
Fibroblast MMP14-Dependent Collagen Processing Is Necessary for Melanoma Growth

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Melanoma Spheroids
2.3. Tumor Grafting Experiments
2.4. Atomic Force Microscopy (AFM)
2.5. Immunoblot
2.6. Immunofluorescence Staining
2.7. Picrosirius Red Staining
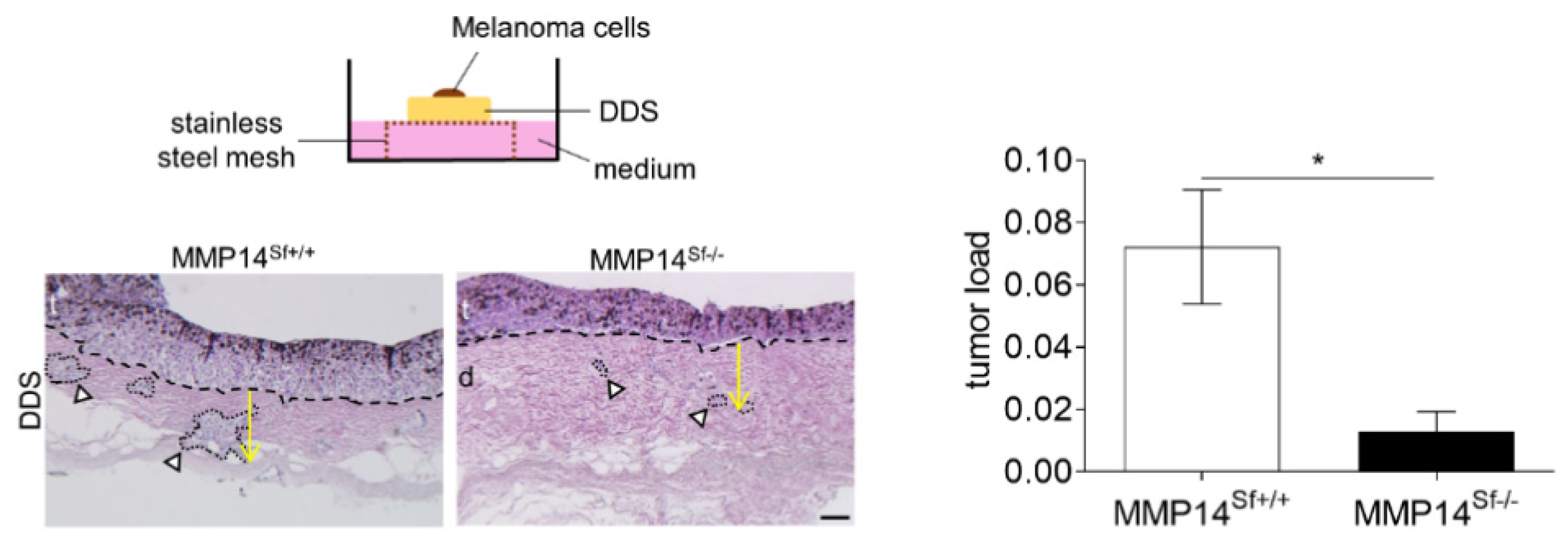
2.8. Invasion Assays Using De-Epidermized Devitalized Skin (DDS)
2.9. Second Harmonic Generation
2.10. RNA Isolation and Real-Time PCR
2.11. Statistics
2.12. BrdU (5-bromo-2′-deoxyuridine) Incorporation Assay
3. Results
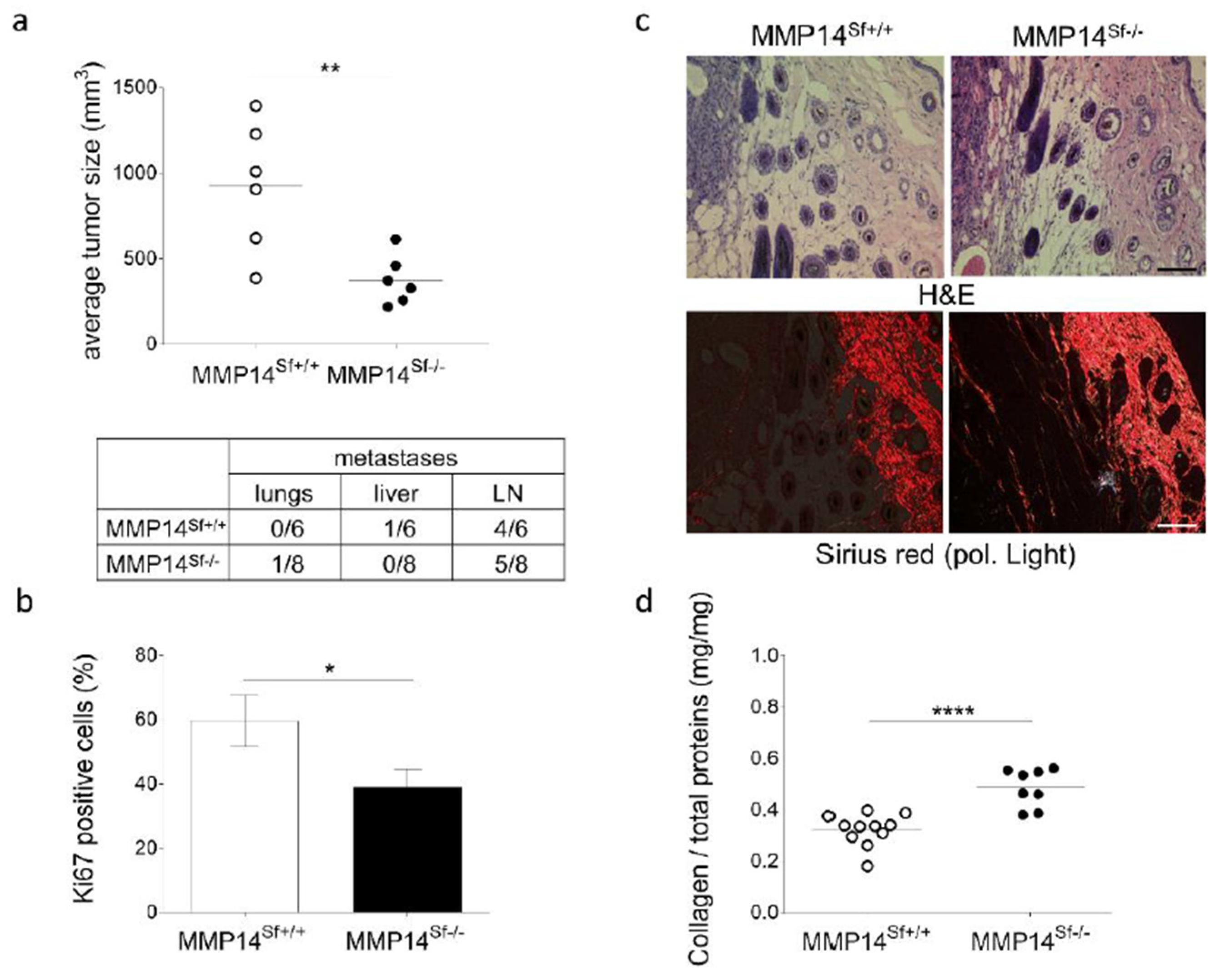
3.1. Reduced Melanoma Growth in MMP14Sf−/− Mice
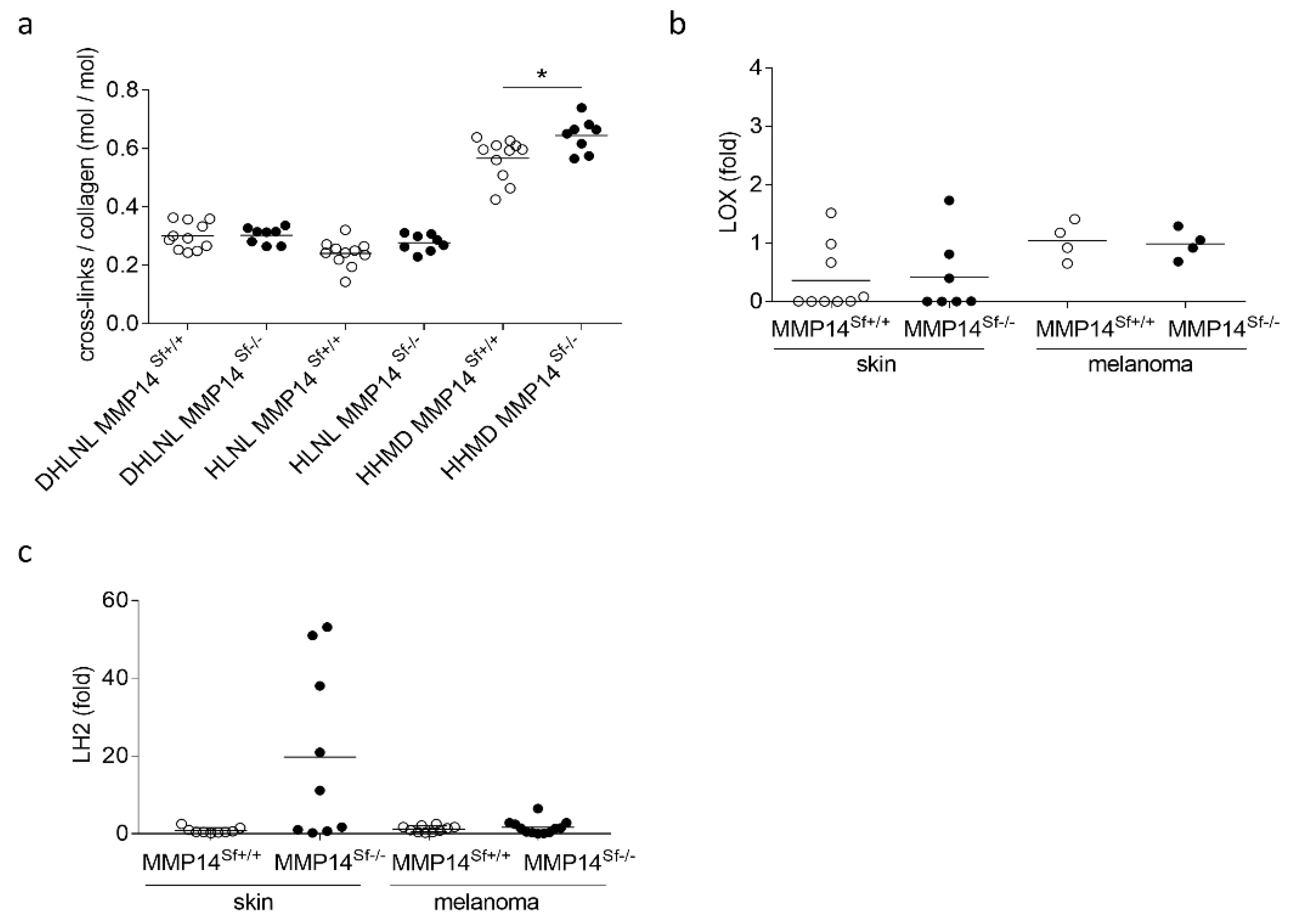
3.2. Alterations in the Peritumoral Matrix of MMP14Sf−/− Mice
3.3. Reduced Area of Melanoma Tumor Nests in Skin Composites
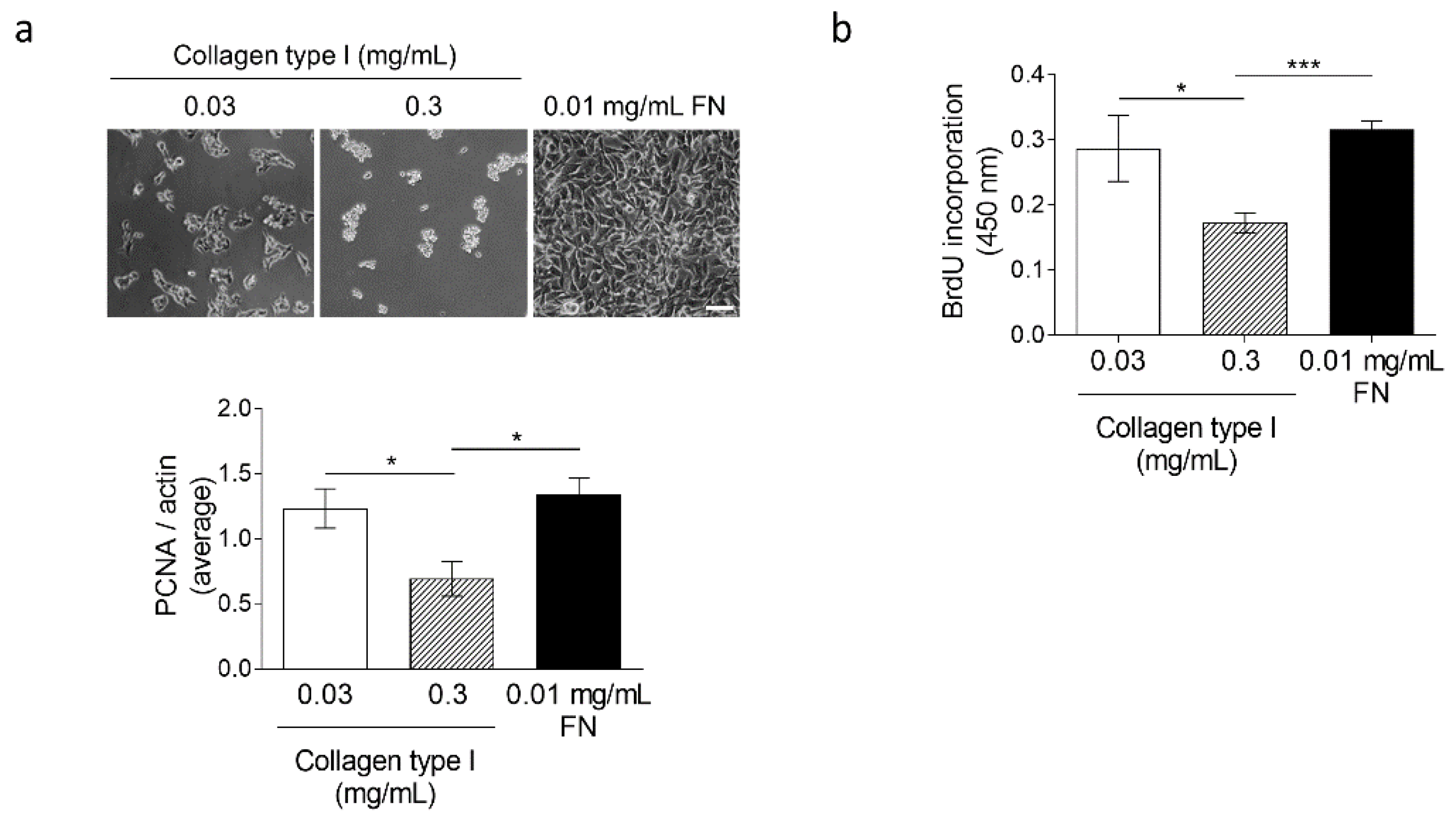
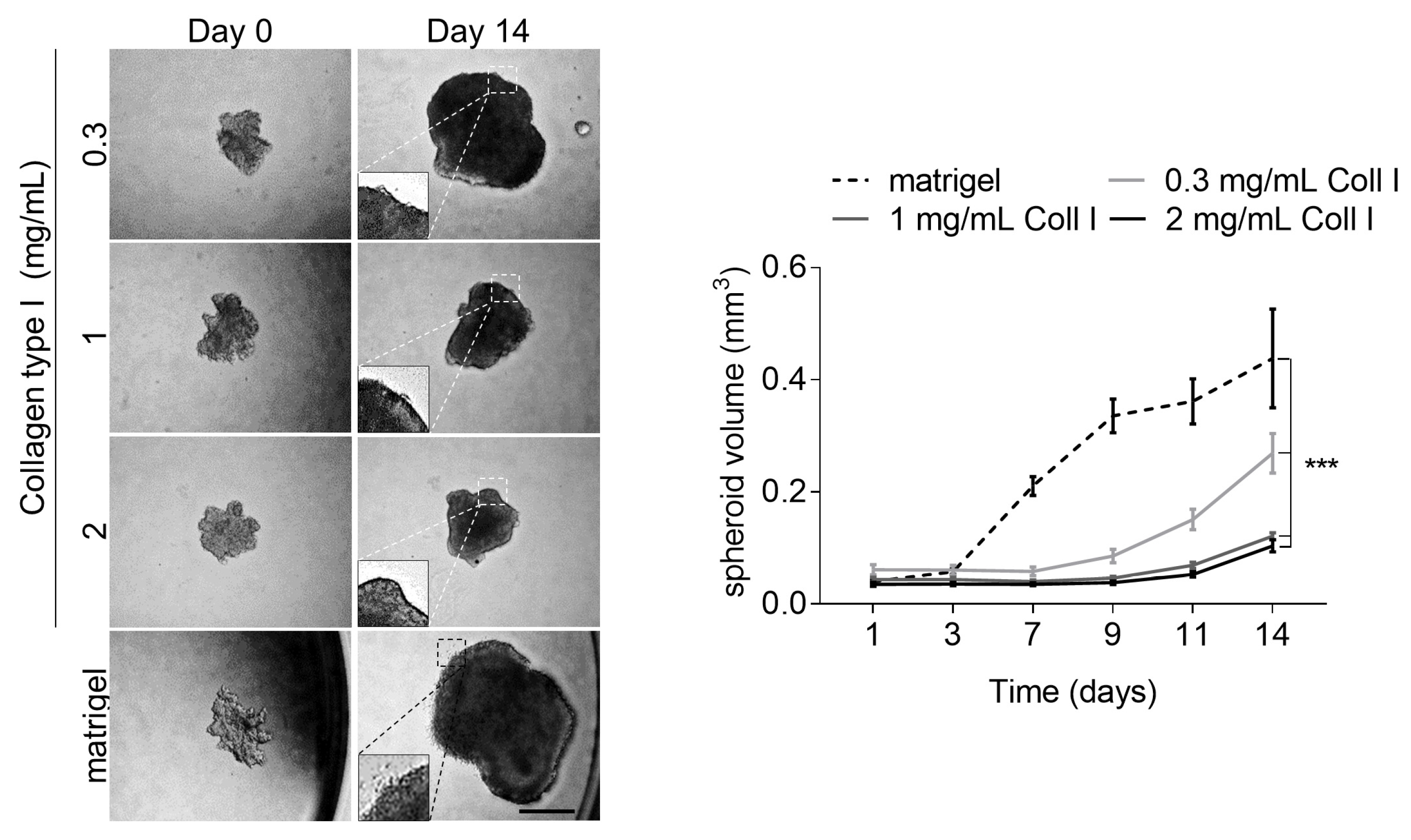
3.4. High Collagen Type I and Tissue Tension Levels Negatively Modulate Melanoma Growth
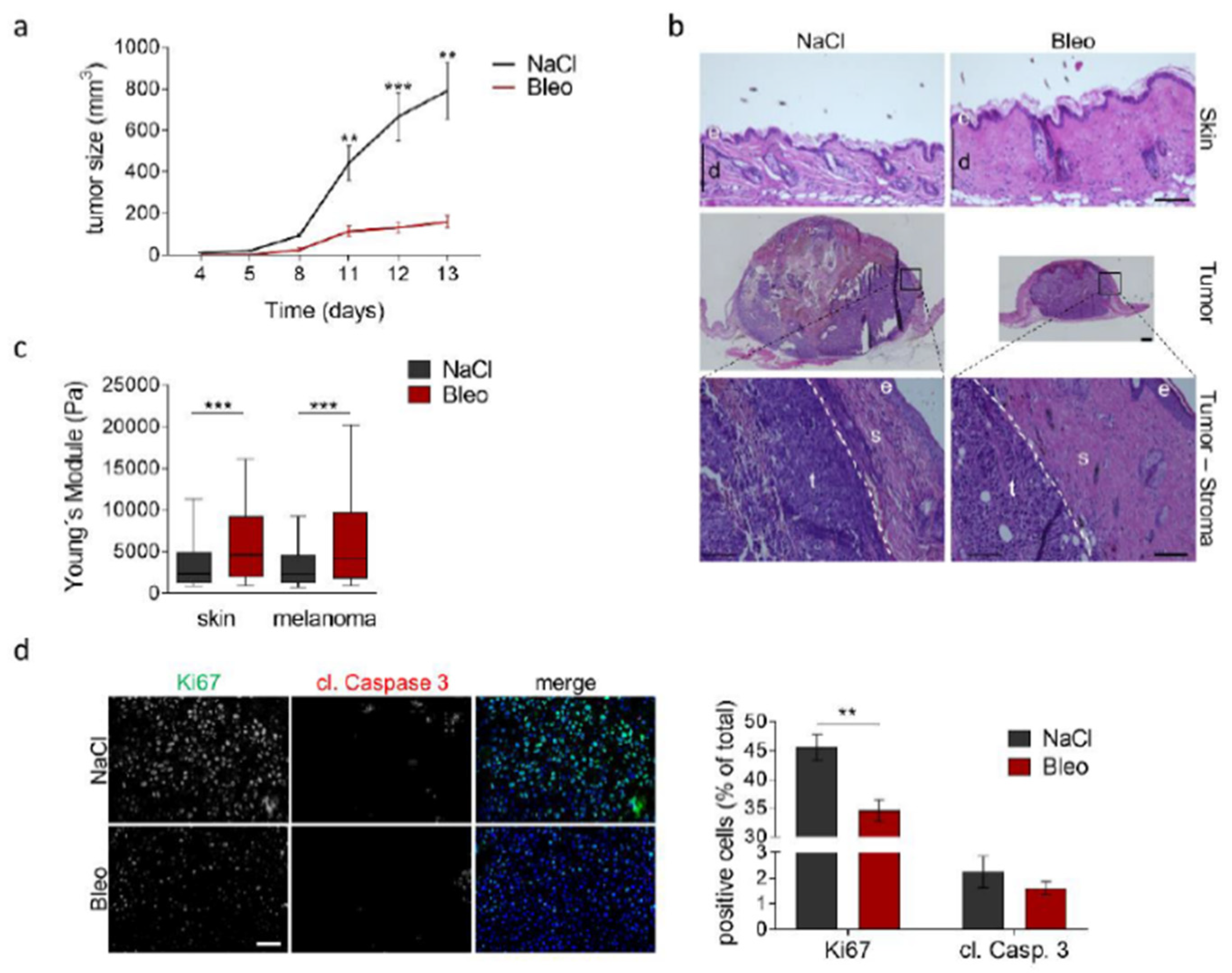
3.5. Increased Tissue Stiffness and Matrix Density Levels Lead to Reduced Melanoma Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clause, K.C.; Barker, T.H. Extracellular matrix signaling in morphogenesis and repair. Curr. Opin. Biotechnol. 