
Whole-Genome Sequencing Identifies PPARGC1A as a Putative Modifier of Cancer Risk in BRCA1/2 Mutation Carriers

 ,
,  , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Whole-Genome Sequencing, Variant Calling, and Variant Filtering
2.3. Statistical Analysis
2.4. Network Propagation and Pathway Enrichment Analysis
2.5. Validation Using PCAWG Breast and Ovarian Cancer Cohorts
2.6. Study Approval
3. Results
3.1. Study Population in the Discovery Stage
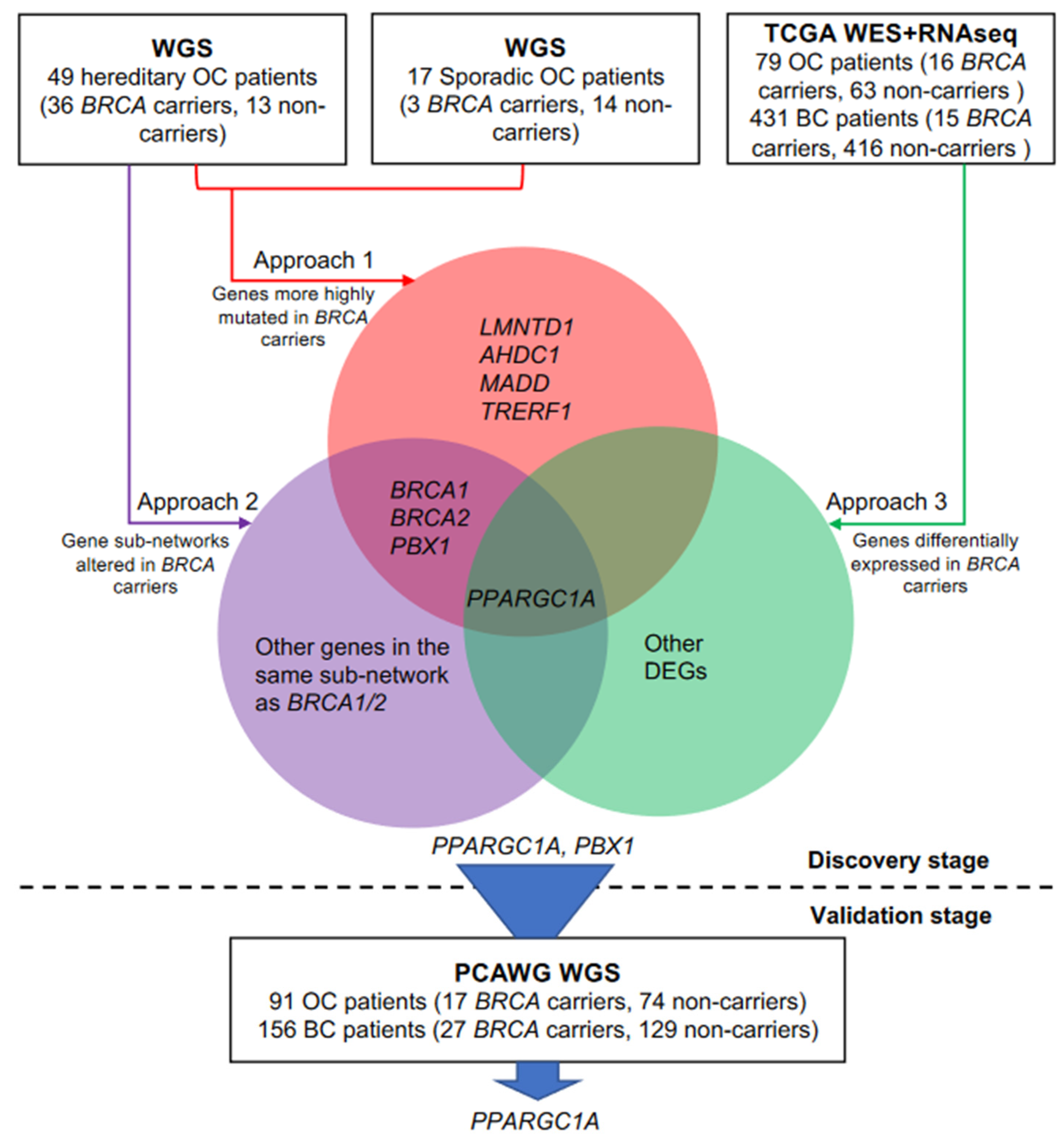
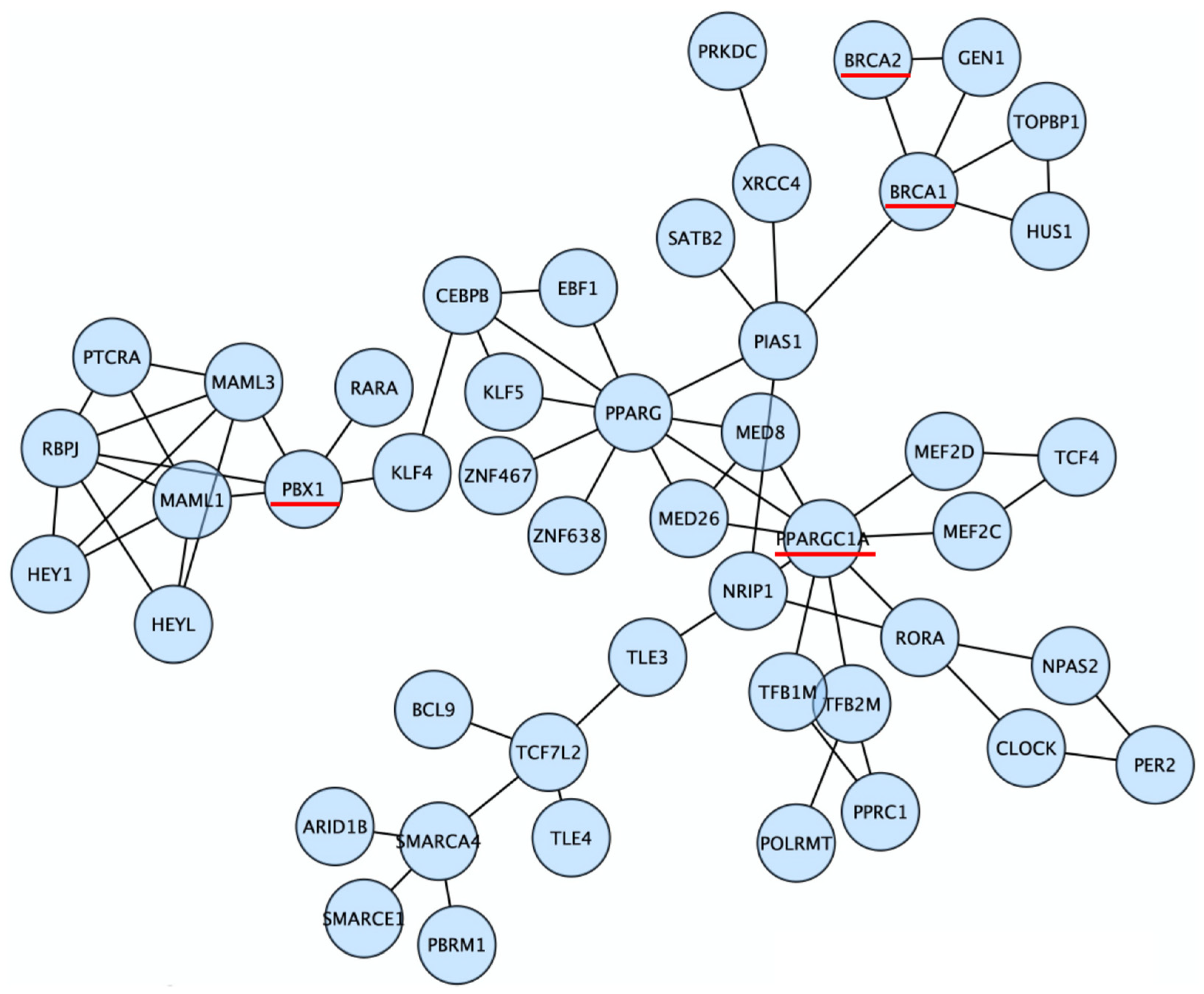
3.2. Discovery of Candidate Genes That Modify Cancer Risks of BRCA1/2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Hereditary OC (36 BRCA Carriers vs. 13 Non-Carriers) | Hereditary OC + Sporadic OC (39 BRCA Carriers vs. 27 Non-Carriers) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| # and Fraction of Mutated BRCA Carriers | # and Fraction of Mutated Non-Carriers | p-Value * | # and Fraction of Mutated BRCA Carriers | # and Fraction of Mutated Non-Carriers | p-Value * | |||||
| BRCA1 | 29 | 0.81 | 0 | 0.00 | 2.95 × 10−7 | 30 | 0.77 | 0 | 0.00 | 3.84 × 10−11 |
| BRCA2 | 10 | 0.28 | 0 | 0.00 | 3.09 × 10−2 | 12 | 0.31 | 0 | 0.00 | 7.94 × 10−4 |
| PPARGC1A | 11 | 0.31 | 0 | 0.00 | 2.06 × 10−2 | 11 | 0.28 | 1 | 0.04 | 9.99 × 10−3 |
| PBX1 | 14 | 0.39 | 1 | 0.08 | 3.49 × 10−2 | 14 | 0.36 | 3 | 0.11 | 2.15 × 10−2 |
| LMNTD1 | 9 | 0.25 | 0 | 0.00 | 4.58 × 10−2 | 9 | 0.23 | 0 | 0.00 | 5.73 × 10−3 |
| AHDC1 | 9 | 0.25 | 0 | 0.00 | 4.58 × 10−2 | 9 | 0.23 | 1 | 0.04 | 3.01 × 10−2 |
| MADD | 9 | 0.25 | 0 | 0.00 | 4.58 × 10−2 | 9 | 0.23 | 1 | 0.04 | 3.01 × 10−2 |
| TRERF1 | 9 | 0.25 | 0 | 0.00 | 4.58 × 10−2 | 10 | 0.26 | 1 | 0.04 | 1.75 × 10−2 |
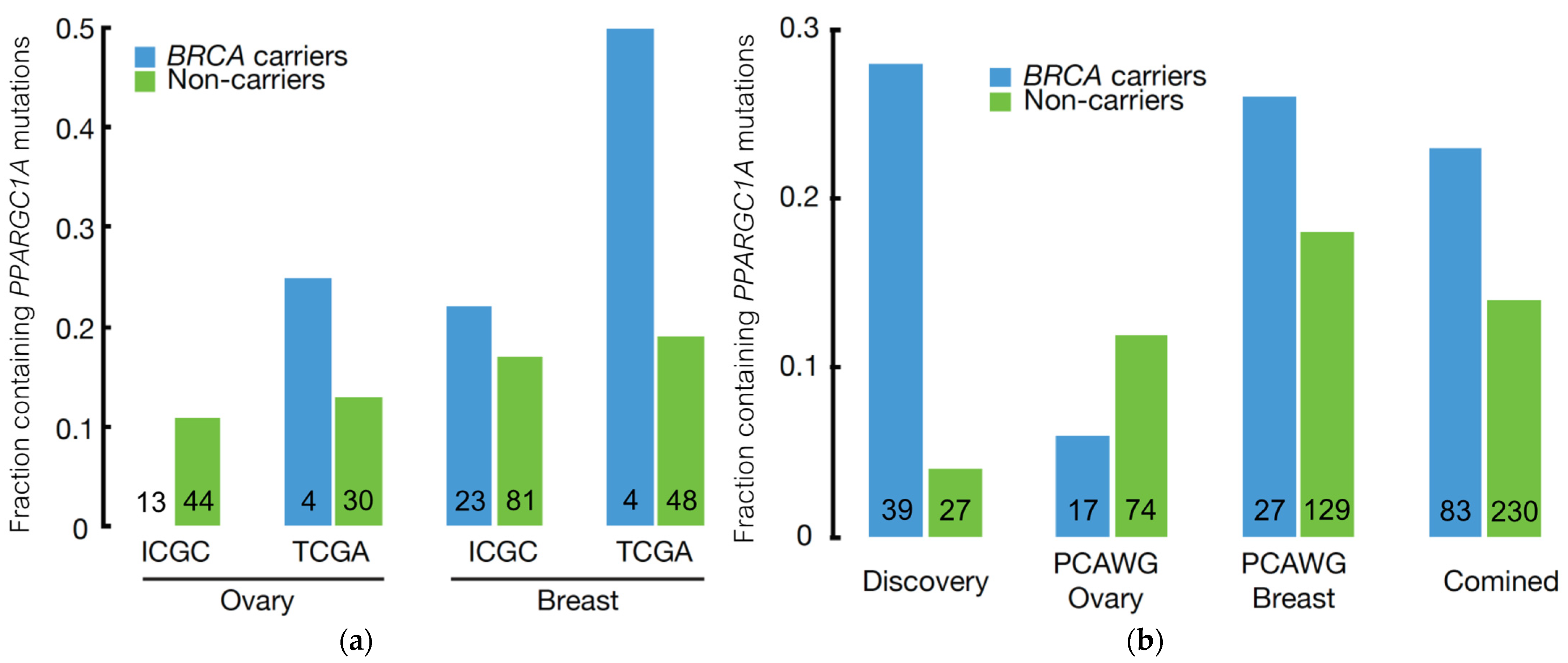
