
IL-33-Induced Transcriptional Activation of LPIN1 Accelerates Breast Tumorigenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Establishment of Stable Cell Lines
2.2. Antibodies and Reagents
2.3. Mammalian Expression Plasmids and Small Interfering RNA
2.4. Cell Proliferation Assay (Bromodeoxyuridine Incorporation)
2.5. RNA Extraction and Real-Time PCR
2.6. Protein Immunoblotting and Immunoprecipitation
2.7. Chromatin Immunoprecipitation
2.8. Anchorage-Independent Cellular Transformation Assay (Soft Agar Assay)
2.9. Tumorigenicity Assay in BALB/c Mice
2.10. Tumor Samples
2.11. Immunohistochemical Staining Analysis
2.12. Quantification and Statistical Analysis
3. Results
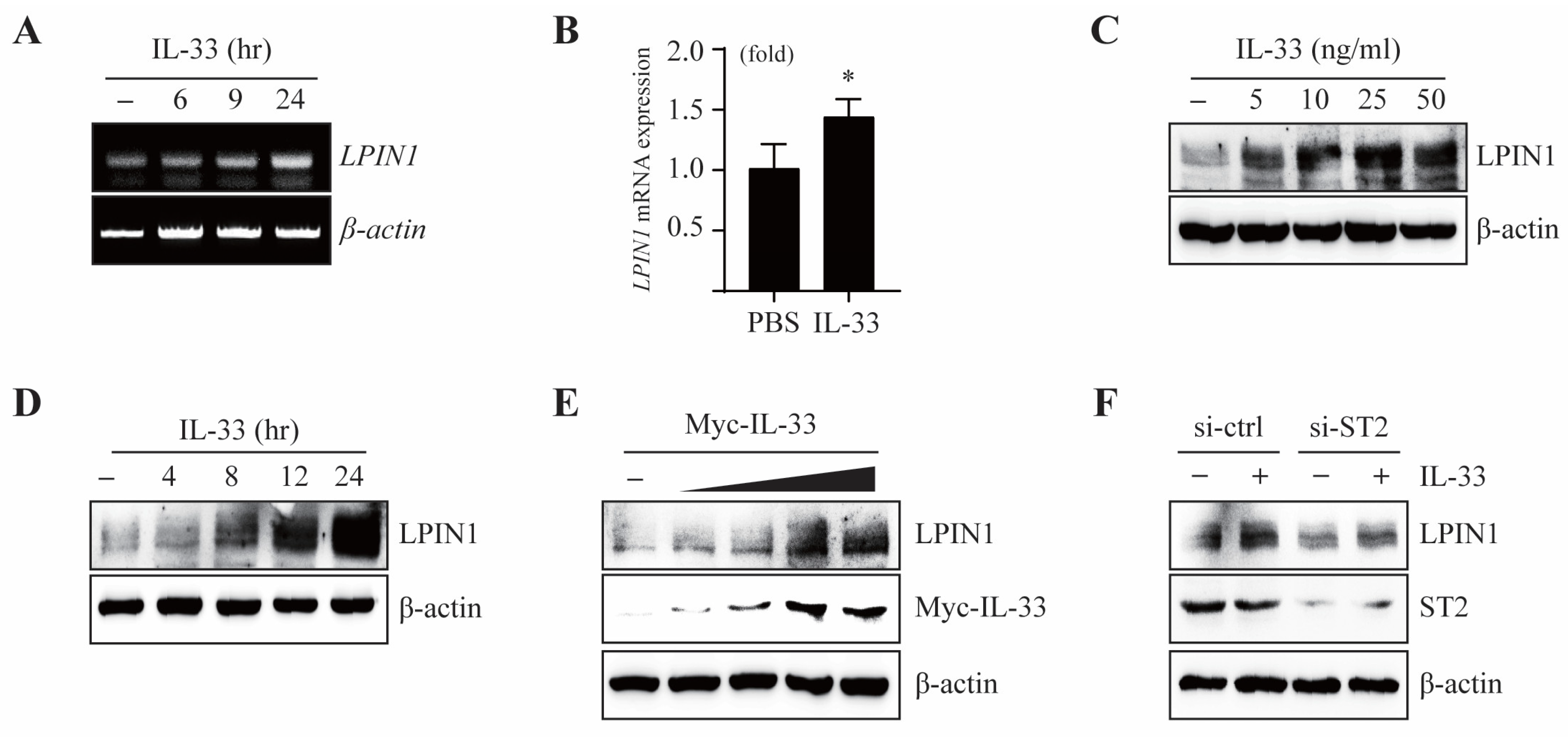
3.1. IL-33 Induces LPIN1 mRNA and Protein Expression
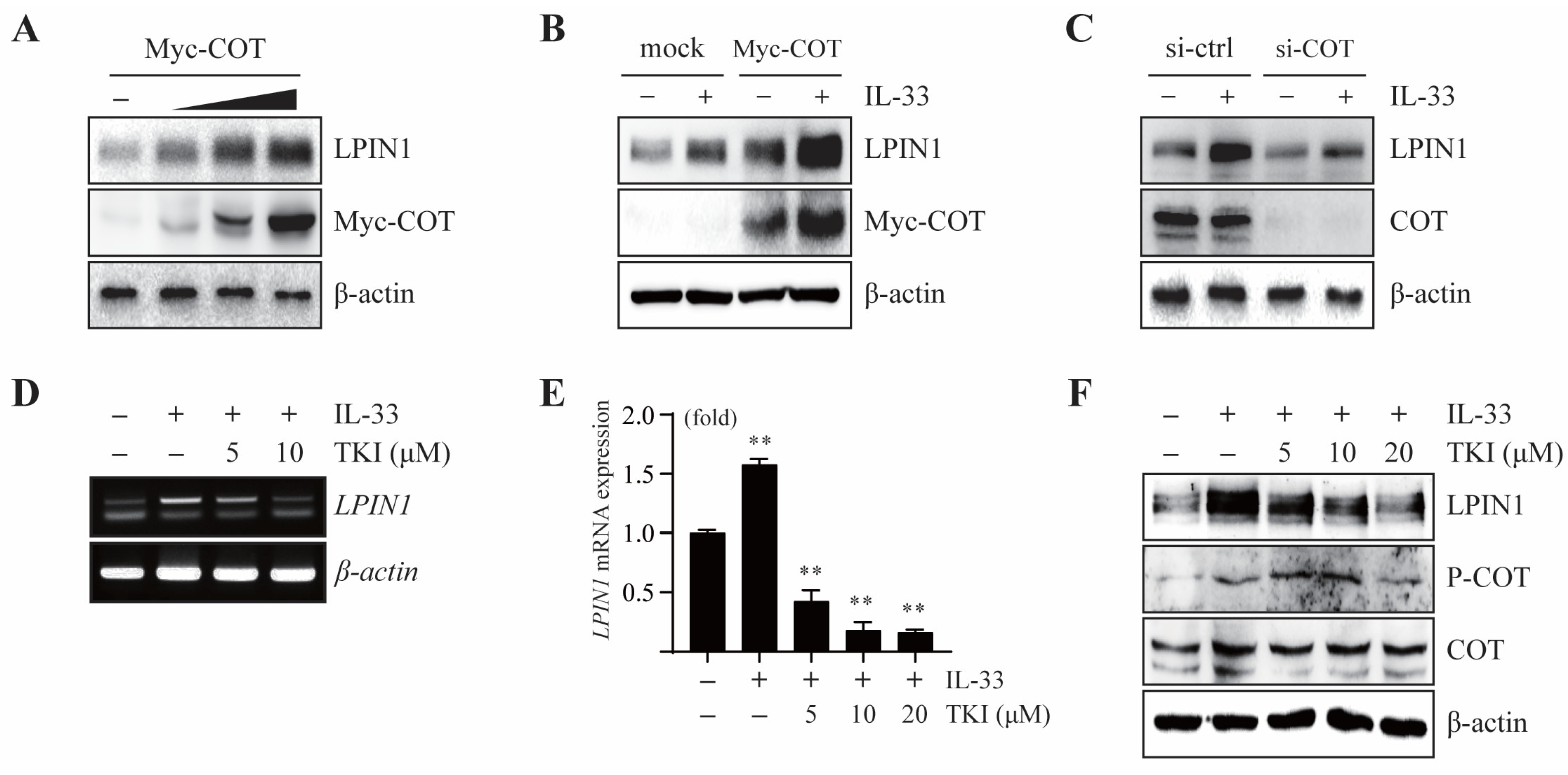
3.2. COT Mediates LPIN1 Expression Induced by IL-33
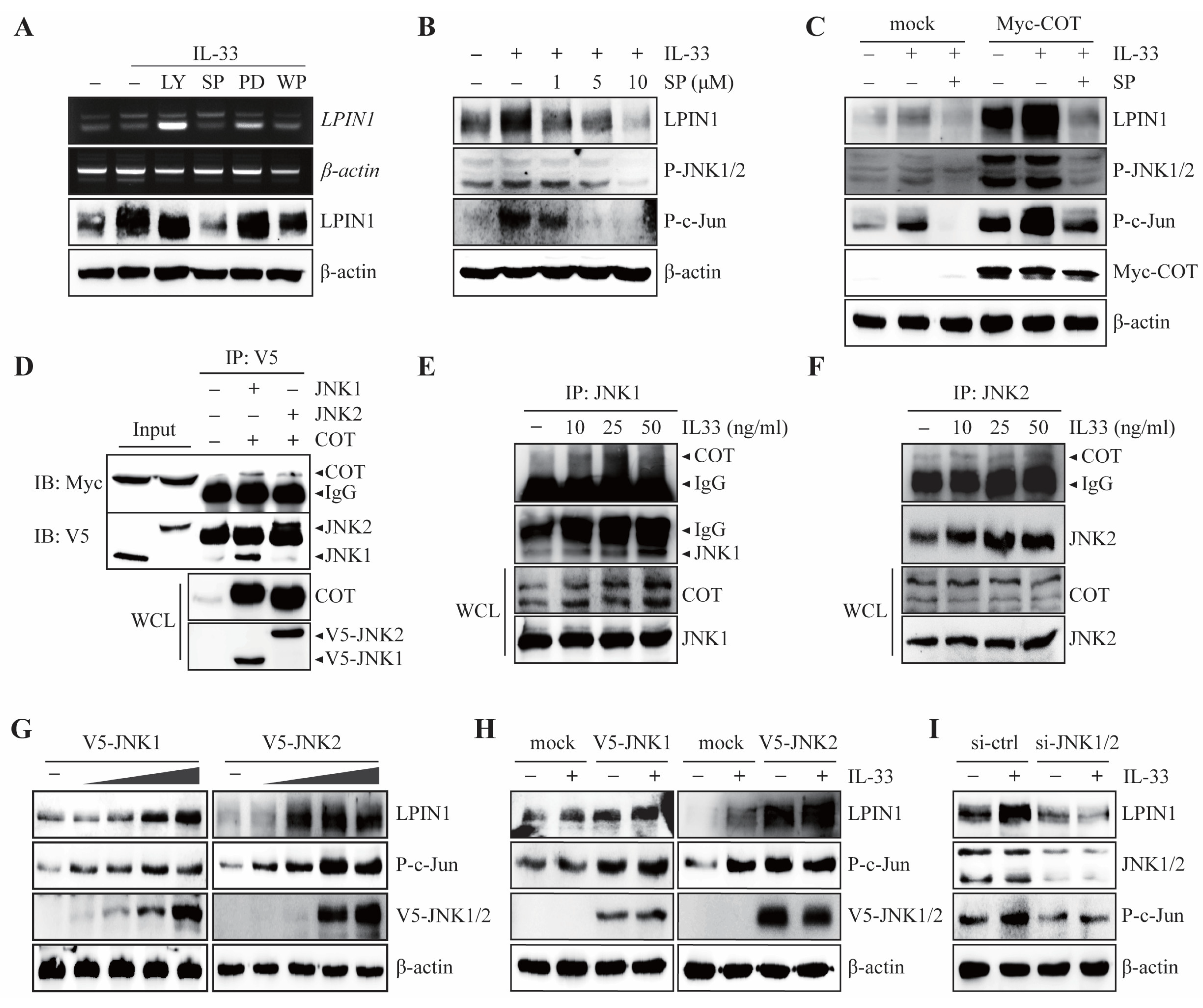
3.3. JNK1/2 Plays a Key Role in IL-33/ST2/COT-Mediated LPIN1 Expression
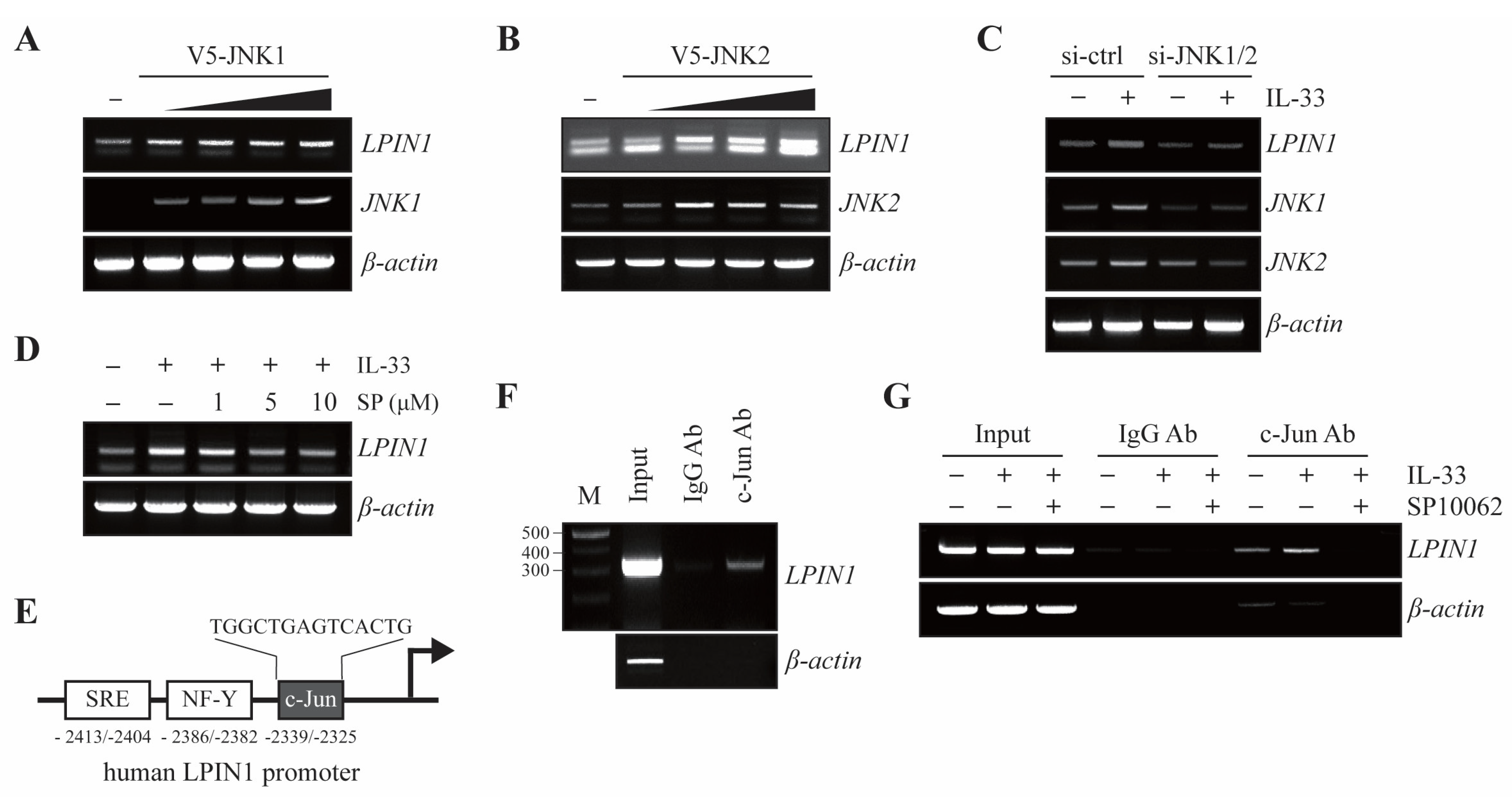
3.4. JNK1/2 Induces LPIN1 Expression through Increased Association of c-Jun with the LPIN1 Promoter
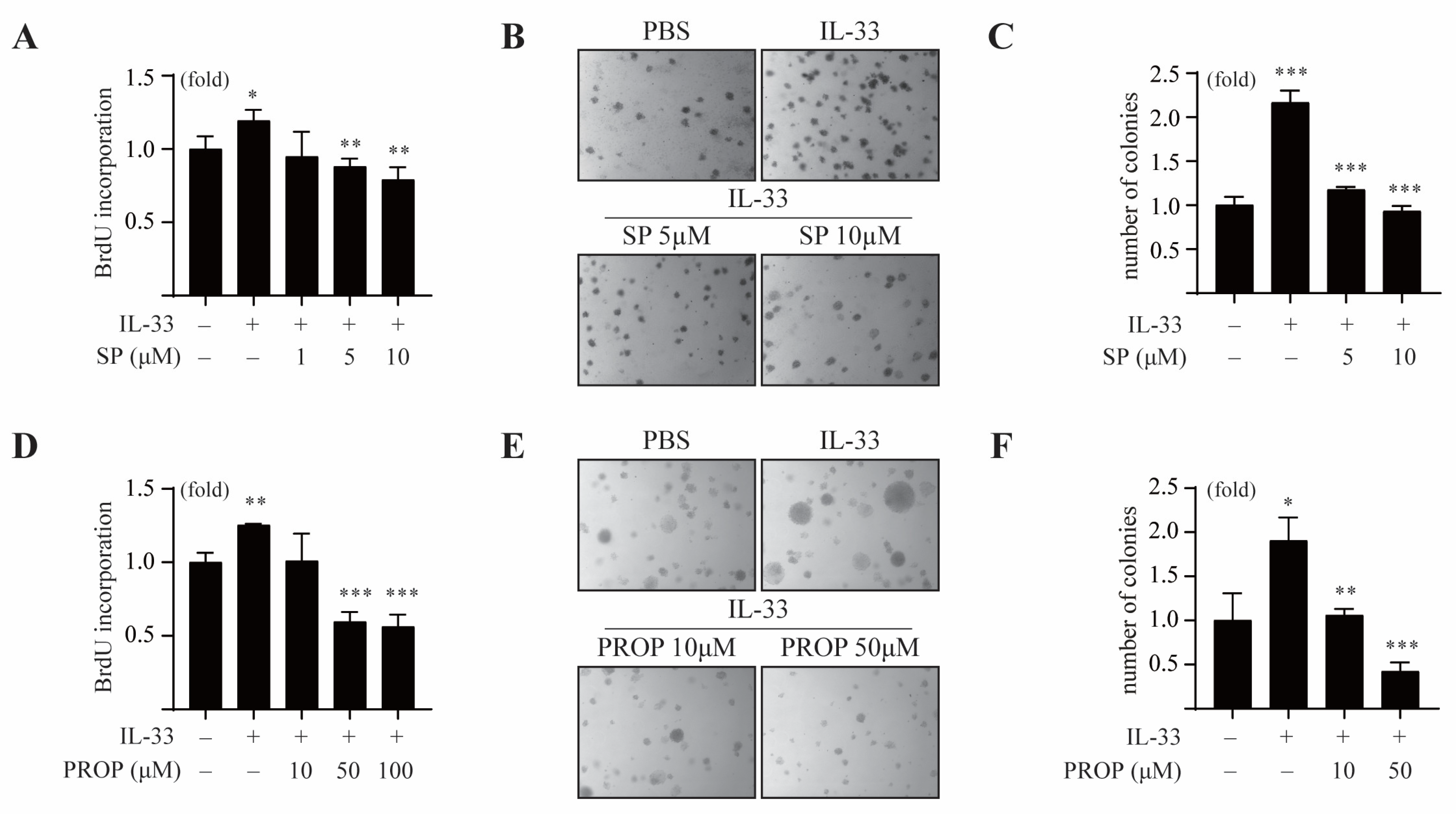
3.5. LPIN1 Overexpression Enhances Breast Cancer Cell Transformation Induced by IL-33
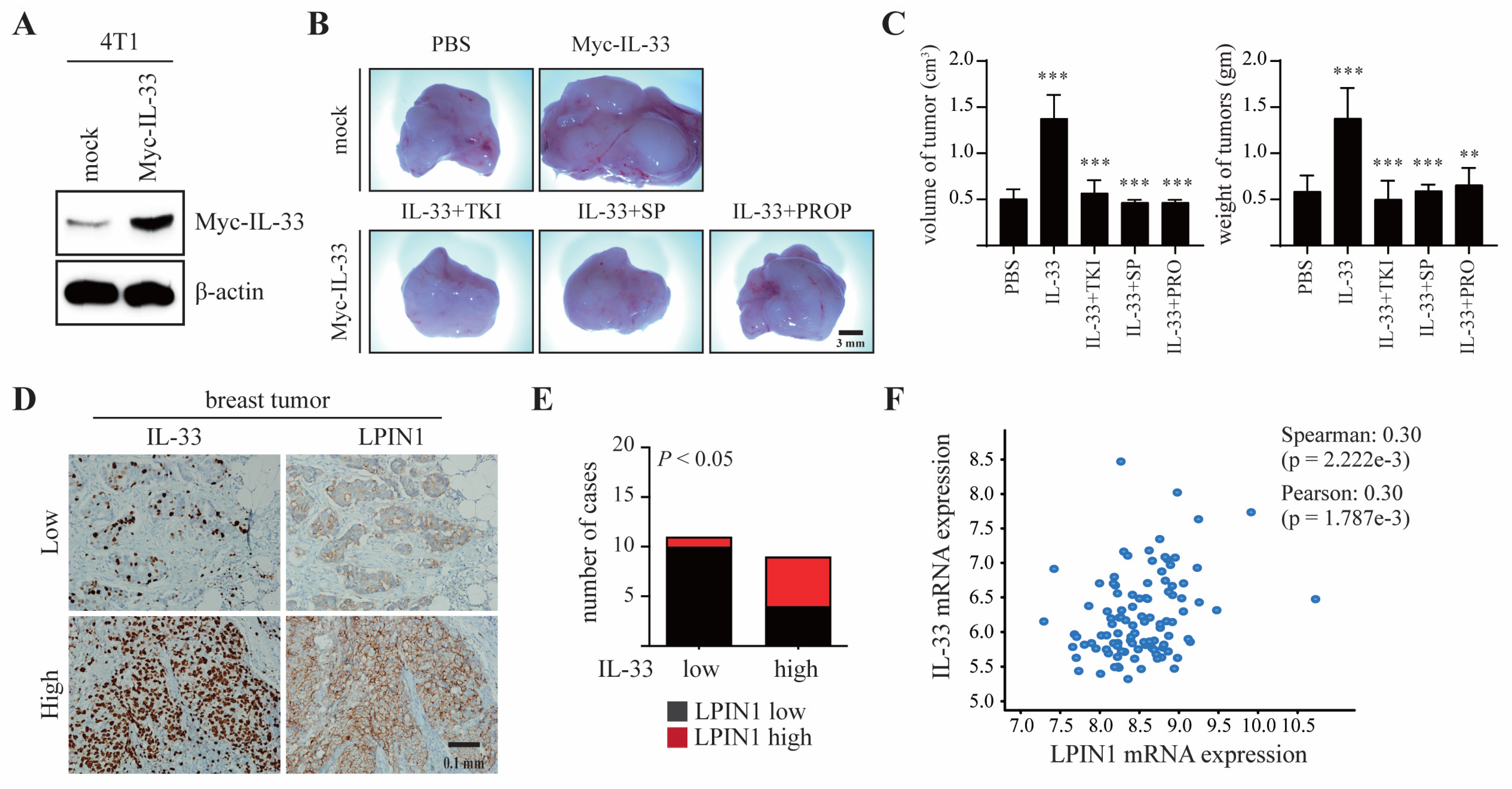