2013, 24, 830–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol. Cancer Res. 2016, 14, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, N.; Mauch, C.; Zigrino, P. Metalloproteinases in melanoma. Eur. J. Cell Biol. 2014, 93, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Fernandez, A.; Soria-Valles, C.; Osorio, F.G.; Gutierrez-Abril, J.; Garabaya, C.; Aguirre, A.; Fueyo, A.; Fernandez-Garcia, M.S.; Puente, X.S.; Lopez-Otin, C. Loss of MT1-MMP causes cell senescence and nuclear defects which can be reversed by retinoic acid. EMBO J. 2015, 34, 1875–1888. [Google Scholar] [CrossRef] [Green Version]
- Holmbeck, K.; Bianco, P.; Caterina, J.; Yamada, S.; Kromer, M.; Kuznetsov, S.A.; Mankani, M.; Robey, P.G.; Poole, A.R.; Pidoux, I.; et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999, 99, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Apte, S.S.; Soininen, R.; Cao, R.; Baaklini, G.Y.; Rauser, R.W.; Wang, J.; Cao, Y.; Tryggvason, K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. USA 2000, 97, 4052–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zigrino, P.; Brinckmann, J.; Niehoff, A.; Lu, Y.; Giebeler, N.; Eckes, B.; Kadler, K.E.; Mauch, C. Fibroblast-Derived MMP-14 Regulates Collagen Homeostasis in Adult Skin. J. Investig. Dermatol. 2016, 136, 1575–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapinska, A.M.; Fields, G.B. The Expanding Role of MT1-MMP in Cancer Progression. Pharmaceuticals 2019, 12, 77. [Google Scholar] [CrossRef] [Green Version]
- Kurschat, P.; Wickenhauser, C.; Groth, W.; Krieg, T.; Mauch, C. Identification of activated matrix metalloproteinase-2 (MMP-2) as the main gelatinolytic enzyme in malignant melanoma by in situ zymography. J. Pathol. 2002, 197, 179–187. [Google Scholar] [CrossRef]