3.3. Independent Validation of BRCA Modifier Candidate Genes
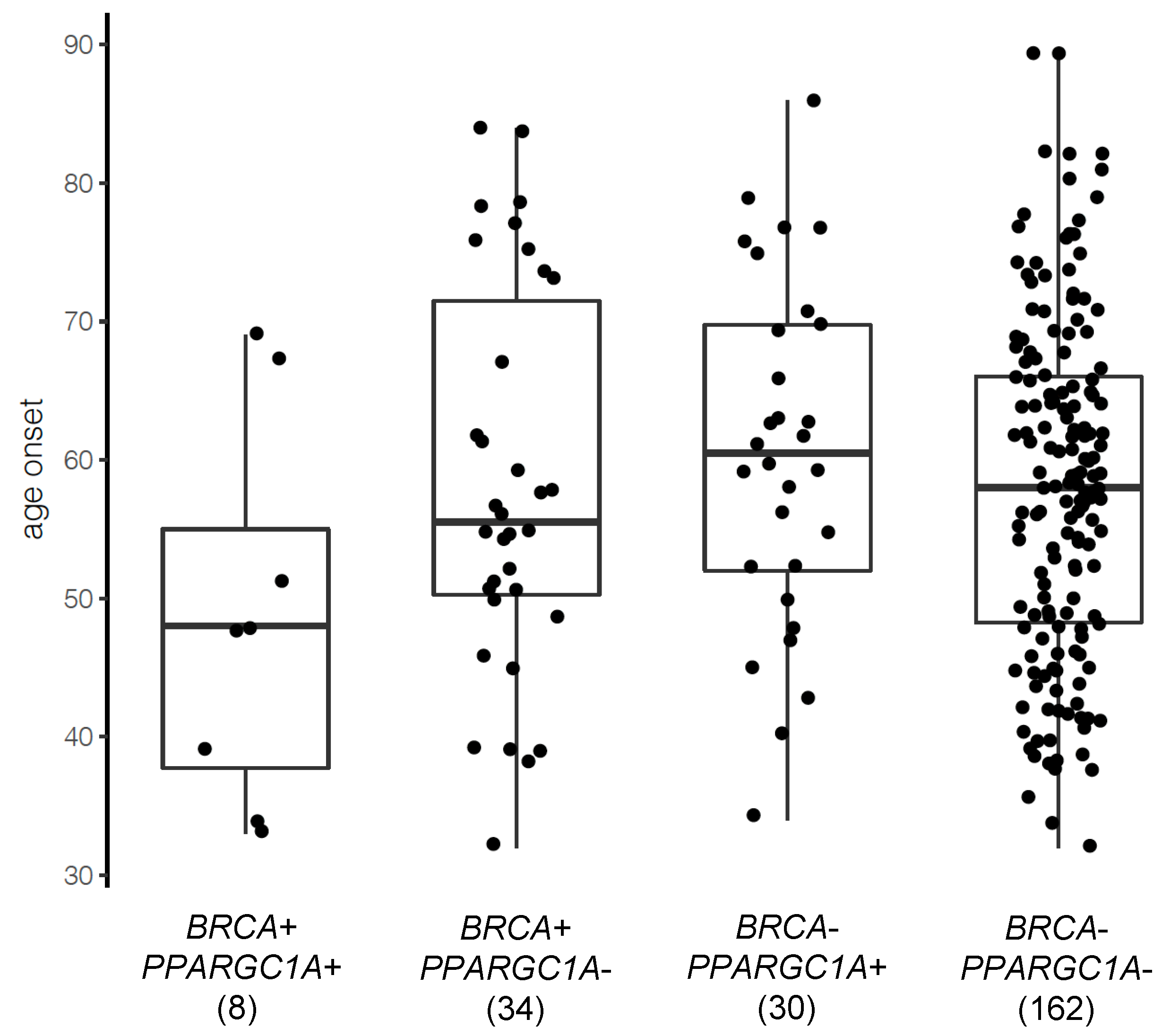
3.4. PPARGC1A as a Novel BRCA Modifier Candidate Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. [Google Scholar]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Mavaddat, N.; Peock, S.; Frost, D.; Ellis, S.; Platte, R.; Fineberg, E.; Evans, D.G.; Izatt, L.; Eeles, R.A.; Adlard, J.; et al. Cancer Risks for BRCA1 and BRCA2 Mutation Carriers: Results from Prospective Analysis of EMBRACE. J. Natl. Cancer Inst. 2013, 105, 812–822. [Google Scholar] [CrossRef] [Green Version]
- LeVasseur, N.; Chia, S. Cancer screening and prevention in BRCA mutation carriers: A missed opportunity? Br. J. Cancer 2019, 121, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27, v103–v110. [Google Scholar] [CrossRef]
- Chenevix-Trench, G.; Milne, R.L.; Antoniou, A.C.; Couch, F.J.; Easton, D.F.; Goldgar, D.E. An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: The Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res. 2007, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Friebel, T.M.; Domchek, S.M.; Rebbeck, T.R. Modifiers of Cancer Risk in BRCA1 and BRCA2 Mutation Carriers: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2014, 106, dju091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, R.L.; Antoniou, A.C. Genetic modifiers of cancer risk for BRCA1 and BRCA2 mutation carriers. Ann. Oncol. 2011, 22 (Suppl. S1), i11–i17. [Google Scholar] [CrossRef] [PubMed]
- Kotsopoulos, J.; Lubinski, J.; Lynch, H.T.; Kim-Sing, C.; Neuhausen, S.; Demsky, R.; Foulkes, W.D.; Ghadirian, P.; Tung, N.; Ainsworth, P.; et al. Oophorectomy after menopause and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Narod, S.A. Modifiers of risk of hereditary breast and