
Characterization of the Immune Response to PD-1 Blockade during Chemoradiotherapy for Head and Neck Squamous Cell Carcinoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
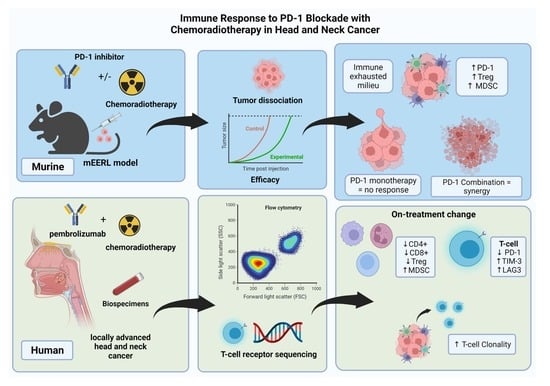
1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Human Subjects
2.3. Human Sample Collection and Processing
2.4. Multicolor Flow Cytometry
2.5. T-Cell Clonality
2.6. Statistical Analysis
3. Results
3.1. PD-1 Blockade Synergizes with Chemoradiotherapy in an HPV+ HNSCC Syngeneic Model
3.2. Circulating T-Cell Populations Decline with an Apparent Rise in Immunosuppressive Monocytes during Chemoradiotherapy despite PD-1 Blockade in HNSCC Patients
3.3. During Chemoradiotherapy, PD-1 Blockade Reduces PD-1 Expressing T-Cell Populations, However, with a Concordant Rise in Other Exhaustive Checkpoint Expression
3.4. Chemoradiotherapy in Combination with PD-1 Blockade Increases Clonal Selection in PBMC T-Cell Repertoire
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, R.; Masuyer, E.; Swaminathan, R.; Ferlay, J.; Whelan, S. Head and Neck Cancer: A Global Perspective on Epidemiology and Prognosis. Anticancer Res. 1998, 18, 4779–4786. [Google Scholar] [PubMed]
- Vokes, E.E.; Agrawal, N.; Seiwert, T.Y. HPV-Associated Head and Neck Cancer. J. Natl. Cancer Inst. 2015, 107, djv344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignon, J.-P.; le Maître, A.; Maillard, E.; Bourhis, J.; on behalf of the MACH-NC Collaborative Group. Meta-Analysis of Chemotherapy in Head and Neck Cancer (MACH-NC): An Update on 93 Randomised Trials and 17,346 Patients. Radiother. Oncol. 2009, 92, 4–14. [Google Scholar] [CrossRef]
- Spanos, W.C.; Nowicki, P.; Lee, D.W.; Hoover, A.; Hostager, B.; Gupta, A.; Anderson, M.E.; Lee, J.H. Immune Response During Therapy With Cisplatin or Radiation for Human Papillomavirus–Related Head and Neck Cancer. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 1137. [Google Scholar] [CrossRef] [Green Version]
- Vermeer, D.W.; Spanos, W.C.; Vermeer, P.D.; Bruns, A.M.; Lee, K.M.; Lee, J.H. Radiation-Induced Loss of Cell Surface CD47 Enhances Immune-Mediated Clearance of Human Papillomavirus-Positive Cancer. Int. J. Cancer 2013, 133, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Lucido, C.; Vermeer, P.; Wieking, B.; Vermeer, D.; Lee, J. CD137 Enhancement of HPV Positive Head and Neck Squamous Cell Carcinoma Tumor Clearance. Vaccines 2014, 2, 841–853. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.J.; Forde, P.M.; Hammers, H.-J.; Emens, L.A.; Taube, J.M.; Topalian, S.L. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin. Oncol. 2015, 42, 587–600. [Google Scholar] [CrossRef] [Green Version]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 Pathway in Tolerance and Autoimmunity: PD-1 Pathway, Tregs, and Autoimmune Diseases. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Kim, J.W.; Eder, J.P. Prospects for Targeting PD-1 and PD-L1 in Various Tumor Types. Oncology 2014, 28 (Suppl. S3), 15–28. [Google Scholar] [PubMed]