3.6. LPIN1 Abundance Is Positively Correlated with IL-33 Expression and Promotes Breast Mammary Tumorigenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Candido, J.; Hagemann, T. Cancer-Related Inflammation. J. Clin. Immunol. 2013, 33, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Coyle, Y.M. The effect of environment on breast cancer risk. Breast Cancer Res. Treat. 2004, 84, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Majeed, W.; Aslam, B.; Javed, I.; Khaliq, T.; Muhammad, F.; Ali, A.; Raza, A. Breast cancer: Major risk factors and recent developments in treatment. Asian Pac. J. Cancer Prev. 2014, 15, 3353–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk factors and preventions of breast cancer. Int. J. Biol. Sci. 2017, 13, 1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, D.; Komninou, D.; Stephenson, G. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes. Rev. 2004, 5, 153–165. [Google Scholar] [CrossRef]
- Stephenson, G.D.; Rose, D.P. Breast cancer and obesity: An update. Nutr. Cancer 2003, 45, 1–16. [Google Scholar] [CrossRef]
- Carmichael, A.R.; Bates, T. Obesity and breast cancer: A review of the literature. Breast 2004, 13, 85–92. [Google Scholar] [CrossRef]
- Carmichael, A. Obesity and prognosis of breast cancer. Obes. Rev. 2006, 7, 333–340. [Google Scholar] [CrossRef]
