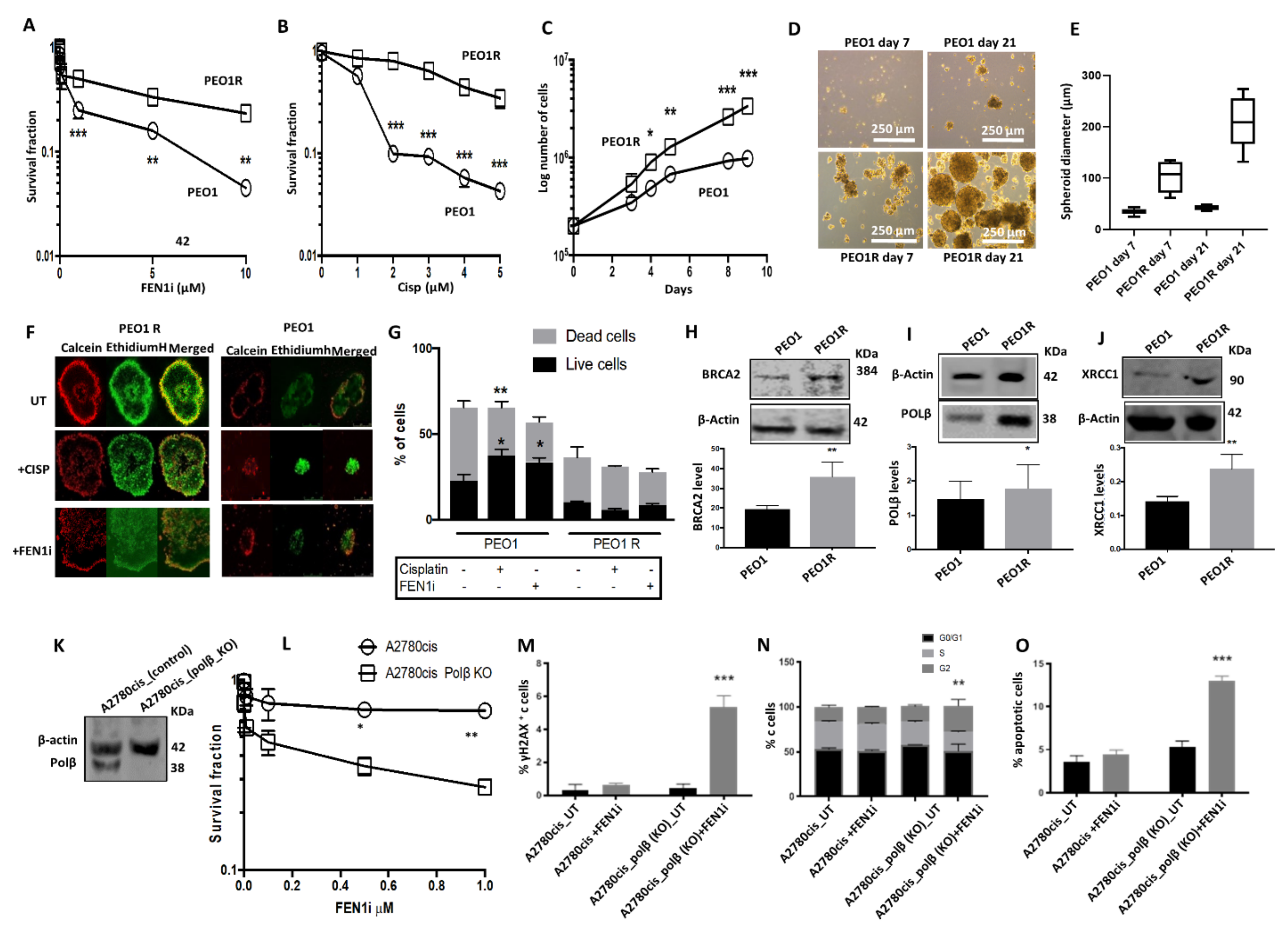
2.7. FEN1i Resistant PEO1R Cells Re-Express BRCA2
2.8. FEN1i Is Synthetically Lethal with POLβ Deficient, but Not with a Deficiency in XRCC1, ATM or MRE11
2.9. High through-Put Screening (HTS) and Identification of FEN1 Inhibitors
3. Discussion
4. Methods
4.1. Clinical Study
FEN1 Protein Level in Ovarian Cancers
4.2. Pre-Clinical Study
4.2.1. Cell Lines and Tissue Culture
4.2.2. Generation of FEN1 Knock-Downs (KD)
4.2.3. CRISPR Editing of FEN1
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopez-Martinez, D.; Liang, C.-C.; Cohn, M.A. Cellular response to DNA interstrand crosslinks: The Fanconi anemia pathway. Cell. Mol. Life Sci. 2017, 73, 3097–3114. [Google Scholar] [CrossRef] [Green Version]
- Spyridon, P.; Basourakos, L.L.; Ana, M.; Aparicio, A.; Paul, G.C.; Jeri, K.; Timothy, C.T. Combination Platinum-based and DNA Damage Response-targeting Cancer Therapy: Evolution and Future Directions. Curr. Med. Chem. 2017, 24, 1586–1606. [Google Scholar]
- Santos, N.A.; Bezerra, C.S.; Martins, N.M.; Curti, C.; Bianchi, M.L.; Santos, A.C. Hydroxyl radical scavenger ameliorates cisplatin-induced nephrotoxicity by preventing oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Cancer Chemother. Pharmacol. 2008, 61, 145–155. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage: From mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Jia, J.; Finger, L.D.; Guo, Z.; Zer, C.; Shen, B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011, 39, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Larsen, E.; Gran, C.; Saether, B.E.; Seeberg, E.; Klungland, A. Proliferation failure and gamma radiation sensitivity of Fen1 null mutant mice at the blastocyst stage. Mol. Cell. Biol. 2003, 23, 5346–5353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucherlapati, M.; Nguyen, A.; Kuraguchi, M.; Yang, K.; Fan, K.; Bronson, R.; Wei, K.; Lipkin, M.; Edelmann, W.; Kucherlapati, R. Tumor progression in Apc(1638N) mice with Exo1 and Fen1 deficiencies. Oncogene 2007, 26, 6297–6306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, E.; Kleppa, L.; Meza, T.J.; Meza-Zepeda, L.A.; Rada, C.; Castellanos, C.G.; Lien, G.F.; Nesse, G.J.; Neuberger, M.S.; Laerdahl, J.K.; et al. Early-onset lymphoma and extensive embryonic apoptosis in two domain-specific Fen1 mice mutants. Cancer Res. 2008, 68, 4571–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Dai, H.; Zhou, M.; Li, M.; Singh, P.; Qiu, J.; Tsark, W.; Huang, Q.; Kernstine, K.; Zhang, X.; et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat. Med. 2007, 13, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhou, C.; Zhou, L.; Peng, L.; Li, D.; Zhang, X.; Zhou, M.; Kuang, P.; Yuan, Q.; Song, X.; et al. Functional FEN1 genetic variants contribute to risk of hepatocellular carcinoma, esophageal cancer, gastric cancer and colorectal cancer. Carcinogenesis 2012, 33, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Guo, H.; Wu, C.; He, Y.; Yu, D.; Zhou, L.; Wang, F.; Xu, J.; Tan, W.; Wang, G.; et al. Functional FEN1 polymorphisms are associated with DNA damage levels and lung cancer risk. Hum. Mutat. 2009, 30, 1320–1328. [Google Scholar] [CrossRef]