- Lyford-Pike, S.; Peng, S.; Young, G.D.; Taube, J.M.; Westra, W.H.; Akpeng, B.; Bruno, T.C.; Richmon, J.D.; Wang, H.; Bishop, J.A.; et al. Evidence for a Role of the PD-1:PD-L1 Pathway in Immune Resistance of HPV-Associated Head and Neck Squamous Cell Carcinoma. Cancer Res. 2013, 73, 1733–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovedi, S.; Adlard, A.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.; Stratford, I.; Poon, E.; Morrow, M.; Stewart, R.; et al. The Anti-Tumor Immune Response Generated by Radiation Therapy May Be Limited by Tumor Cell Adaptive Resistance and Can Be Circumvented by PD-L1 Blockade. J. Immunother. Cancer 2014, 2, O9. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and Anti–PD-L1 Treatment Synergistically Promote Antitumor Immunity in Mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Qin, X.; Liu, C.; Zhou, Y.; Wang, G. Cisplatin Induces Programmed Death-1-Ligand 1(PD-L1) over-Expression in Hepatoma H22 Cells via Erk /MAPK Signaling Pathway. Cell. Mol. Biol. 2010, 56, 1366–1372. [Google Scholar]
- Tran, L.; Allen, C.T.; Xiao, R.; Moore, E.; Davis, R.; Park, S.-J.; Spielbauer, K.; Van Waes, C.; Schmitt, N.C. Cisplatin Alters Antitumor Immunity and Synergizes with PD-1/PD-L1 Inhibition in Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Res. 2017, 5, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Parikh, F.; Duluc, D.; Imai, N.; Clark, A.; Misiukiewicz, K.; Bonomi, M.; Gupta, V.; Patsias, A.; Parides, M.; Demicco, E.G.; et al. Chemoradiotherapy-Induced Upregulation of PD-1 Antagonizes Immunity to HPV-Related Oropharyngeal Cancer. Cancer Res. 2014, 74, 7205–7216. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and Is Associated with Lung Cancer Progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 Expression Is Associated with Tumor Antigen–Specific CD8+ T Cell Dysfunction in Melanoma Patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Jie, H.-B.; Gildener-Leapman, N.; Li, J.; Srivastava, R.M.; Gibson, S.P.; Whiteside, T.L.; Ferris, R.L. Intratumoral Regulatory T Cells Upregulate Immunosuppressive Molecules in Head and Neck Cancer Patients. Br. J. Cancer 2013, 109, 2629–2635. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.-W.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Ma, S.-R.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. LAG-3 Confers Poor Prognosis and Its Blockade Reshapes Antitumor Response in Head and Neck Squamous Cell Carcinoma. Oncoimmunology 2016, 5, e1239005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesaro, Inc. A Phase 1 Dose Escalation and Cohort Expansion Study of TSR-033, an Anti-LAG-3 Monoclonal Antibody, Alone and in Combination with an Anti-PD-1 in Patients With Advanced Solid Tumors; Clinical trial registration NCT03250832; clinicaltrials.gov, 2022. Available online: http://clinicaltrials.gov/ct2/show/NCT03250832 (accessed on 13 May 2022).