- Blücher, C.; Stadler, S.C. Obesity and breast cancer: Current insights on the role of fatty acids and lipid metabolism in promoting breast cancer growth and progression. Front. Endocrinol. 2017, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Grunt, T.W. Interacting cancer machineries: Cell signaling, lipid metabolism, and epigenetics. Trends Endocrinol. Metab. 2018, 29, 86–98. [Google Scholar] [CrossRef]
- Cheng, M.; Bhujwalla, Z.M.; Glunde, K. Targeting Phospholipid Metabolism in Cancer. Front. Oncol. 2016, 6, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.E.; Finck, B.N. Dual function lipin proteins and glycerolipid metabolism. Trends Endocrinol. Metab. 2011, 22, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reue, K.; Brindley, D.N. Thematic Review Series: Glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism. J. Lipid Res. 2008, 49, 2493–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Weng, Y.; Bai, Y.; Wang, Z.; Wang, S.; Zhu, J.; Zhang, F. Lipin-1 determines lung cancer cell survival and chemotherapy sensitivity by regulation of endoplasmic reticulum homeostasis and autophagy. Cancer Med. 2018, 7, 2541–2554. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, F.; Tay, L.W.R.; Boroda, S.; Nian, W.; Levental, K.R.; Levental, I.; Harris, T.E.; Chang, J.T.; Du, G. Lipin-1 regulation of phospholipid synthesis maintains endoplasmic reticulum homeostasis and is critical for triple-negative breast cancer cell survival. FASEB