- Kurschat, P.; Zigrino, P.; Nischt, R.; Breitkopf, K.; Steurer, P.; Klein, C.E.; Krieg, T.; Mauch, C. Tissue inhibitor of matrix metalloproteinase-2 regulates matrix metalloproteinase-2 activation by modulation of membrane-type 1 matrix metalloproteinase activity in high and low invasive melanoma cell lines. J. Biol. Chem. 1999, 274, 21056–21062. [Google Scholar] [CrossRef] [Green Version]
- Zigrino, P.; Drescher, C.; Mauch, C. Collagen-induced proMMP-2 activation by MT1-MMP in human dermal fibroblasts and the possible role of alpha2beta1 integrins. Eur. J. Cell Biol. 2001, 80, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.B.; Westphal, J.R.; Zendman, A.J.; Becker, J.C.; Ruiter, D.J.; van Muijen, G.N. Expression and activation of matrix metalloproteinase-2 (MMP-2) and its co-localization with membrane-type 1 matrix metalloproteinase (MT1-MMP) correlate with melanoma progression. J. Pathol. 2000, 191, 245–256. [Google Scholar] [CrossRef]
- Thakur, V.; Bedogni, B. The membrane tethered matrix metalloproteinase MT1-MMP at the forefront of melanoma cell invasion and metastasis. Pharmacol. Res. 2016, 111, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Zigrino, P.; Kuhn, I.; Bäuerle, T.; Zamek, J.; Fox, J.W.; Neumann, S.; Licht, A.; Schorpp-Kistner, M.; Angel, P.; Mauch, C. Stromal expression of MMP-13 is required for melanoma invasion and metastasis. J. Investig. Dermatol. 