- Nguyen, L.T.; Ohashi, P.S. Clinical Blockade of PD1 and LAG3--Potential Mechanisms of Action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, A.C.; Strand, G.L.; Nowicki, P.N.; Anderson, M.E.; Vermeer, P.D.; Klingelhutz, A.J.; Bossler, A.D.; Pottala, J.V.; Hendriks, W.; Lee, J.H. Impaired PTPN13 Phosphatase Activity in Spontaneous or HPV-Induced Squamous Cell Carcinomas Potentiates Oncogene Signaling through the MAP Kinase Pathway. Oncogene 2009, 28, 3960–3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, A.C.; Spanos, W.C.; Harris, G.F.; Anderson, M.E.; Klingelhutz, A.J.; Lee, J.H. The Role of Human Papillomavirus 16 E6 in Anchorage-Independent and Invasive Growth of Mouse Tonsil Epithelium. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanos, W.C.; Geiger, J.; Anderson, M.E.; Harris, G.F.; Bossler, A.D.; Smith, R.B.; Klingelhutz, A.J.; Lee, J.H. Deletion of the PDZ Motif of HPV16 E6 Preventing Immortalization and Anchorage-Independent Growth in Human Tonsil Epithelial Cells. Head Neck 2008, 30, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Uphoff, C.C.; Drexler, H.G. Comparative PCR Analysis for Detection of Mycoplasma Infections in Continuous Cell Lines. In Vitro Cell. Dev. Biol. Anim. 2002, 38, 79–85. [Google Scholar] [CrossRef]
- Powell, S.F.; Gold, K.A.; Gitau, M.M.; Sumey, C.J.; Lohr, M.M.; McGraw, S.C.; Nowak, R.K.; Jensen, A.W.; Blanchard, M.J.; Fischer, C.D.; et al. Safety and Efficacy of Pembrolizumab With Chemoradiotherapy in Locally Advanced Head and Neck Squamous Cell Carcinoma: A Phase IB Study. J. Clin. Oncol. 2020, 38, 2427–2437. [Google Scholar] [CrossRef]
- Donahue, R.N.; Lepone, L.M.; Grenga, I.; Jochems, C.; Fantini, M.; Madan, R.A.; Heery, C.R.; Gulley, J.L.; Schlom, J. Analyses of the Peripheral Immunome Following Multiple Administrations of Avelumab, a Human IgG1 Anti-PD-L1 Monoclonal Antibody. J. Immunother. Cancer 2017, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupar, R.; Hautmann, M.G.; Pathak, R.R.; Varier, I.; McLaren, C.; Gaag, D.; Hellerbrand, C.; Evert, M.; Laban, S.; Idel, C.; et al. Immunometabolic Determinants of Chemoradiotherapy Response and Survival in Head and Neck Squamous Cell Carcinoma. Am. J. Pathol. 2018, 188, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilones, K.A.; Vanpouille-Box, C.; Demaria, S. Combination of Radiotherapy and Immune Checkpoint Inhibitors. Semin. Radiat. Oncol. 2015, 25, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef]
- Wargo, J.A.; Reuben, A.; Cooper, Z.A.; Oh, K.S.; Sullivan, R.J. Immune Effects of Chemotherapy, Radiation, and Targeted Therapy and Opportunities for Combination With Immunotherapy. Semin. Oncol. 2015, 42, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and Clinical Activity of Pembrolizumab for Treatment of Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-012): An Open-Label, Multicentre, Phase 1b Trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Cohen, E.E.W. Pembrolizumab versus Methotrexate, Docetaxel, or Cetuximab for Recurrent or Metastatic Head-and-Neck Squamous Cell Carcinoma (KEYNOTE-040): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Meng, X.; Kong, L.; Mu, D.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor-Infiltrating Lymphocytes, Forkhead Box P3, Programmed Death Ligand-1, and Cytotoxic T Lymphocyte–Associated Antigen-4 Expressions before and after Neoadjuvant Chemoradiation in Rectal Cancer. Transl. Res. 2015, 166, 721–732.e1. [Google Scholar] [CrossRef]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, S.; Kim, S.S.; Pang, J.; Gold, K.A.; Gutkind, J.S.; Califano, J.A.; Mell, L.K.; Cohen, E.E.W.; Sharabi, A.B. Immune Modulation of Head and Neck Squamous Cell Carcinoma and the Tumor Microenvironment by Conventional Therapeutics. Clin. Cancer Res. 2019, 25, 4211–4223. [Google Scholar] [CrossRef] [Green Version]