J. 2017, 31, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Brohée, L.; Demine, S.; Willems, J.; Arnould, T.; Colige, A.C.; Deroanne, C.F. Lipin-1 regulates cancer cell phenotype and is a potential target to potentiate rapamycin treatment. Oncotarget 2015, 6, 11264. [Google Scholar] [CrossRef]
- Dinarvand, N.; Khanahmad, H.; Hakimian, S.M.; Sheikhi, A.; Rashidi, B.; Bakhtiari, H.; Pourfarzam, M. Expression and clinicopathological significance of lipin-1 in human breast cancer and its association with p53 tumor suppressor gene. J. Cell. Physiol. 2020, 235, 5835–5846. [Google Scholar] [CrossRef]
- Schwartz, C.; O′Grady, K.; Lavelle, E.C.; Fallon, P.G. Interleukin 33: An innate alarm for adaptive responses beyond Th2 immunity–emerging roles in obesity, intestinal inflammation, and cancer. Eur. J. Immunol. 2016, 46, 1091–1100. [Google Scholar] [CrossRef]
- Miller, A.M.; Liew, F.Y. The IL-33/ST2 pathway—A new therapeutic target in cardiovascular disease. Pharmacol. Ther. 2011, 131, 179–186. [Google Scholar] [CrossRef]
- Palmer, G.; Gabay, C. Interleukin-33 biology with potential insights into human diseases. Nat. Rev. Rheumatol. 2011, 7, 321. [Google Scholar] [CrossRef]
- Saluja, R.; Hawro, T.; Eberle, J.; Church, M.; Maurer, M. Interleukin-33 promotes the proliferation of mouse mast cells through ST2/MyD88 and p38 MAPK-dependent and Kit-independent pathways. J. Biol. Regul. Homeost. Agents 2014, 28, 575–585. [Google Scholar] [PubMed]