ovarian cancer. Nat. Rev. Cancer 2002, 2, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Wang, X.; McGuffog, L.; Lee, A.; Olswold, C.; Kuchenbaecker, K.B.; Soucy, P.; Fredericksen, Z.; Barrowdale, D.; Dennis, J.; et al. Genome-Wide Association Study in BRCA1 Mutation Carriers Identifies Novel Loci Associated with Breast and Ovarian Cancer Risk. PLoS Genet. 2013, 9, e1003212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudet, M.M.; Kuchenbaecker, K.B.; Vijai, J.; Klein, R.J.; Kirchhoff, T.; McGuffog, L.; Barrowdale, D.; Dunning, A.M.; Lee, A.; Dennis, J.; et al. Identification of a BRCA2-specific modifier locus at 6p24 related to breast cancer risk. PLoS Genet. 2013, 9, e1003173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, A.C.; Wang, X.; Fredericksen, Z.S.; McGuffog, L.; Tarrell, R.; Sinilnikova, O.M.; Healey, S.; Morrison, J.; Kartsonaki, C.; Lesnick, T.; et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat. Genet. 2010, 42, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Kartsonaki, C.; Gayther, S.A.; Pharoah, P.D.P.; Sinilnikova, O.M.; Beesley, J.; Chen, X.; McGuffog, L.; Healey, S.; Couch, F.J.; et al. Genetic variation at 9p22.2 and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2011, 103, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Zhang, J.; Chen, Y.; Hu, Q.; Shen, H.; Huang, R.-Y.; Liu, Q.; Kaur, J.; Long, M.; Battaglia, S.; et al. Whole-exome sequencing of ovarian cancer families uncovers putative predisposition genes. Int. J. Cancer 2020, 146, 2147–2155. [Google Scholar] [CrossRef]