- Sendo, S.; Saegusa, J.; Morinobu, A. Myeloid-Derived Suppressor Cells in Non-Neoplastic Inflamed Organs. Inflamm. Regen. 2018, 38, 19. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive Resistance to Anti-PD1 Therapy by Tim-3 Upregulation Is Mediated by the PI3K-Akt Pathway in Head and Neck Cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.Y.; Ferris, R.L.; Beck, J.T.; Harrington, K.; Haddad, R.I.; Bourhis, J.; Tahara, M.; Geraldes, M.; Nuyten, D.S.A.; Goldberg, Z.; et al. JAVELIN Head and Neck 100: A Phase 3 Trial of Avelumab in Combination with Chemoradiotherapy (CRT) vs CRT for 1st-Line Treatment of Locally Advanced Squamous Cell Carcinoma of the Head and Neck (LA SCCHN). J. Clin. Oncol. 2017, 35, TPS6093. [Google Scholar] [CrossRef]
- Machiels, J.P.H.; Licitra, L.; Rischin, D.; Waldron, J.; Burtness, B.; Gregoire, V.; Shekar, T.; Brown, H.M.; Cheng, J.D.; Siu, L.L. KEYNOTE-412: Pembrolizumab (Pembro) in Combination with Chemoradiation versus Chemoradiation Alone in Locally Advanced Head and Neck Squamous Cell Carcinoma (LA-HNSCC). J. Clin. Oncol. 2017, 35, TPS6090. [Google Scholar] [CrossRef]
- Cohen, E.E.; Ferris, R.L.; Psyrri, A.; Haddad, R.; Tahara, M.; Bourhis, J.; Harrington, K.J.; Chang, P.M.-H.; Lin, J.-C.; Razaq, M.; et al. 910O Primary Results of the Phase III JAVELIN Head & Neck 100 Trial: Avelumab plus Chemoradiotherapy (CRT) Followed by Avelumab Maintenance vs CRT in Patients with Locally Advanced Squamous Cell Carcinoma of the Head and Neck (LA SCCHN). Ann. Oncol. 2020, 31, S658. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total Patients |
|---|---|
| (N = 35) | |
| Median Age, Years | 61.6 |
| Range | (38–81) |
| Sex | |
| Male | 30 (85.7%) |
| Female | 5 (14.3%) |
| Race | |
| White, Non-Hispanic | 35 (100%) |
| Primary Site | |
| Oropharynx | 22 (62.8%) |
| Larynx | 9 (25.7%) |
| Hypopharynx | 4 (11.5%) |
| TNM Stage (AJCC 7th ed.) [35] | |
| III | 8 (22.8%) |
| IVA | 27 (77.2%) |
| T0–1 * | 8 (22.8%) |
| T2 | 11 (31.4%) |
| T3 | 11 (31.4%) |
| T4 | 5 (14.4%) |
| N0 | 3 (8.6%) |
| N1 | 4 (11.4%) |
| N2 | 28 (80.0%) |
| p16 (HPV) Status | |
| Positive | 21 (60.0%) |
| Negative | 14 (40.0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callejas-Valera, J.L.; Vermeer, D.W.; Lucido, C.T.; Williamson, C.; Killian, M.; Vermeer, P.D.; Spanos, W.C.; Powell, S.F. Characterization of the Immune Response to PD-1 Blockade during Chemoradiotherapy for Head and Neck Squamous Cell Carcinoma. Cancers 2022, 14, 2499. https://doi.org/10.3390/cancers14102499
Callejas-Valera JL, Vermeer DW, Lucido CT, Williamson C, Killian M, Vermeer PD, Spanos WC, Powell SF. Characterization of the Immune Response to PD-1 Blockade during Chemoradiotherapy for Head and Neck Squamous Cell Carcinoma. Cancers. 2022; 14(10):2499. https://doi.org/10.3390/cancers14102499
Chicago/Turabian StyleCallejas-Valera, Juan L., Daniel W. Vermeer, Christopher T. Lucido, Caitlin Williamson, Marisela Killian, Paola D. Vermeer, William C. Spanos, and Steven F. Powell. 2022. "Characterization of the Immune Response to PD-1 Blockade during Chemoradiotherapy for Head and Neck Squamous Cell Carcinoma" Cancers 14, no. 10: 2499. https://doi.org/10.3390/cancers14102499
APA StyleCallejas-Valera, J. L., Vermeer, D. W., Lucido, C. T., Williamson, C., Killian, M., Vermeer, P. D., Spanos, W. C., & Powell, S. F. (2022). Characterization of the Immune Response to PD-1 Blockade during Chemoradiotherapy for Head and Neck Squamous Cell Carcinoma. Cancers, 14(10), 2499. https://doi.org/10.3390/cancers14102499