- Kim, J.; Lim, S.; Kim, G.; Yun, H.; Ahn, S.; Choi, H. Interleukin-33/ST2 axis promotes epithelial cell transformation and breast tumorigenesis via upregulation of COT activity. Oncogene 2015, 34, 4928–4938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasmer, M.-H.; Krebs, P. The role of IL-33-dependent inflammation in the tumor microenvironment. Front. Immunol. 2017, 7, 682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, G.; Vasanthakumar, R.; Mohan, V.; Babu, S.; Aravindhan, V. Increased IL-12 and decreased IL-33 serum levels are associated with increased Th1 and suppressed Th2 cytokine profile in patients with diabetic nephropathy (CURES-134). Int. J. Clin. Exp. Pathol. 2014, 7, 8008. [Google Scholar]
- Yin, H.; Li, X.-Y.; Jin, X.-B.; Zhang, B.-B.; Gong, Q.; Yang, H.; Zheng, F.; Gong, F.-L.; Zhu, J.-Y. IL-33 prolongs murine cardiac allograft survival through induction of TH2-type immune deviation. Transplantation 2010, 89, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Huang, Y.; Su, Q.; Lin, Q.; Liu, W.; Luo, J.; Yu, B.; He, Y.; Qian, H.; Liu, Y. Potential of IL-33 for preventing the kidney injury via regulating the lipid metabolism in gout patients. J. Diabetes Res. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese Jr, R.V. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Chen, Y.; Rui, B.-B.; Tang, L.-Y.; Hu, C.-M. Lipin family proteins-key regulators in lipid metabolism. Ann. Nutr. Metab. 2015, 66, 10–18. [Google Scholar] [CrossRef]
- Phan, J.; Péterfy, M.; Reue, K. Lipin expression preceding peroxisome proliferator-activated receptor-γ is critical for adipogenesis in vivo and in vitro. J. Biol. Chem. 2004, 279, 29558–29564. [Google Scholar] [CrossRef] [Green Version]
- Phan, J.; Reue, K. Lipin, a lipodystrophy and obesity gene. Cell Metab. 2005, 1, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Peña, M.M.O. The role of IL-33/ST2 pathway in tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef] [Green Version]
- Vougioukalaki, M.; Kanellis, D.C.; Gkouskou, K.; Eliopoulos, A.G. Tpl2 kinase signal transduction in inflammation and cancer. Cancer Lett. 2011, 304, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Lee, J.-H.; Lee, S.H.; Do, S.-I.; Cho, S.-D.; Forslund, O.; Inn, K.-S.; Lee, J.-S.; Deng, F.-M.; Melamed, J.; et al. TPL2 Is an Oncogenic Driver in Keratocanthoma and Squamous Cell Carcinoma. Cancer Res. 2016, 76, 6712–6722. [Google Scholar] [CrossRef] [Green Version]