- Tayo, B.O.; DiCioccio, R.A.; Liang, Y.; Trevisan, M.; Cooper, R.S.; Lele, S.; Sucheston, L.; Piver, S.M.; Odunsi, K. Complex Segregation Analysis of Pedigrees from the Gilda Radner Familial Ovarian Cancer Registry Reveals Evidence for Mendelian Dominant Inheritance. PLoS ONE 2009, 4, e5939. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Mohiyuddin, M.; Mu, J.C.; Li, J.; Bani Asadi, N.; Gerstein, M.B.; Abyzov, A.; Wong, W.H.; Lam, H.Y.K. MetaSV: An accurate and integrative structural-variant caller for next generation sequencing. Bioinformatics 2015, 31, 2741–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, Z.N.; Osborne, E.J.; Cone, K.R.; Kennedy, B.J.; Domyan, E.T.; Shapiro, M.D.; Elde, N.C.; Yandell, M. Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput. Biol. 2015, 11, e1004572. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Q.; Yao, S.; Kwan, M.L.; Roh, J.M.; Zhao, H.; Ambrosone, C.B.; Kushi, L.H.; Liu, S.; Zhu, Q. SeqSQC: A Bioconductor Package for Evaluating the Sample Quality of Next-generation Sequencing Data. Genom. Proteom. Bioinform. 2019, 17, 211–218. [Google Scholar] [CrossRef]
- Leiserson, M.D.M.; Vandin, F.; Wu, H.-T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Conway, J.R.; Camp, S.Y.; Witkowski, L.; Kofman, E.; Reardon, B.; Han, S.; Moore, N.; Elmarakeby, H.; Salari, K.; et al. Detection of Pathogenic Variants with Germline Genetic Testing Using Deep Learning vs Standard Methods in Patients with Prostate Cancer and Melanoma. JAMA 2020, 324, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Davidson, N.R.; Demircioğlu, D.; Fonseca, N.A.; He, Y.; Kahles, A.; Lehmann, K.-V.; Liu, F.; Shiraishi, Y.; Soulette, C.M.; et al. Genomic basis for RNA alterations in cancer. Nature 2020, 578, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirtenberger, M.; Tchatchou, S.; Hemminki, K.; Schmutzhard, J.; Sutter, C.; Schmutzler, R.K.; Meindl, A.; Wappenschmidt, B.; Kiechle, M.; Arnold, N.; et al. Associations of genetic variants in the estrogen receptor coactivators PPARGC1A, PPARGC1B and EP300 with familial breast cancer. Carcinogenesis 2006, 27, 2201–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Permuth-Wey, J.; Chen, Y.A.; Tsai, Y.