2009, 129, 2686–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikova, V.O.; Bolshakov, S.V.; Walker, C.; Ananthaswamy, H.N. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene 2004, 23, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley, K.S.; Haass, N.K.; Brafford, P.A.; Lioni, M.; Flaherty, K.T.; Herlyn, M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol. Cancer Ther. 2006, 5, 1136–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Takagawa, S.; Katayama, I.; Yamazaki, K.; Hamazaki, Y.; Shinkai, H.; Nishioka, K. Animal model of sclerotic skin. I: Local injections of bleomycin induce sclerotic skin mimicking scleroderma. J. Investig. Dermatol. 1999, 112, 456–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennhöfer, R.; Kurschat, P.; Zigrino, P.; Klose, A.; Bosserhoff, A.; van Muijen, G.; Krieg, T.; Mauch, C.; Hunzelmann, N. Invasion of melanoma cells into dermal connective tissue in vitro: Evidence for an important role of cysteine proteases. Int. J. Cancer 2003, 106, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Natal, R.A.; Vassallo, J.; Paiva, G.R.; Pelegati, V.B.; Barbosa, G.O.; Mendonça, G.R.; Bondarik, C.; Derchain, S.F.; Carvalho, H.F.; Lima, C.S.; et al. Collagen analysis by second-harmonic generation microscopy predicts outcome of luminal breast cancer. Tumor Biol. 2018, 40, 1010428318770953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezakhaniha, R.; Agianniotis, A.; Schrauwen, J.T.; Griffa, A.; Sage, D.; Bouten, C.V.; van