- Eliopoulos, A.G.; Wang, C.C.; Dumitru, C.D.; Tsichlis, P.N. TPL2 transduces CD40 and TNF signals that activate ERK and regulates IgE induction by CD40. EMBO J. 2003, 22, 3855–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Choi, H.S. Cot/Tpl2 is required for neoplastic cell transformation induced by interleukin-22: A possible mechanism in breast carcinogenesis. In Proceedings of the American Association for the Cancer Research (AACR) 104th Annual Meeting, Washington, DC, USA, 6–10 April 2013. [Google Scholar]
- Varin, E.; Wojtusciszyn, A.; Broca, C.; Muller, D.; Ravier, M.; Ceppo, F.; Renard, E.; Tanti, J.; Dalle, S. Inhibition of the MAP3 kinase Tpl2 protects rodent and human β-cells from apoptosis and dysfunction induced by cytokines and enhances anti-inflammatory actions of exendin-4. Cell Death Dis. 2016, 7, e2065. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Gallagher, E. From JNK to pay dirt: Jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 2005, 57, 283–295. [Google Scholar] [CrossRef]
- Albanese, C.; Johnson, J.; Watanabe, G.; Eklund, N.; Vu, D.; Arnold, A.; Pestell, R.G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995, 270, 23589–23597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.; Katiyar, S.; Willmarth, N.E.; Liu, M.; Ma, X.; Flomenberg, N.; Lisanti, M.P.; Pestell, R.G. c-Jun induces mammary epithelial cellular invasion and breast cancer stem cell expansion. J. Biol. Chem. 2010, 285, 8218–8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Han, B.S.; Kim, W.K.; Lee, S.C.; Oh, K.-J.; Bae, K.-H. c-Jun regulates adipocyte differentiation via the KLF15-mediated mode. Biochem. Biophys. Res. Commun. 2016, 469, 552–558. [Google Scholar]