-Y.; Chen, Z.; Qu, X.; Lancaster, J.M.; Stockwell, H.; Dagne, G.; Iversen, E.; Risch, H.; et al. Inherited variants in mitochondrial biogenesis genes may influence epithelial ovarian cancer risk. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1131–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignatelli, M.; Cocca, C.; Santos, A.; Perez-Castillo, A. Enhancement of BRCA1 gene expression by the peroxisome proliferator-activated receptor γ in the MCF-7 breast cancer cell line. Oncogene 2003, 22, 5446–5450. [Google Scholar] [CrossRef] [Green Version]
- Mastropasqua, F.; Girolimetti, G.; Shoshan, M. PGC1α: Friend or Foe in Cancer? Genes 2018, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Gravel, S.-P. Deciphering the Dichotomous Effects of PGC-1α on Tumorigenesis and Metastasis. Front. Oncol. 2018, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.-H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173. [Google Scholar] [CrossRef] [Green Version]
- McGuirk, S.; Audet-Delage, Y.; St-Pierre, J. Metabolic Fitness and Plasticity in Cancer Progression. Trends Cancer 2020, 6, 49–61. [Google Scholar] [CrossRef]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [Green Version]
- Van Vugt, M.A.T.M.; Parkes, E.E. When breaks get hot: Inflammatory signaling in BRCA1/2-mutant cancers. Trends Cancer 2022, 8, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Alsop, K.; Etemadmoghadam, D.; Hondow, H.; Mikeska, T.; Dobrovic, A.; deFazio, A.; Smyth, G.K.; Levine, D.A.; Mitchell, G.; et