- Kim, I.S. Down-regulation of human FEN-1 gene expression during differentiation of promyelocytic leukemia cells. Exp. Mol. Med. 1998, 30, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Girard, L.; Sekine, I.; Sunaga, N.; Ramirez, R.D.; Kamibayashi, C.; Minna, J.D. Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer. Oncogene 2003, 22, 7243–7246. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Sohn, H.Y.; Yoon, S.Y.; Oh, J.H.; Yang, J.O.; Kim, J.H.; Song, K.S.; Rho, S.M.; Yoo, H.S.; Kim, Y.S.; et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin. Cancer Res. 2005, 11, 473–482. [Google Scholar]
- Krause, A.; Combaret, V.; Iacono, I.; Lacroix, B.; Compagnon, C.; Bergeron, C.; Valsesia-Wittmann, S.; Leissner, P.; Mougin, B.; Puisieux, A. Genome-wide analysis of gene expression in neuroblastomas detected by mass screening. Cancer Lett. 2005, 225, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Yang, M.; Dai, H.; Yu, D.; Huang, Q.; Tan, W.; Kernstine, K.H.; Lin, D.; Shen, B. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol. Cancer Res. 2008, 6, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.; Russell, R.; Albarakati, N.; Maloney, D.J.; Dorjsuren, D.; Rueda, O.M.; Moseley, P.; Mohan, V.; Sun, H.; Abbotts, R.; et al. Genomic and protein expression analysis reveals flap endonuclease 1 (FEN1) as a key biomarker in breast and ovarian cancer. Mol. Oncol. 2014, 8, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Keymeulen, S.; Nelson, R.; Tong, T.R.; Yuan, Y.C.; Yun, X.; Liu, Z.; Lopez, J.; Raz, D.J.; Kim, J.Y. Overexpression of Flap Endonuclease 1 Correlates with Enhanced Proliferation and Poor Prognosis of Non-Small-Cell Lung Cancer. Am. J. Pathol. 2018, 188, 242–251. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Luo, L.; Zhu, H.; Yang, H.; Zhang, Y.; Wu, H.; Sun, H.; Jiang, F.; Kathera, C.S.; Liu, L.; et al. FEN1 promotes tumor progression and confers cisplatin resistance in non-small-cell lung cancer. Mol. Oncol. 2017, 11, 640–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Kong, J.; Qiu, Y.; Yang, X.; Wang, W.; Yan, L. Identification of core genes and outcomes in hepatocellular carcinoma by bioinformatics analysis. J. Cell. Biochem. 2018, 120, 10069–10081. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Ye, W.; Cai, L.; Meng, X.; Ke, G.; Huang, C.; Peng, Z.; Yu, Y.; Golden, J.A.; Tartakoff, A.M.; et al. The roles of multiple importins for nuclear import of murine aristaless-related homeobox protein. J. Biol. Chem. 2009, 284, 20428–20439. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.; Bono, F.; Jinek, M.; Conti, E. Structural biology of nucleocytoplasmic transport. Annu. Rev. Biochem. 2007, 76, 647–671. [Google Scholar] [CrossRef]
- Qiu, J.; Li, X.; Frank, G.; Shen, B. Cell cycle-dependent and DNA damage-inducible nuclear localization of FEN-1 nuclease is consistent with its dual functions in DNA replication and repair. J. Biol. Chem. 2001, 276, 4901–4908. [Google Scholar] [CrossRef] [Green Version]
- Dorjsuren, D.; Kim, D.; Maloney, D.J.; Wilson, D.M., 3rd; Simeonov, A. Complementary non-radioactive assays for investigation of human flap endonuclease 1 activity. Nucleic Acids Res. 2011, 39, e11. [Google Scholar] [CrossRef] [Green Version]