- Schummer, P.; Kuphal, S.; Vardimon, L.; Bosserhoff, A.K.; Kappelmann, M. Specific c-Jun target genes in malignant melanoma. Cancer Biol. Ther. 2016, 17, 486–497. [Google Scholar] [CrossRef] [Green Version]
- Nateri, A.S.; Spencer-Dene, B.; Behrens, A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 2005, 437, 281–285. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-Y.; Kim, G.; Lim, S.-C.; Choi, H.-S. IL-33-Induced Transcriptional Activation of LPIN1 Accelerates Breast Tumorigenesis. Cancers 2021, 13, 2174. https://doi.org/10.3390/cancers13092174
Kim J-Y, Kim G, Lim S-C, Choi H-S. IL-33-Induced Transcriptional Activation of LPIN1 Accelerates Breast Tumorigenesis. Cancers. 2021; 13(9):2174. https://doi.org/10.3390/cancers13092174
Chicago/Turabian StyleKim, Jin-Young, Garam Kim, Sung-Chul Lim, and Hong-Seok Choi. 2021. "IL-33-Induced Transcriptional Activation of LPIN1 Accelerates Breast Tumorigenesis" Cancers 13, no. 9: 2174. https://doi.org/10.3390/cancers13092174
APA StyleKim, J. -Y., Kim, G., Lim, S. -C., & Choi, H. -S. (2021). IL-33-Induced Transcriptional Activation of LPIN1 Accelerates Breast Tumorigenesis. Cancers, 13(9), 2174. https://doi.org/10.3390/cancers13092174