al. Nonequivalent gene expression and copy number alterations in high-grade serous ovarian cancers with BRCA1 and BRCA2 mutations. Clin. Cancer Res. 2013, 19, 3474–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlpine, J.N.; Porter, H.; Köbel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieser, V.; Gaugg, I.; Fleischer, M.; Shivalingaiah, G.; Wenzel, S.; Sprung, S.; Lax, S.F.; Zeimet, A.G.; Fiegl, H.; Marth, C. BRCA1/2 and TP53 mutation status associates with PD-1 and PD-L1 expression in ovarian cancer. Oncotarget 2018, 9, 17501–17511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruand, M.; Barras, D.; Mina, M.; Ghisoni, E.; Morotti, M.; Lanitis, E.; Fahr, N.; Desbuisson, M.; Grimm, A.; Zhang, H.; et al. Cell-autonomous inflammation of BRCA1-deficient ovarian cancers drives both tumor-intrinsic immunoreactivity and immune resistance via STING. Cell Rep. 2021, 36, 109412. [Google Scholar] [CrossRef]
- McCarthy, A.; Savage, K.; Gabriel, A.; Naceur, C.; Reis-Filho, J.S.; Ashworth, A. A mouse model of basal-like breast carcinoma with metaplastic elements. J. Pathol. 2007, 211, 389–398. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.J.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, p53, and Brca1 or Brca2 Cooperate in Inducing Metastatic Serous Epithelial Ovarian Cancer. Cancer Res. 2012, 72, 4141–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from highthroughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Liu, X.; White, S.; Peng, B.; Johnson, A.D.; Brody, J.A.; Li, A.H.; Huang, Z.; Carroll, A.; Wei, P.; Gibbs, R.; et al. WGSA: An annotation pipeline for human genome sequencing studies. J. Med. Genet. 