de Vosse, F.N.; Unser, M.; Stergiopulos, N. Experimental investigation of collagen waviness and orientation in the arterial adventitia using confocal laser scanning microscopy. Biomech. Model. Mechanobiol. 2012, 11, 461–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, M.W.; Keely, P.J. Why the stroma matters in breast cancer: Insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adhes. Migr. 2012, 6, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinckmann, J.; Neess, C.M.; Gaber, Y.; Sobhi, H.; Notbohm, H.; Hunzelmann, N.; Fietzek, P.P.; Muller, P.K.; Risteli, J.; Gebker, R.; et al. Different pattern of collagen cross-links in two sclerotic skin diseases: Lipodermatosclerosis and circumscribed scleroderma. J. Investig. Dermatol. 2001, 117, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Terajima, M.; Yang, Y.; Sun, L.; Ahn, Y.H.; Pankova, D.; Puperi, D.S.; Watanabe, T.; Kim, M.P.; Blackmon, S.H.; et al. Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. J. Clin. Investig. 2015, 125, 1147–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trackman, P.C. Enzymatic and non-enzymatic functions of the lysyl oxidase family in bone. Matrix Biol. 2016, 52–54, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Slot, A.J.; Zuurmond, A.M.; Bardoel, A.F.; Wijmenga, C.; Pruijs, H.E.; Sillence, D.O.; Brinckmann, J.; Abraham, D.J.; Black, C.M.; Verzijl, N.; et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 2003, 278, 40967–40972. [Google Scholar] [CrossRef] [Green Version]