- Exell, J.C.; Thompson, M.J.; Finger, L.D.; Shaw, S.J.; Debreczeni, J.; Ward, T.A.; McWhirter, C.; Sioberg, C.L.; Martinez Molina, D.; Abbott, W.M.; et al. Cellularly active N-hydroxyurea FEN1 inhibitors block substrate entry to the active site. Nat. Chem. Biol. 2016, 12, 815–821. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Fatah, T.; Sultana, R.; Abbotts, R.; Hawkes, C.; Seedhouse, C.; Chan, S.; Madhusudan, S. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int. J. Cancer 2013, 132, 2778–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Barros, A.C.; Takeda, A.A.; Chang, C.W.; Kobe, B.; Fontes, M.R. Structural basis of nuclear import of flap endonuclease 1 (FEN1). Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulkes, W.D.; Shuen, A.Y. In brief: BRCA1 and BRCA2. J. Pathol. 2013, 230, 347–349. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Sitz, J.; Grapton, D.; Orthwein, A. BRCA2 functions: From DNA repair to replication fork stabilization. Endocr. Relat. Cancer 2016, 23, T1–T17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, J.S.; Baldeyron, C.; Carreira, A. Molding BRCA2 function through its interacting partners. Cell Cycle 2015, 14, 3389–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, K.N.; Domchek, S.M. Cancer treatment according to BRCA1 and BRCA2 mutations. Nat. Rev. Clin. Oncol. 2012, 9, 520–528. [Google Scholar] [CrossRef]
- Ramus, S.J.; Gayther, S.A. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol. Oncol. 2009, 3, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Dizon, D.S. PARP inhibitors for targeted treatment in ovarian cancer. Lancet 2017, 390, 1929–1930. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.A.; McHugh, P.J.; Durant, S.T. Small molecule inhibitors uncover synthetic genetic interactions of human flap endonuclease 1 (FEN1) with DNA damage response genes. PLoS ONE 2017, 12, e0179278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e6. [Google Scholar] [CrossRef] [Green Version]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331.e3. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesquita, K.A.; Ali, R.; Doherty, R.; Toss, M.S.; Miligy, I.; Alblihy, A.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M., III; et al. FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers. Cancers 2021, 13, 1866. https://doi.org/10.3390/cancers13081866
Mesquita KA, Ali R, Doherty R, Toss MS, Miligy I, Alblihy A, Dorjsuren D, Simeonov A, Jadhav A, Wilson DM III, et al. FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers. Cancers. 2021; 13(8):1866. https://doi.org/10.3390/cancers13081866
Chicago/Turabian StyleMesquita, Katia A., Reem Ali, Rachel Doherty, Michael S. Toss, Islam Miligy, Adel Alblihy, Dorjbal Dorjsuren, Anton Simeonov, Ajit Jadhav, David M. Wilson, III, and et al. 2021. "FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers" Cancers 13, no. 8: 1866. https://doi.org/10.3390/cancers13081866
APA StyleMesquita, K. A., Ali, R., Doherty, R., Toss, M. S., Miligy, I., Alblihy, A., Dorjsuren, D., Simeonov, A., Jadhav, A., Wilson, D. M., III, Hickson, I., Tatum, N. J., Rakha, E. A., & Madhusudan, S. (2021). FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers. Cancers, 13(8), 1866. https://doi.org/10.3390/cancers13081866