2016, 53, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.E.; Pratt, H.E.; Purcaro, M.J.; Weng, Z. A curated benchmark of enhancer-gene interactions for evaluating enhancer-target gene prediction methods. Genome Biol. 2020, 21, 17. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic. Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNAseq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e314. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Q.; Wang, J.; Yu, H.; Hu, Q.; Bateman, N.W.; Long, M.; Rosario, S.; Schultz, E.; Dalgard, C.L.; Wilkerson, M.D.; et al. Whole-Genome Sequencing Identifies PPARGC1A as a Putative Modifier of Cancer Risk in BRCA1/2 Mutation Carriers. Cancers 2022, 14, 2350. https://doi.org/10.3390/cancers14102350
Zhu Q, Wang J, Yu H, Hu Q, Bateman NW, Long M, Rosario S, Schultz E, Dalgard CL, Wilkerson MD, et al. Whole-Genome Sequencing Identifies PPARGC1A as a Putative Modifier of Cancer Risk in BRCA1/2 Mutation Carriers. Cancers. 2022; 14(10):2350. https://doi.org/10.3390/cancers14102350
Chicago/Turabian StyleZhu, Qianqian, Jie Wang, Han Yu, Qiang Hu, Nicholas W. Bateman, Mark Long, Spencer Rosario, Emily Schultz, Clifton L. Dalgard, Matthew D. Wilkerson, and et al. 2022. "Whole-Genome Sequencing Identifies PPARGC1A as a Putative Modifier of Cancer Risk in BRCA1/2 Mutation Carriers" Cancers 14, no. 10: 2350. https://doi.org/10.3390/cancers14102350
APA StyleZhu, Q., Wang, J., Yu, H., Hu, Q., Bateman, N. W., Long, M., Rosario, S., Schultz, E., Dalgard, C. L., Wilkerson, M. D., Sukumar, G., Huang, R.-Y., Kaur, J., Lele, S. B., Zsiros, E., Villella, J., Lugade, A., Moysich, K., Conrads, T. P., ... Odunsi, K. (2022). Whole-Genome Sequencing Identifies PPARGC1A as a Putative Modifier of Cancer Risk in BRCA1/2 Mutation Carriers. Cancers, 14(10), 2350. https://doi.org/10.3390/cancers14102350