- Jang, I.; Beningo, K.A. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Henriet, P.; Zhong, Z.D.; Brooks, P.C.; Weinberg, K.I.; DeClerck, Y.A. Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27KIP1. Proc. Natl. Acad. Sci. USA 2000, 97, 10026–10031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miskolczi, Z.; Smith, M.P.; Rowling, E.J.; Ferguson, J.; Barriuso, J.; Wellbrock, C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene 2018, 37, 3166–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planska, D.; Burocziova, M.; Strnadel, J.; Horak, V. Immunohistochemical Analysis of Collagen IV and Laminin Expression in Spontaneous Melanoma Regression in the Melanoma-Bearing Libechov Minipig. Acta Histochem. Cytochem. 2015, 48, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taloni, A.; Alemi, A.A.; Ciusani, E.; Sethna, J.P.; Zapperi, S.; La Porta, C.A. Mechanical properties of growing melanocytic nevi and the progression to melanoma. PLoS ONE 2014, 9, e94229. [Google Scholar] [CrossRef] [Green Version]
- Zigrino, P.; Mauch, C. Proteases in Melanoma. In Melanoma Development. Molecular Biology, Genetics and Clinical Application, 2nd ed.; Springer Book: Vienna, Austria, 2016. [Google Scholar]
- Hofmann, U.B.; Westphal, J.R.; Van Muijen, G.N.; Ruiter, D.J. Matrix metalloproteinases in human melanoma. J. Investig. Dermatol. 2000, 115, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nia, H.T.; Munn, L.L.; Jain, R.K. Physical traits of cancer. Science 2020, 370, eaaz0868. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Papalazarou, V.; Salmeron-Sanchez, M.; Machesky, L.M. Tissue engineering the cancer microenvironment-challenges and opportunities. Biophys. Rev. 2018, 10, 1695–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, K.M. Non-enzymatic glycation of collagen in aging and diabetes. Proc. Soc. Exp. Biol. Med. 1991, 196, 17–29. [Google Scholar] [CrossRef]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Kraning-Rush, C.M.; Reinhart-King, C.A. Controlling matrix stiffness and topography for the study of tumor cell migration. Cell Adhes. Migr. 2012, 6, 274–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [Green Version]
- Ilina, O.; Friedl, P. Mechanisms of collective cell migration at a glance. J. Cell Sci. 2009, 122, 3203–3208. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Knäuper, V.; Bailey, L.; Worley, J.R.; Soloway, P.; Patterson, M.L.; Murphy, G. Cellular activation of proMMP-13 by MT1-MMP depends on the C-terminal domain of MMP-13. FEBS Lett. 2002, 532, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef]
- Busam, K.J. Desmoplastic melanoma. Clin. Lab. Med. 2011, 31, 321–330. [Google Scholar] [CrossRef]
- Blessing, K.; McLaren, K.M. Histological regression in primary cutaneous melanoma: Recognition, prevalence and significance. Histopathology 1992, 20, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Bekisch, B.; Uerlich, M.; Haller, O.; Bieber, T.; Tüting, T. Type I interferon-associated recruitment of cytotoxic lymphocytes: A common mechanism in regressive melanocytic lesions. Am. J. Clin. Pathol. 2005, 124, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.L.; Nguyen, A.M.; Roder, D.; Roberts-Thomson, P. Risk of cancer in patients with scleroderma: A population based cohort study. Ann. Rheum. Dis. 2003, 62, 728–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pach, E.; Brinckmann, J.; Rübsam, M.; Kümper, M.; Mauch, C.; Zigrino, P. Fibroblast MMP14-Dependent Collagen Processing Is Necessary for Melanoma Growth. Cancers 2021, 13, 1984. https://doi.org/10.3390/cancers13081984
Pach E, Brinckmann J, Rübsam M, Kümper M, Mauch C, Zigrino P. Fibroblast MMP14-Dependent Collagen Processing Is Necessary for Melanoma Growth. Cancers. 2021; 13(8):1984. https://doi.org/10.3390/cancers13081984
Chicago/Turabian StylePach, Elke, Jürgen Brinckmann, Matthias Rübsam, Maike Kümper, Cornelia Mauch, and Paola Zigrino. 2021. "Fibroblast MMP14-Dependent Collagen Processing Is Necessary for Melanoma Growth" Cancers 13, no. 8: 1984. https://doi.org/10.3390/cancers13081984
APA StylePach, E., Brinckmann, J., Rübsam, M., Kümper, M., Mauch, C., & Zigrino, P. (2021). Fibroblast MMP14-Dependent Collagen Processing Is Necessary for Melanoma Growth. Cancers, 13(8), 1984. https://doi.org/10.3390/cancers13081984